Abstract
Enantiomers (2S, 4S)- and (2R, 4R)-3-(naphthalene-1-ylmethyl) pentane-2,4-diols were synthesized by the reduction of (Z)-4-hydroxy-3-(naphthalene-1-ylmethyl) pent-3-en-2-one with NaBH4 in methanol (MeOH). Crystallization in dichloromethane of this racemic mixture led to simple crystals with a crystalline habit with similar morphologies; however, in a group of them, it was possible to find a barely observable difference that allowed determining a crystal structure for each of the enantiomers, the 2S,4S, and the 2R,4R.
1. Introduction
Compounds containing the β-diketone function play an essential role in nature, being part of the structure of metabolites such as curcumin, a molecule to which countless biological properties are attributed, among which the anti-cancer activity stands out [1,2,3,4,5]. Through condensation with aldehydes, β-diketones are valuable synthetic intermediates that serve as building blocks for the synthesis of derivatives such as benzylidenes that show activities against breast cancer (MCF-7) and leukemia (K562), among others [6]. Furthermore, reducing the β-diketone function leads to 1,3–diols furnishing relevant synthetic scaffolds and providing a basic skeleton of the remarkable molecules v.gr. Rifamycin B (used for the treatment of HIV), Rosuvastatin (used to prevent cardiovascular diseases), and Compactin (used as a hypolipidemic agent) [7].
However, the 1,3-diols obtained after the reduction of the β-diketone function are isomeric mixtures whose separation can be a complex but necessary task since the convenience of making available optically pure compounds is well recognized [8]. In this regard, synthesizing aliphatic molecules of the 1,3-glycol type and its derivatives and separating their stereoisomers is a recurring problem. For this reason, one of the strategies used is chemical derivation to facilitate their separation [9]. Itoh et al. described the separation of various 1,1-Bis(1-Hydroxyalkyl)cyclopropanes via the formation of 1,3-dioxane derivatives by acetalization of the diols. In addition, Bianchini et al. described the obtention of a new optically pure compound (R)-(R)-3-benzyl-2,4-bis-(diphenylphosphino)pentane [10]. It should be noted that the obtention of this compound was achieved by prior synthesis of the molecule (S)-(S)-PhCH2CH(CH(OH)CH3)2 by enantioselective reduction of 3-benzyl-2,4-pentanedione with [((S)-BINAP)Ru(p-cymene)Cl]Cl. Chemoenzymatic transformation is another recently used method for high-yield preparations of enantiomeric compounds. For instance, Bäckvall et al. reported the use of Candida Antarctica lipase B to transform α-substituted β-hydroxy ketones with yields up to 95% with good diastereoisomeric proportions. The direct crystallization of enantiomers from a racemic mixture is a method that receives constant attention. However, reports of successful cases of the efficient crystallization of enantiomers are scarce. Thus, trial and error remain a common practice in this field. [11] Thus, in this work, we describe the spontaneous resolution of 1,3-diols by crystallizing their enantiomers (R, R, and S, S). A discussion of the supramolecular arrangement and their Hirshfeld plots is carried out regarding their crystallographic characteristics.
2. Results and Discussion
2.1. X-ray Diffraction
The compounds 4a and 4b were synthesized by reduction with NaBH4 in MeOH of 3 at 0 °C (Scheme 1) [12,13]. As indicated in Scheme 1, the reduction reaction gave rise to the formation of compounds 4a and 4b; however, they were not the only species obtained. Two other diastereomers corresponding to the 1,3-diol meso compound with the relative configuration 2S, 3r, 4R (2S, 3s, 4R), and additionally, a hydroxy ketone resulting from the reduction of a single carbonyl carbon, with a relative configuration syn or anti were obtained.
