Abstract
Isoetin derivatives are a rare class of flavonoids with a rather erratic occurrence across the plant kingdom. The Cichorieae tribe of the Asteraceae family has proven to be a rich source and a centre of chemical diversity of this class of compounds. Here, we describe the chromatographic isolation and mainly NMR-based structure elucidation of a previously undescribed isoetin derivative from Leontodon hispidus L. (Asteraceae, Cichorieae). The chemophenetic relevance is discussed briefly.
1. Introduction
The flavonoid isoetin was first described in 1974 from species of the genus Isoetes (Isoetaceae, Isoetales, Lycopodiopsida) [1]. Since then, surprisingly few derivatives of this flavonoid have been found, all in all totalling 14 derivatives and an additional 14 compounds when methyl ethers of isoetin are included [1]. The currently known natural derivatives of isoetin feature the following substituents: α-arabinose, α-glucose, β-glucose, β-glucuronic acid, β-xylose, 4′-O-acetylxylose, O-methyl ethers, and p-coumaroyl moieties [1]. HPLC-MS studies have revealed the existence of additional isoetin derivatives in leaves of Leontodon hispidus L. Here, we describe the isolation and structure elucidation of one of these compounds, representing a previously undescribed natural product.
2. Results
2.1. Isolation of Compound 1
In polar extracts of L. hispidus leaves, 28 (!) putative isoetin derivatives were detected using UHPLC-HRMS (data not shown). Thus, more isoetin derivatives seem to be occurring in L. hispidus than have been described previously [1].
One of these compounds (Figure 1) with a molecular formula (and exact mass of the [M-H]− ion) of C26H28O16 (m/z = 595.1310) 1 was isolated. Isolation of this compound was accomplished using successive extraction with solvents of different polarities, liquid–liquid partitioning, Sephadex LH-20 column chromatography, and semi-preparative reversed-phase HPLC.
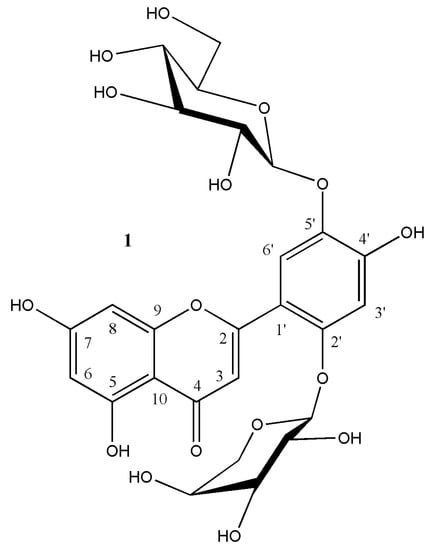
Figure 1.
Isoetin 2′-O-α-l-arabinopyranoside-5′-O-β-d-glucopyranoside 1 from Leontodon hispidus.
2.2. Structure Elucidation of Compound 1
Compound 1 displayed NMR data (Table 1) and mass data (see above) congruent with those of an isoetin O-hexoside-O-pentoside. HMBC cross peaks form the anomeric protons of the sugar moieties to the isoetin moiety, indicating substitution in positions O-2′/O-4′ and O-5′. A combination of 2D NMR experiments (HH-COSY, HSQC, HSQC-TOCSY, and HMBC) enabled us to assign individual proton and carbon signals to the two sugar moieties.

Table 1.
1H NMR and 13C NMR data of isoetin derivative 1.
Signals for the sugar moiety attached in position O-2′/O-4′ were congruent with an arabinopyranose moiety, while signals of the sugar moiety attached in position O-5′ were indicating a glucopyranose moiety. Positions O-2′/O-4′ were not distinguishable by HMBC spectroscopy (Figure 2), because both C-2′ and C-4′ had the same distance from the protons in positions C-3′ and C-6′: two bonds and three bonds, respectively.
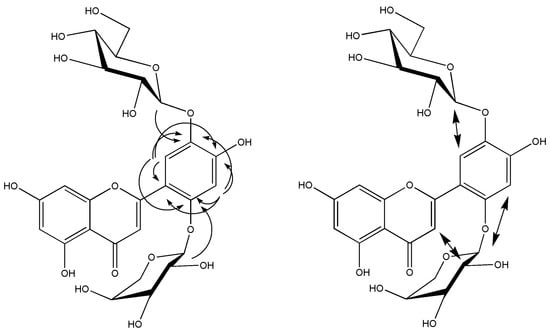
Figure 2.
