
Synthesis of Methyl 2-((4R)-3-Acryloyl-4-phenyloxazolidin-2-yl)acetates

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
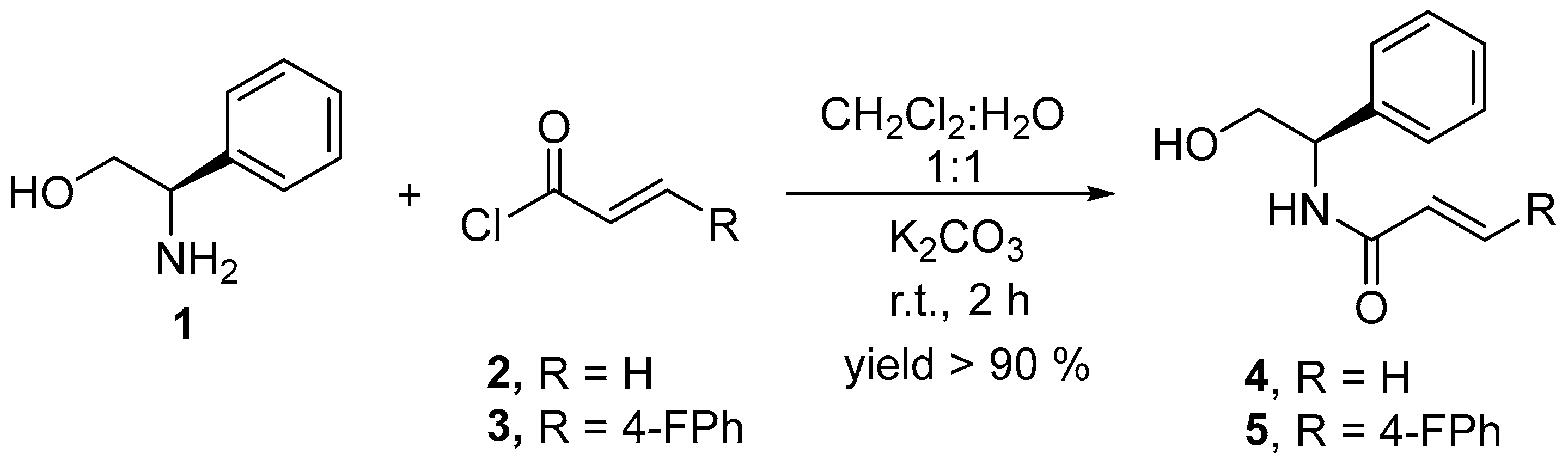
2.1. Synthesis of Chiral Acrylamides
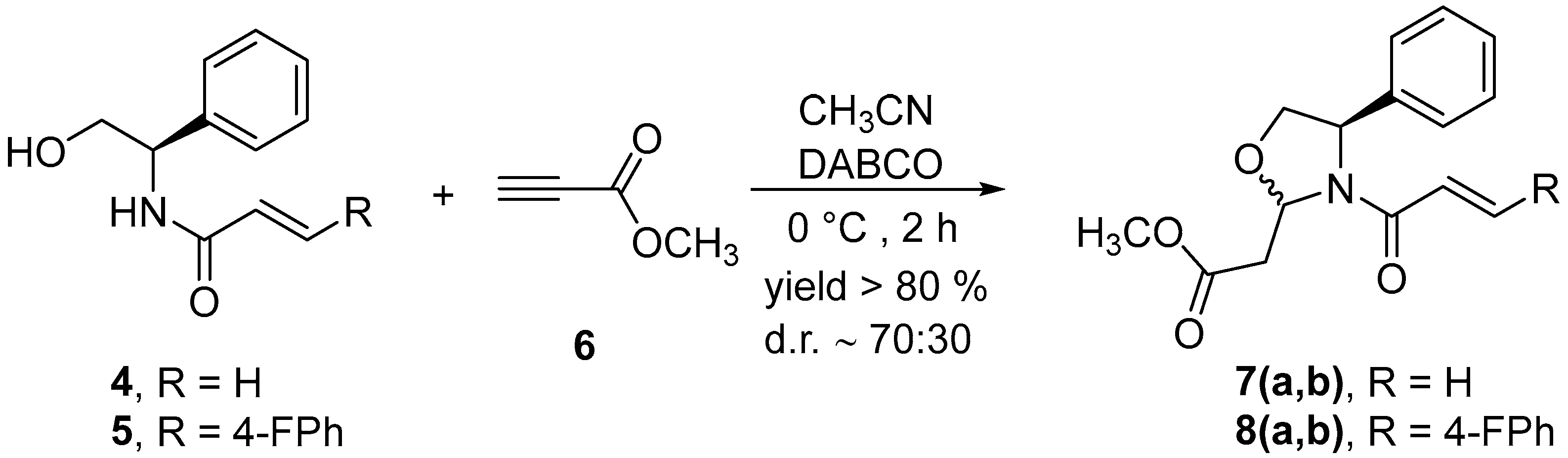
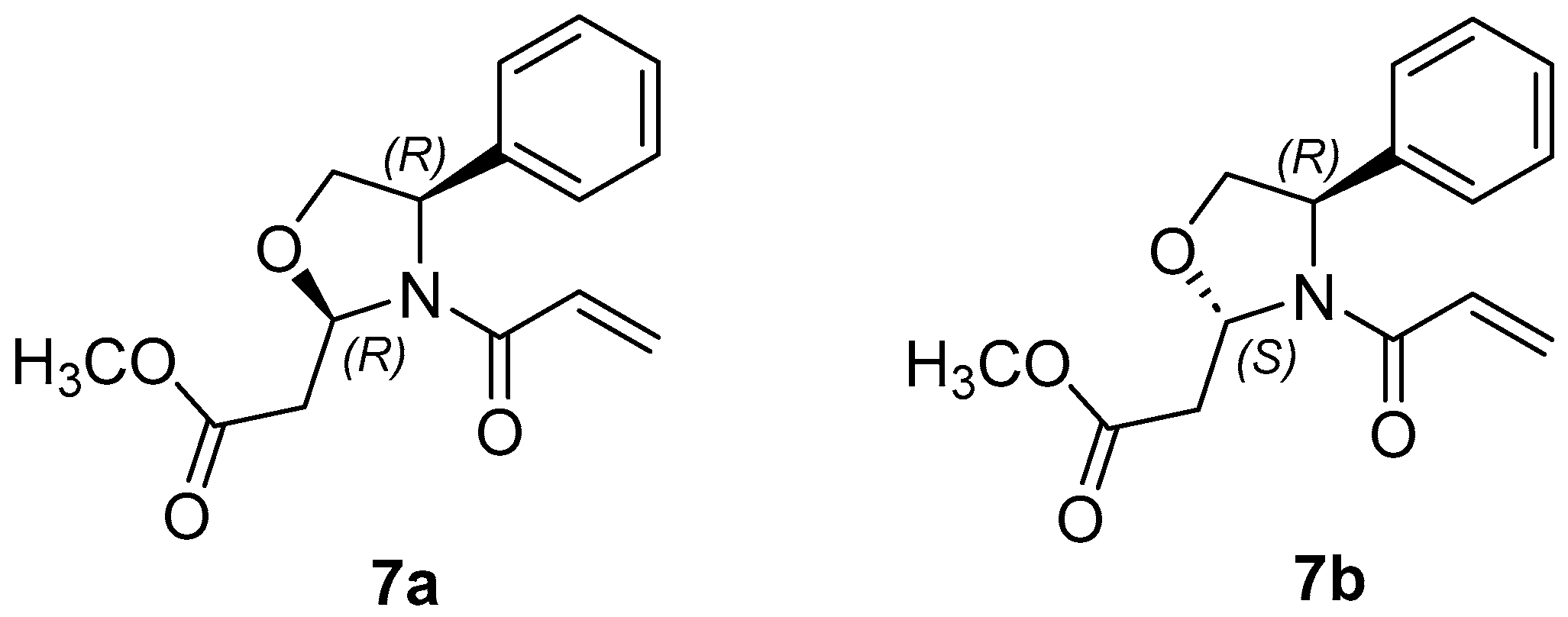
2.2. Synthesis of Methyl 2-(4R)-3-Acryloyl-4-phenyloxazolidin-2-yl)acetates, 7 and 8
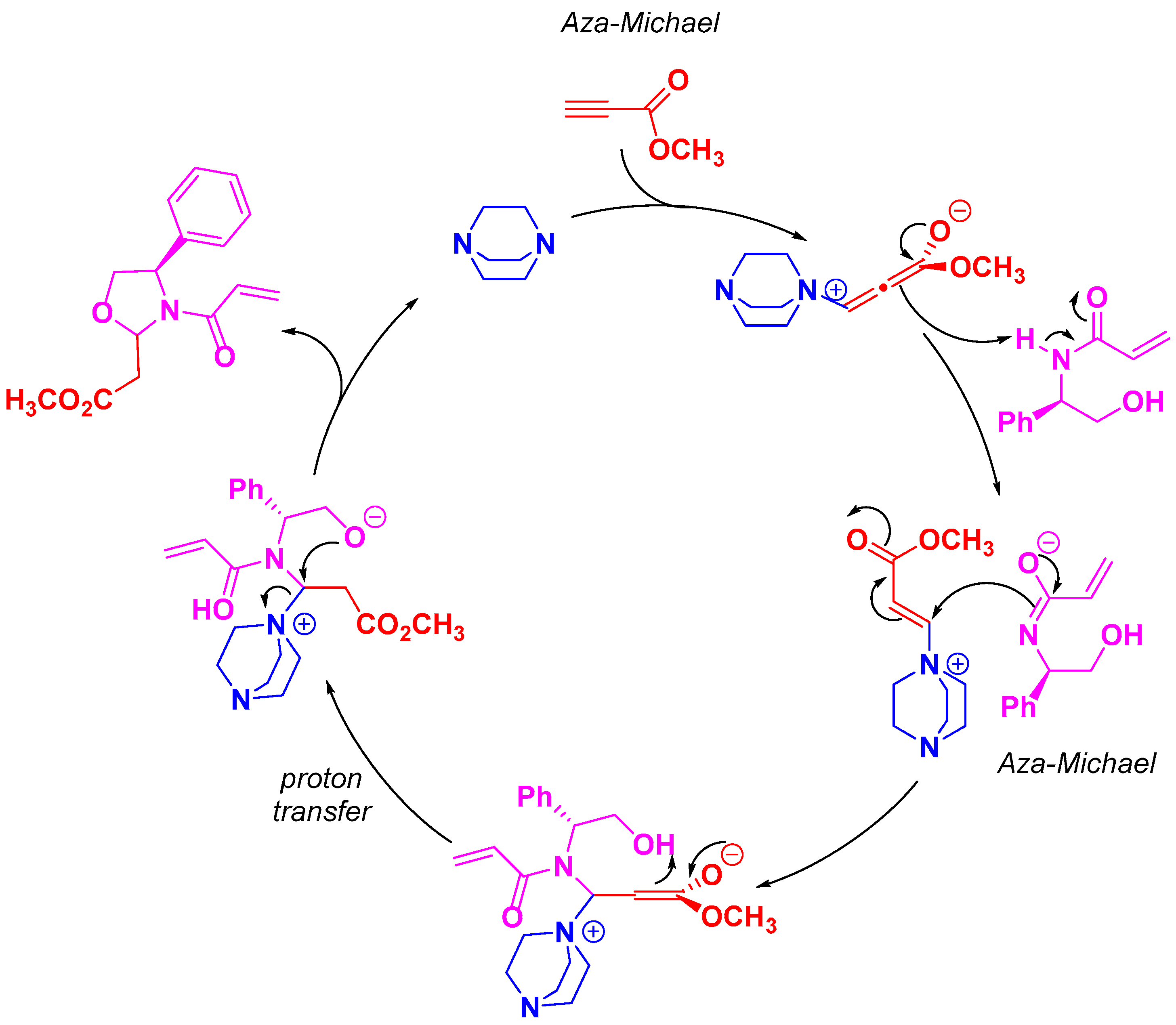
3. Discussion
4. Materials and Methods
4.1. General
4.2. Synthesis of Chiral Acrylamides
4.3. Synthesis of Methyl 2-(4R)-3-Acryloyl-4-phenyloxazolidin-2-yl)acetates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cordero, F.M.; Giomi, D.; Lascialfari, L. Five-Membered Ring Systems. With O and N Atoms. In Progress in Heterocyclic Chemistry; Elsevier Ltd.: Amsterdam, The Netherlands, 2013; Volume 25, pp. 291–317. [Google Scholar] [CrossRef]
- Qian, X.; Xu, X.; Li, Z.; Li, Z.; Song, G. Syntheses, Structures and Bioactivities of Fluorine-Containing Phenylimino-Thia(Oxa)Zolidine Derivatives as Agricultural Bioregulators. J. Fluor. Chem. 2004, 125, 1609–1620. [Google Scholar] [CrossRef]
- Morales-Monarca, G.-H.; Gnecco, D.; Terán, J.L. Diastereoselective Functionalization of Chiral N-Acyl-1,3-oxazolidines and Their Applications in the Synthesis of Bioactive Molecules. Eur. J. Org. Chem. 2022, 33. [Google Scholar] [CrossRef]