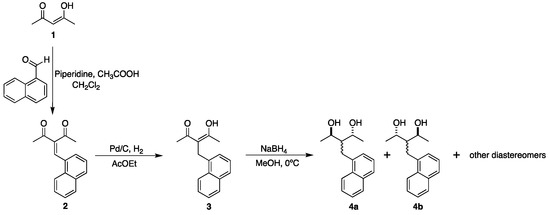
Scheme 1.
Synthesis of enantiomers 4a and 4b.
A portion of the diastereomeric mixture (racemate 4a/4b, 1,3-diol meso, and hydroxy ketone) was chromatographed using a ternary mixture of hexane-dichloromethane-methanol (Hex-CH2Cl2-MeOH) in a 60:35:5 ratio. The fraction containing 4a/4b spontaneously afforded the corresponding monocrystal. This was possible despite being a racemate and using a non-chiral and common organic solvent such as dichloromethane. The crystal structure of compounds 4a and 4b was carried out in single-crystal X-ray diffraction analysis (Figure 1). The crystallographic data are shown in Table 1, demonstrating that both enantiomers had similar cell parameters and crystallized in the orthorhombic P212121 space group (see Supplementary Materials). The principal intramolecular interaction observed in both enantiomers is the hydrogen bond (Figure 2) with an OH···O distance of 1.881 and 1.915 Å for 4a and 4b, respectively.
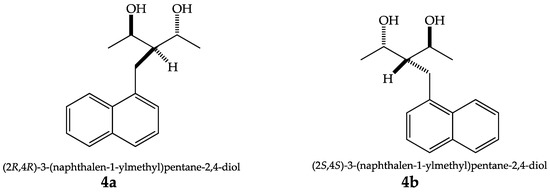
Figure 1.
Absolute configuration of compounds 4a and 4b.

Table 1.
Crystallographic data of compounds 4a and 4b.
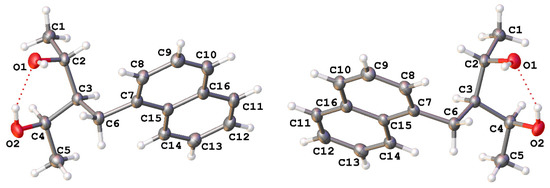
Figure 2.
Crystal structure of compounds 4a (left) and 4b (right). Displacement ellipsoids are drawn at a 50% probability level. The intramolecular interactions are shown as dashed red lines.
To understand the underlying intermolecular interactions that stabilized the crystal, we further examined its crystal structure, revealing two important intermolecular bondings; an OH···H and a C-H···π interaction was observed in both compounds, as reported in Table 2 and Table 3. It should be emphasized that a closer look at the crystal structures presented the same molecular and supramolecular arrangements; for this reason, we only describe 4a. The first interaction was (O2···H1-O1); this was different from the intramolecular hydrogen bonding mentioned above in which both hydroxyl groups acted as acceptor and donor, forming intermolecular interactions that extended in one direction (Figure 3a). The second interaction was from naphthalene–naphthalene interactions (C9-H9···Cg, 2.856 Å), as depicted in Figure 3b. All intermolecular distances and angles were within the expected range reported previously [14,15,16].
Table 2.
Intermolecular interactions in compound 4a.
Table 3.
Intermolecular interactions in compound 4b.
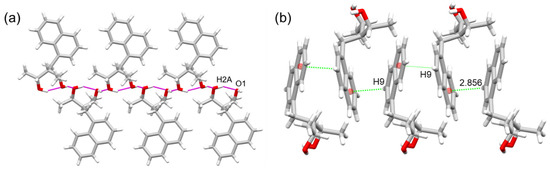
Figure 3.
Intermolecular interactions of compound 4a; (a) presence of OH··· and (b) CH···π-type interactions.