Relevant HMBC (left) and ROESY (right) signals observed for isoetin 2′-O-α-l-arabinopyranoside-5′-O-β-d-glucopyranoside.
A ROESY (Figure 2) experiment showed strong cross peaks between the signals assignable to H-3′ of the isoetin moiety and the anomeric proton of the arabinose moiety and between the signals assignable to H-6′ of the isoetin moiety and the anomeric proton of the glucose moiety. Additionally, a weak cross peak between the signals assignable to H-3 of the isoetin moiety and H-2″ of the arabinose moiety was detected, thus indicating that the arabinose moiety was attached via the O-2′ of the isoetin moiety.
To prove the identity of the sugar moiety and to distinguish between d- and l-arabinose and d- and l-glucose, respectively, we used gas chromatography after hydrolysis and octylation employing the method described in detail previously [2]. As standard reference compounds, we used d-arabinose and l-arabinose (both from Sigma Aldrich, St. Louis, MO, USA), d-xylose (Merck), l-xylose (Sigma Aldrich), d-glucose (Merck), and l-glucose (Sigma). Retention times and relative intensities of the obtained signals for the hydrolysis products of compound 1 were congruent with the ones obtained for l-arabinose and d-glucose. Conclusively, compound 1 was identified as isoetin 2′-O-α-l-arabinopyranoside-5′-O-β-d-glucopyranoside, a previously undescribed isoetin derivative.
3. Discussion
The detection of a whole series of isoetin derivatives in the leaves of L. hispidus shows that many more derivatives of this rather rare flavonoid exist in the plant kingdom. Using the currently available detection and separation capabilities, all previously reported sources of isoetin might serve as starting points for a dedicated search for these natural compounds. Indeed, in the early literature, the structures of flavonoid glycosides after hydrolysis were often only partially elucidated, and the flavonoid aglycone and the sugar moieties were separately identified.
We will in the near future focus our attention on new isoetin derivatives from the Cichorieae tribe of the Asteraceae family, and in particular on the genera Hypochaeris and Leontodon.
4. Materials and Methods
4.1. Plant Material
Leaves of L. hispidus were collected in July 2018 in a meadow (local vernacular name “desert”) above Rino di Sonico, Provincia di Brescia, Lombardia, Italy; N 46°09′11.0″, E 10°21′30.0″; 740 m a.m.s.l. Plant identities were determined using the latest edition of Flora d’Italia [3,4].
Voucher specimens are preserved in the private herbarium of CZ (CZ-20180726A-2) and of the herbarium of Kiel University (KIEL0005016).
4.2. Plant Material
LC-MS-grade formic acid was purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). LC-MS-grade MeCN, n-hexane, H2O, gradient-grade MeOH, and other (analytical-grade) solvents were obtained from VWR International GmbH (Darmstadt, Germany). The water used for isolation was doubly distilled in-house. DMSO-d6 for NMR spectroscopy was purchased from Euriso-top GmbH (Saarbrücken, Germany).
4.3. Isolation of Isoetin Derivative 1
Finely ground leaves of L. hispidus (184 g) were pre-extracted with ethyl acetate to remove apolar compounds. This step was performed using maceration with 500 mL of ethyl acetate five times and for 24 h for each maceration step and yielded 7.65 g of crude extract after evaporation in vacuo. The remaining plant material was re-macerated with 500 mL of a mixture of methanol–acetone–water (3/1/1, v/v/v) again for 24 h for each maceration step and five times. The crude extract obtained after evaporation in vacuo (18.6 g) was ultra-sonicated with a 200 mL mixture of hexane, ethyl acetate, methanol, and water (1/1/1/1, v/v/v/v). After separation of the water and the organic phase, the water phase was washed for three times with a 100 mL mixture of hexane and ethyl acetate (1/1, v/v) to completely remove remaining traces of chlorophyll. The remaining water phase after evaporation (17.2 g) was re-dissolved in 400 mL of a mixture of n-butanol and water (3/1, v/v). After phase separation, the water phase was again partitioned with 100 mL of n-butanol three consecutive times. After evaporation in vacuo, this resulted in 7.41 g of n-butanol layer and 9.74 g of water layer.