- Carbonnelle, A.-C.; Gotta, V.; Roussi, G. β-Amino alcohol-N-oxides as precursors of chiral oxazolidines: Synthesis of (R)-(-)-cryptostyline I. Heterocycles 1993, 36, 1763–1769. [Google Scholar] [CrossRef]
- Pytkowicz, J.; Stéphany, O.; Marinkovic, S.; Inagaki, S.; Brigaud, T. Straightforward Synthesis of Enantiopure (R)- and (S)-Trifluoroalaninol. Org. Biomol. Chem. 2010, 8, 4540–4542. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.F.; Wang, R.; Liu, L.; Da, C.S.; Yan, W.J.; Xu, Z.Q. Enantioselective Alkynylation of Aromatic Aldehydes Catalyzed by New Chiral Oxazolidine Ligands. Tetrahedron Lett. 2005, 46, 863–865. [Google Scholar] [CrossRef]
- Pichon-Barré, D.; Zhang, Z.; Cador, A.; Vives, T.; Roisnel, T.; Baslé, O.; Jarrige, L.; Cavallo, L.; Falivene, L.; Mauduit, M. Chiral Oxazolidines Acting as Transient Hydroxyalkyl-Functionalized N-Heterocyclic Carbenes: An Efficient Route to Air Stable Copper and Gold Complexes for Asymmetric Catalysis. Chem. Sci. 2022, 13, 8773–8780. [Google Scholar] [CrossRef] [PubMed]
- Khrapova, A.V.; Saroyants, L.V.; Yushin, M.Y.; Zukhairaeva, A.S.; Velikorodov, A.V. Prospects of Using Pharmacologically Active Compounds for the Creation of Antimycobacterial Drugs. Pharm. Chem. J. 2022, 55, 1108–1114. [Google Scholar] [CrossRef]