2.2. Hirshfeld Surface
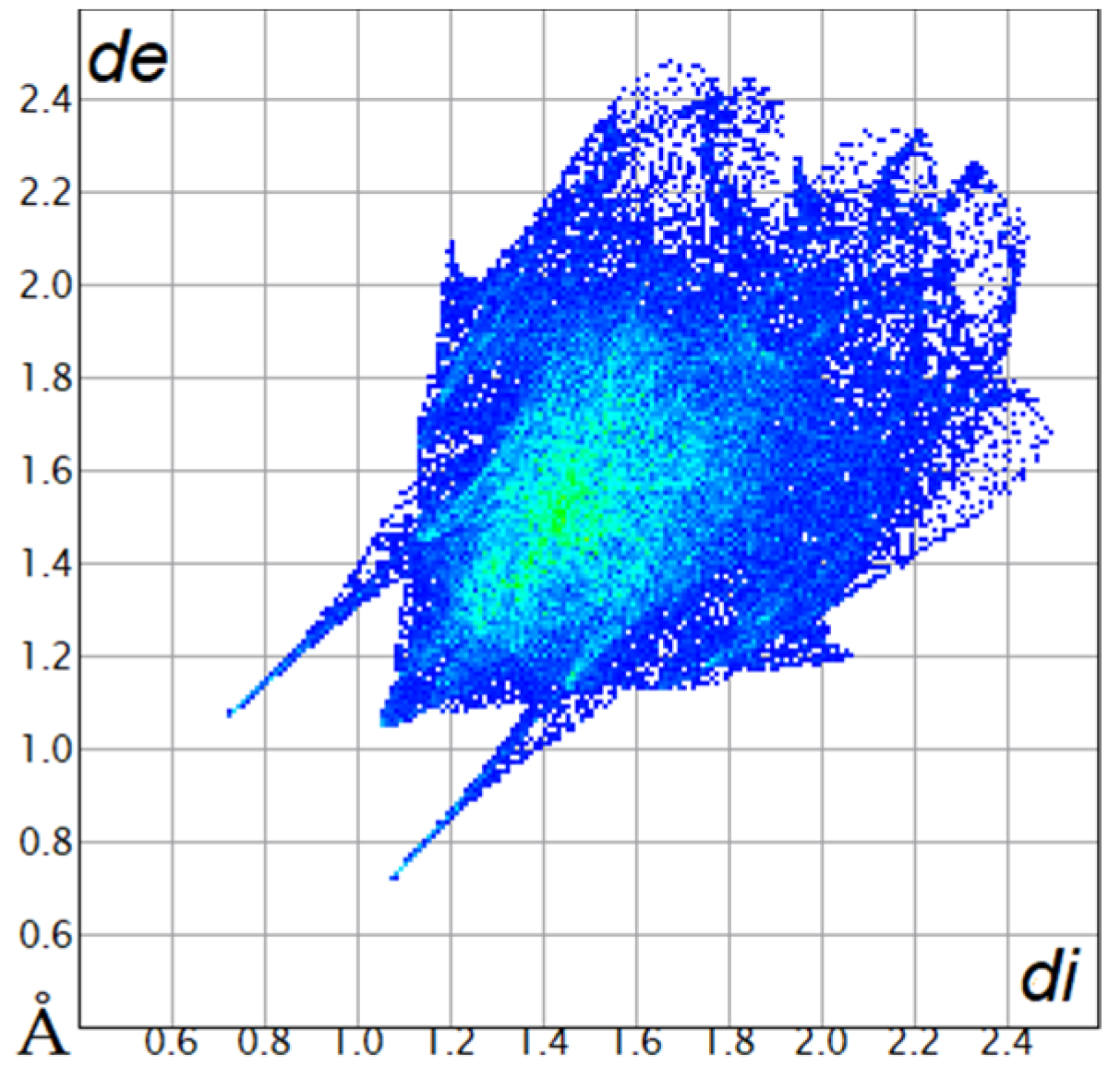