Compound 1 was contained in the n-butanol layer. Compound 1 was further enriched with Sephadex LH-20 column chromatography (column length: 100 cm, column diameter: 3 cm) using methanol as an eluent. Collective fractions 5 (517 mg) contained compound 1. Compound 1 was further enriched with Sephadex column chromatography (column length: 150 cm, column diameter: 2 cm) using a mixture of methanol–acetone–water (3/1/1, v/v/v) as the eluent. The enriched fraction containing compound 1 (120 mg) was then dissolved in 200 µL of dimethyl sulfoxide and finally purified with semi-preparative HPLC using a Waters e2695 module with an Alliance 2998 photodiode array detector and a WFC III fraction collector. The column used was a Phenomenex Aqua 5 µm 125 Å column (250 mm × 10 mm). The flow rate was set to 3.0 mL, the detection wavelength to 350 nm, and compound 1 was purified using an isocratic mixture of 85% water with 0.1% formic acid (solvent A) and 15% acetonitrile (solvent B) to obtain an enriched fraction of 18.9 mg, for which a second semi-preparative HPLC isolation step was performed, and using an isocratic mixture with 10% solvent B, 8.2 mg of pure compound 1 was isolated.
4.4. UHPLC-HRMS
Separation was achieved on a reversed-phase column Waters Cortecs C18 (2.7 µm, 2.1 × 100 mm) column maintained at 40 °C. The binary mobile phase consisted of A. 0.1% formic acid in water and B. 0.1% formic acid in acetonitrile. The run time was 33 min. The following gradient was utilized: the mobile phase was held at 5% B for 1 min, gradually increased to 30% B over 19 min, increased gradually to 50% B over 5 min, increased gradually to 70% B over 5 min, and finally increased gradually to 95% over 3 min. The system was then adjusted to the initial condition of 5% B and equilibrated over 4 min. The flow rate and the injection volume were set to 300 µL/min and 1 µL, respectively. The effluents were connected on-line with a Q Exactive Plus Orbitrap Mass Spectrometer, with which the compounds were detected.
Mass analyses were carried out on a Q Exactive Plus Mass Spectrometer (ThermoFisher Scientific, Inc., Waltham, MA, USA) equipped with a heated electrospray ionization (HESI-II) probe (ThermoScientific). The tune parameters were as follows: spray voltage −2.5 kV; sheath gas flow rate 38; auxiliary gas flow rate 12; spare gas flow rate 0; capillary temperature 320 °C; probe heater temperature 320 °C; and S-lens RF level 50. Acquisition was achieved in full-scan MS and data-dependent MS2 modes. Full-scan spectra over the m/z range of 100 to 1500 were acquired in negative ionization mode at a resolution of 70,000. Other instrument parameters for full MS mode were set as follows: automatic gain control (AGC) target 3e6, maximum injection time (IT) 100 ms, number of scan ranges 1. For DD-MS2 mode, instrument parameters were as follows: microscans 1, resolution 17,500, AGC target 1e5, maximum IT 50 ms, MSX count 1, Top5, isolation window 2.0 m/z, stepped normalized collision energy (NCE) 10, 20, and 60 eV. Data acquisition and processing were carried out with Xcalibur 4.0 software (ThermoScientific).
4.5. NMR Spectroscopy
One-dimensional (1H, 13C) and two-dimensional (HSQC, HSQC-TOCSY, HMBC, COSY, NOESY, ROESY) NMR spectra (see Supplementary Materials) were recorded on a Bruker Avance III 400 NMR spectrometer operating at 400 MHz for the proton channel and 100 MHz for the 13C channel with a 5 mm PABBO broadband probe with a z-gradient unit at 298 K (Bruker BioSpin GmbH, Rheinstetten, Germany).
5. Conclusions
As already discussed elsewhere [5], even the comparatively well-investigated European flora still proves to be a rich source of undescribed and potentially bioactive natural products. L. hispidus, the title species of the current account, also yielded hypocretenolides [6], a structurally well-defined and chemophenetically very restrictedly occurring [7] class of sesquiterpene lactones with pronounced bioactivity [8,9]. Isoetin has an overall rare occurrence within the plant kingdom, however, in phylogenetically very distant groups. This seems to make them a priori very bad chemophenetic markers. However, when zooming in on the groups of occurrence, such as the Cichorieae tribe, some interesting patterns still emerge. For example, within the genus Hieracium s.l., isoetin derivatives are the most indicative marker compound for distinguishing between the genera/subgenera Hieracium (traces or absence of isoetin 4′-O-glucuronide) and Pilosella (isoetin 4′-O-glucuronide, one of the main flavonoids in flowering heads of many taxa) [10].