- Santos, R.V.C.; Cunha, E.G.C.; de Mello, G.S.V.; Rizzo, J.Â.; de Oliveira, J.F.; de Lima, M.D.C.A.; Pitta, I.D.R.; Pitta, M.G.D.R.; Rêgo, M.J.B.M. New Oxazolidines Inhibit the Secretion of Ifn-γ and Il-17 by Pbmcs from Moderate to Severe Asthmatic Patients. Med. Chem. 2021, 17, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, E.D. The Oxazolidines. Chem. Rev. 1953, 53, 309–352. Available online: https://pubs.acs.org/doi/pdf/10.1021/cr60165a005?casa_token=c5gTUV5TSy0AAAAA:bJvrnoQca-SUnvzNdfG9X9fOpZLUAFQehdKNJiUWRpCWP2uqbDa3cPfzhH6toVcZawywHXc9T4n5_ctv_Q (accessed on 17 July 2024). [CrossRef]
- Reyes-Bravo, E.; Gnecco, D.; Juárez, J.R.; Orea, M.L.; Bernès, S.; Aparicio, D.M.; Terán, J.L. Diastereoselective Synthesis of New Zwitterionic Bicyclic Lactams, Scaffolds for Construction of 2-Substituted-4-Hydroxy Piperidine and Its Pipecolic Acid Derivatives. RSC Adv. 2022, 12, 4187–4190. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Buzzetti, L.; Puriņš, M.; Waser, J. Palladium-Catalyzed Trans-Hydroalkoxylation: Counterintuitive Use of an Aryl Iodide Additive to Promote C-H Bond Formation. ACS Catal. 2022, 12, 7565–7570. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhang, Y.; Zhang, Z.; Chen, F.; Huang, L. Copper-Catalyzed Annulation/A 3 -Coupling Cascade: Diastereodivergent Synthesis of Sterically Hindered Monocyclic Oxazolidines Bearing Multiple Stereocenters. Eur. J. Org. Chem. 2019, 2019, 1931–1939. [Google Scholar] [CrossRef]