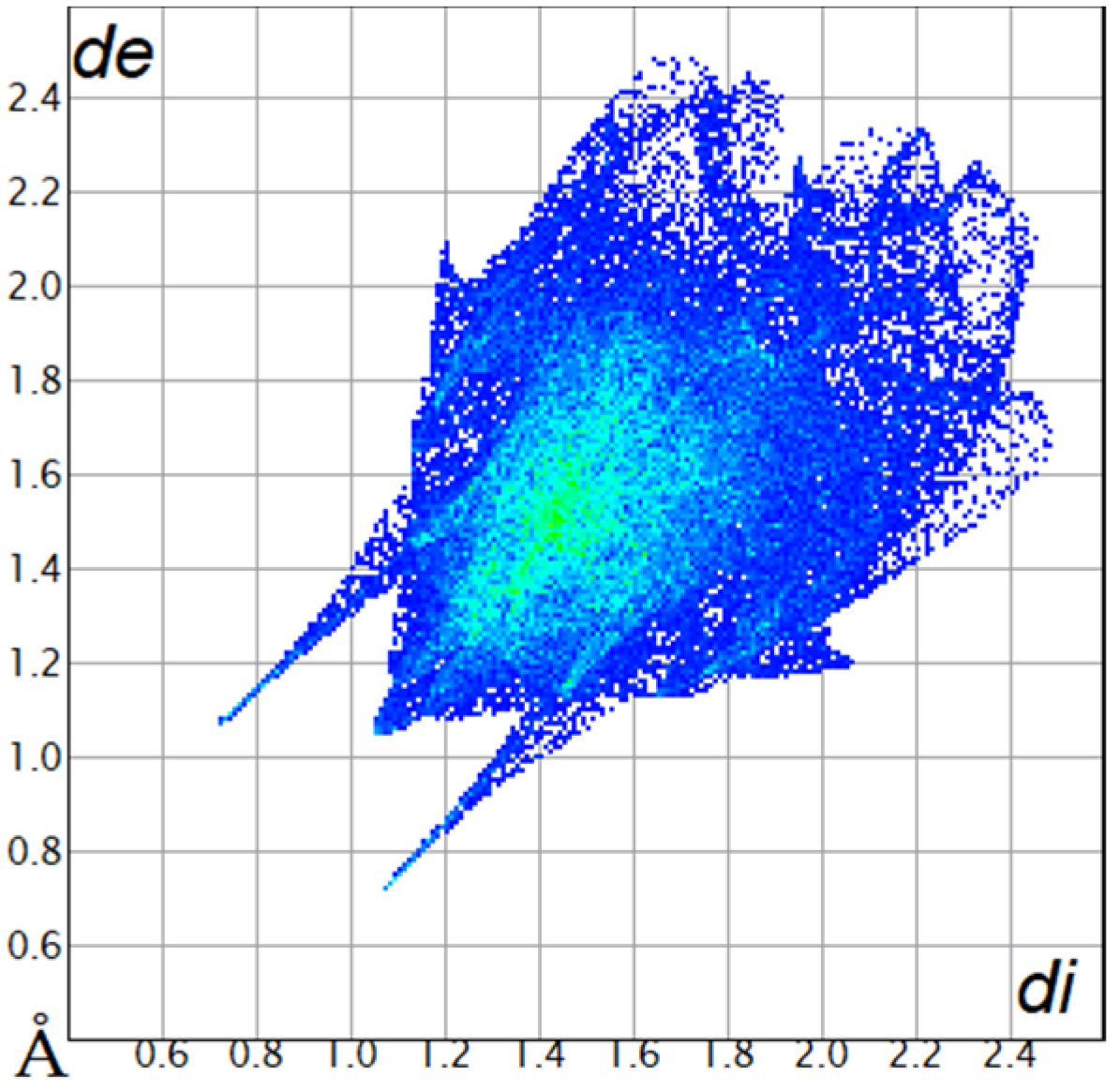