Supplementary Materials
The following supporting information can be downloaded online, Figure S1. HRMS/MS spectrum of compound 1; Figure S2. 1H NMR spectrum of compound 1; Figure S3. 13CNMR spectrum of compound 1; Figure S4. COSY spectrum of compound 1; Figure S5. HSQC spectrum of compound 1; Figure S6. HMBC spectrum of compound 1; Figure S7. HSQC-TOCSY spectrum of compound 1; Figure S8. ROESY spectrum of compound 1.
Author Contributions
Conceptualization, C.Z.; methodology, C.Z. and D.Z.-D.; formal analysis, C.Z.; investigation, F.F., D.Z.-D. and M.G.P.; resources, C.Z. and D.Z.-D.; data curation, C.Z., D.Z.-D., F.F. and M.G.P.; writing—original draft preparation, C.Z.; writing—review and editing, C.Z.; visualization, C.Z.; supervision, C.Z. and M.G.P.; project administration, C.Z.; funding acquisition, C.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
The authors wish to thank Ulrich Girreser and his team for their expert NMR support, Serhat Sezai Çiçek for fruitful discussions, and Moira Madeo for her help in collecting the plant material as well as for the permit to harvest L. hispidus on her property.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zidorn, C. Isoetin and its derivatives: Analytics, chemosystematics, and bioactivities. Biochem. Syst. Ecol. 2015, 61, 402–412. [Google Scholar] [CrossRef]
- Uddin, M.J.; Çiçek, S.S.; Willer, J.; Shulha, O.; Abdalla, M.A.; Sönnichsen, F.; Girreser, U.; Zidorn, C. Phenylpropanoid glycosides and flavonoid glycosides from the leaves of Clerodendrum infortunatum (Lamiaceae). Biochem. Syst. Ecol. 2020, 92, 104131. [Google Scholar] [CrossRef]
- Zidorn, C. Leontodon; Flora d’Italia, 2nd ed.; Pignatti, S., Ed.; Edagricole: Bologna, Italy, 2018; Volume 3, pp. 1063–1070. [Google Scholar]
- Zidorn, C. Leontodon; Flora d’Italia, 2nd ed.; Pignatti, S., Ed.; Edagricole: Bologna, Italy, 2019; Volume 4, pp. 899–901. [Google Scholar]
- Zidorn, C. Bioprospecting of plant natural products in Schleswig-Holstein (Germany) I: Chemodiversity of the Cichorieae tribe (Asteraceae) in Schleswig-Holstein. Phytochem. Rev. 2019, 18, 1223–1253. [Google Scholar] [CrossRef]
- Zidorn, C.; Ellmerer-Müller, E.P.; Stuppner, H. Guaian-5,12-olides from Leontodon hispidus. Phytochemistry 1998, 49, 797–800. [Google Scholar] [CrossRef]
- Enke, N.; Gemeinholzer, B.; Zidorn, C. Molecular and phytochemical systematics of the subtribe Hypochaeridinae (Asteraceae, Cichorieae). Org. Divers. Evol. 2012, 12, 1–16. [Google Scholar] [CrossRef]
- Zidorn, C.; Stuppner, H.; Tiefenthaler, M.; Konwalinka, G. Cytotoxic activities of hypocretenolides from Leontodon hispidus. J. Nat. Prod. 1999, 62, 984–987. [Google Scholar] [CrossRef] [PubMed]
- Zidorn, C.; Dirsch, V.; Rüngeler, P.; Sosa, S.; Della Loggia, R.; Merfort, I.; Pahl, H.L.; Vollmar, A.; Stuppner, H. Anti-inflammatory activities of hypocretenolides from Leontodon hispidus. Planta Med. 1999, 65, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Zidorn, C.; Gottschlich, G.; Stuppner, H. Chemosystematic investigations on phenolics from flowerheads of Central European taxa of Hieracium (Asteraceae). Plant Syst. Evol. 2002, 231, 39–58. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).