- Agami, C.; Dechoux, L.; Hebbe, S. Asymmetric Synthesis of Nitrogen Heterocycles by Reaction of Chiral β-Enaminocarbonyl Substrates with Acrylate Derivatives. Tetrahedron Lett. 2002, 43, 2521–2523. [Google Scholar] [CrossRef]
- Aparicio, D.M.; Gnecco, D.; Juárez, J.R.; Orea, M.L.; Mendoza, A.; Waksman, N.; Salazar, R.; Fores-Alamo, M.; Terán, J.L. Diastereoselective Synthesis of Aryl and Alkyl Trans-Glycidic Amides from Pseudoephedrine-Derived Sulfonium Salt. Chemospecific Exo-Tet Ring Closure for Morpholin-3-Ones. Tetrahedron 2012, 68, 10252–10256. [Google Scholar] [CrossRef]
- Mola, L.; Font, J.; Bosch, L.; Caner, J.; Costa, A.M.; Etxebarría-Jardí, G.; Pineda, O.; De Vicente, D.; Vilarrasa, J. Nucleophile-Catalyzed Additions to Activated Triple Bonds. Protection of Lactams, Imides, and Nucleosides with MocVinyl and Related Groups. J. Org. Chem. 2013, 78, 5832–5842. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, D.; López-Tosco, S.; Cruz-Acosta, F.; Méndez-Abt, G.; García-Tellado, F. Acetylides from Alkyl Propiolates as Building Blocks for C3 Homologation. Angew. Chem. In. Ed. 2009, 48, 2090–2098. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| entry | Base | Yield 7(a,b) | d.r. 7(a,b) |