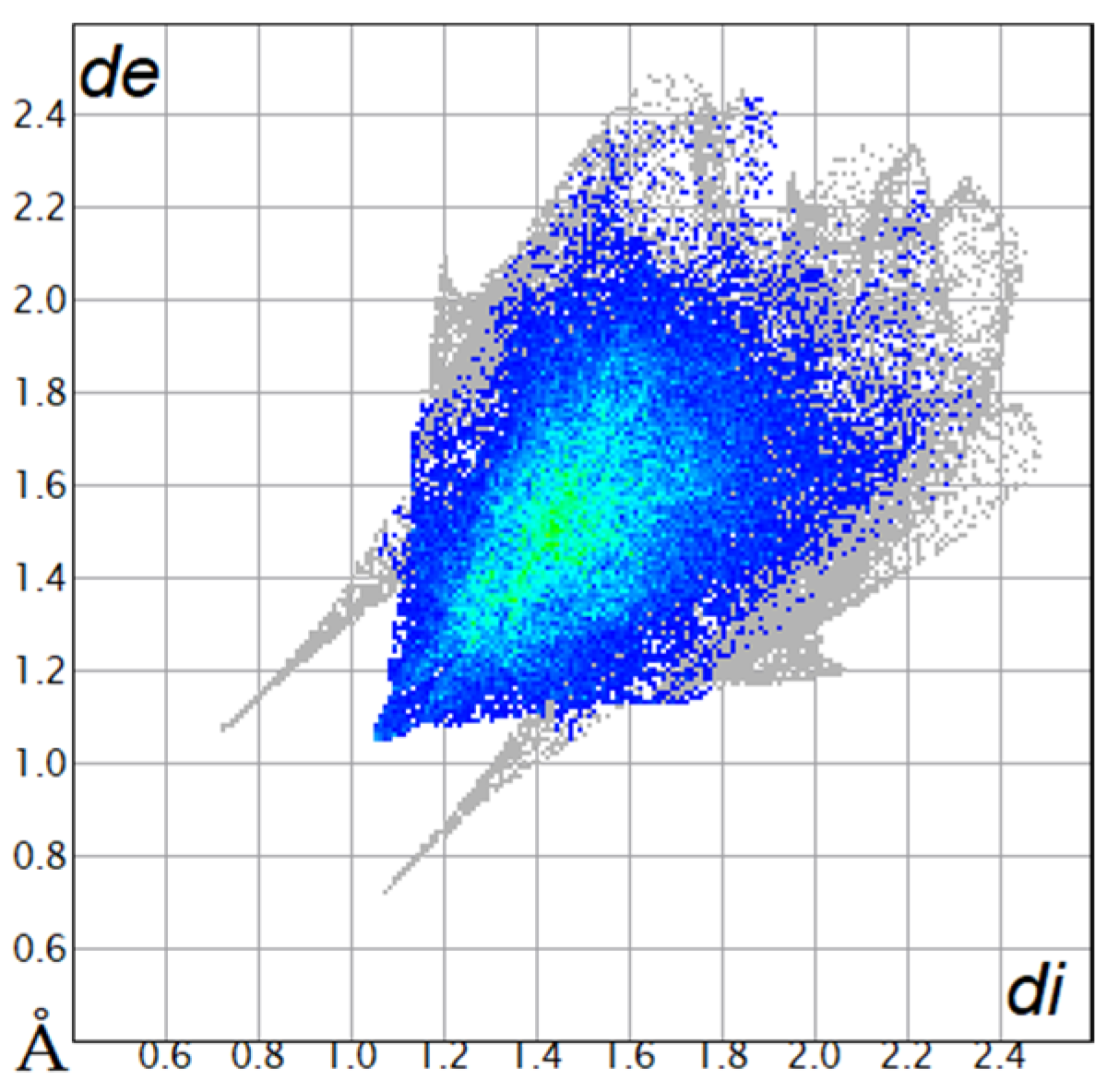