|---|---|---|---|
| 1 | DABCO (10 mol%) | 86% | 74:26 1 |
| 2 | DMAP (1 eq.) | not observed | ----- |
| 3 | DBU (10 mol%) | not observed | ----- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilotzi Xahuentitla, H.; Ortega Montes, J.G.; Gnecco Medina, D.H.; Terán Vázquez, J.L.; Hernández Núñez, E.; Orea Flores, M.L. Synthesis of Methyl 2-((4R)-3-Acryloyl-4-phenyloxazolidin-2-yl)acetates. Molbank 2024, 2024, M1903. https://doi.org/10.3390/M1903
Pilotzi Xahuentitla H, Ortega Montes JG, Gnecco Medina DH, Terán Vázquez JL, Hernández Núñez E, Orea Flores ML. Synthesis of Methyl 2-((4R)-3-Acryloyl-4-phenyloxazolidin-2-yl)acetates. Molbank. 2024; 2024(4):M1903. https://doi.org/10.3390/M1903
Chicago/Turabian StylePilotzi Xahuentitla, Hugo, Jesús Guadalupe Ortega Montes, Dino Hernán Gnecco Medina, Joel Luis Terán Vázquez, Emanuel Hernández Núñez, and Maria Laura Orea Flores. 2024. "Synthesis of Methyl 2-((4R)-3-Acryloyl-4-phenyloxazolidin-2-yl)acetates" Molbank 2024, no. 4: M1903. https://doi.org/10.3390/M1903
APA StylePilotzi Xahuentitla, H., Ortega Montes, J. G., Gnecco Medina, D. H., Terán Vázquez, J. L., Hernández Núñez, E., & Orea Flores, M. L. (2024). Synthesis of Methyl 2-((4R)-3-Acryloyl-4-phenyloxazolidin-2-yl)acetates. Molbank, 2024(4), M1903. https://doi.org/10.3390/M1903