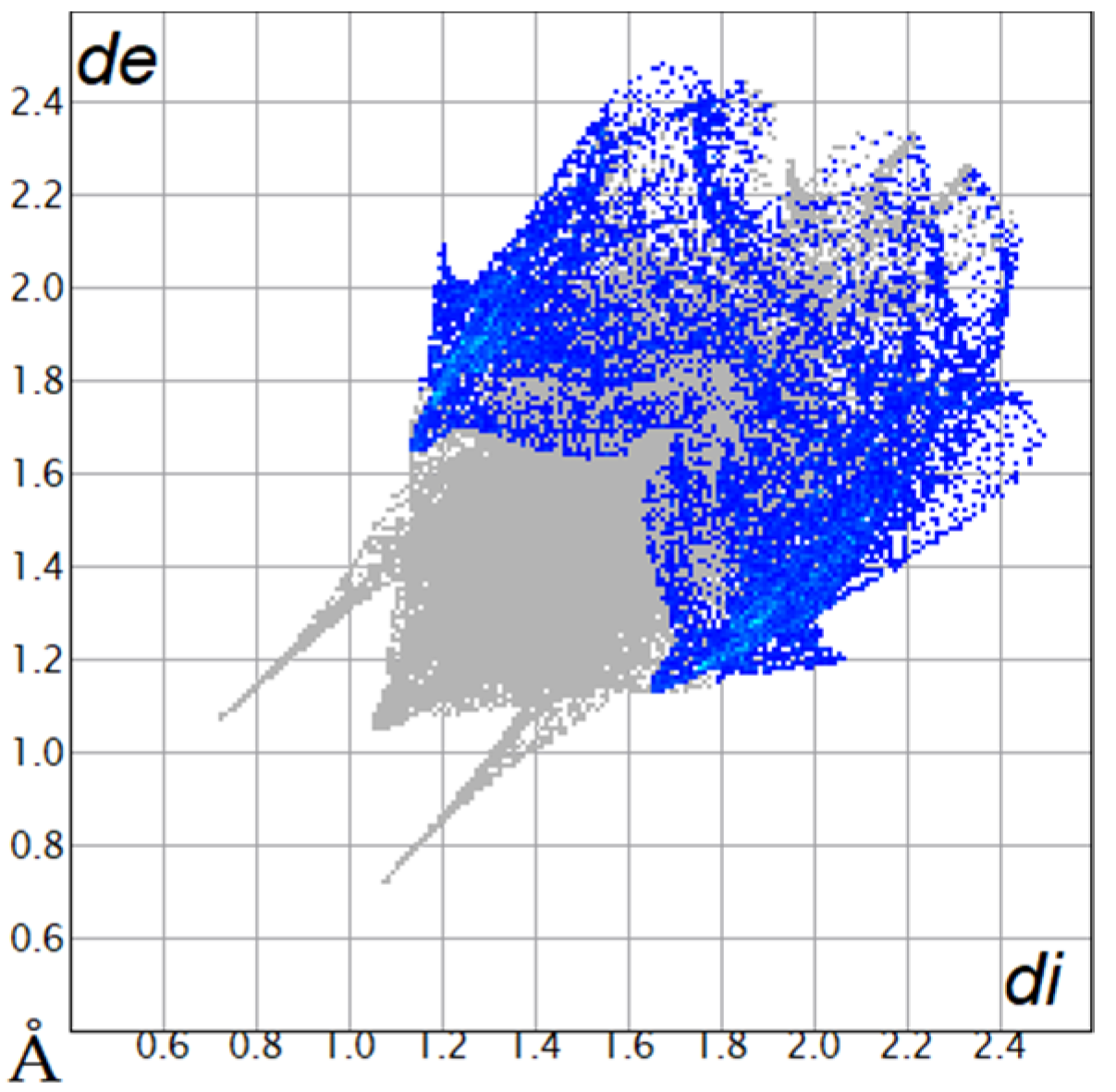
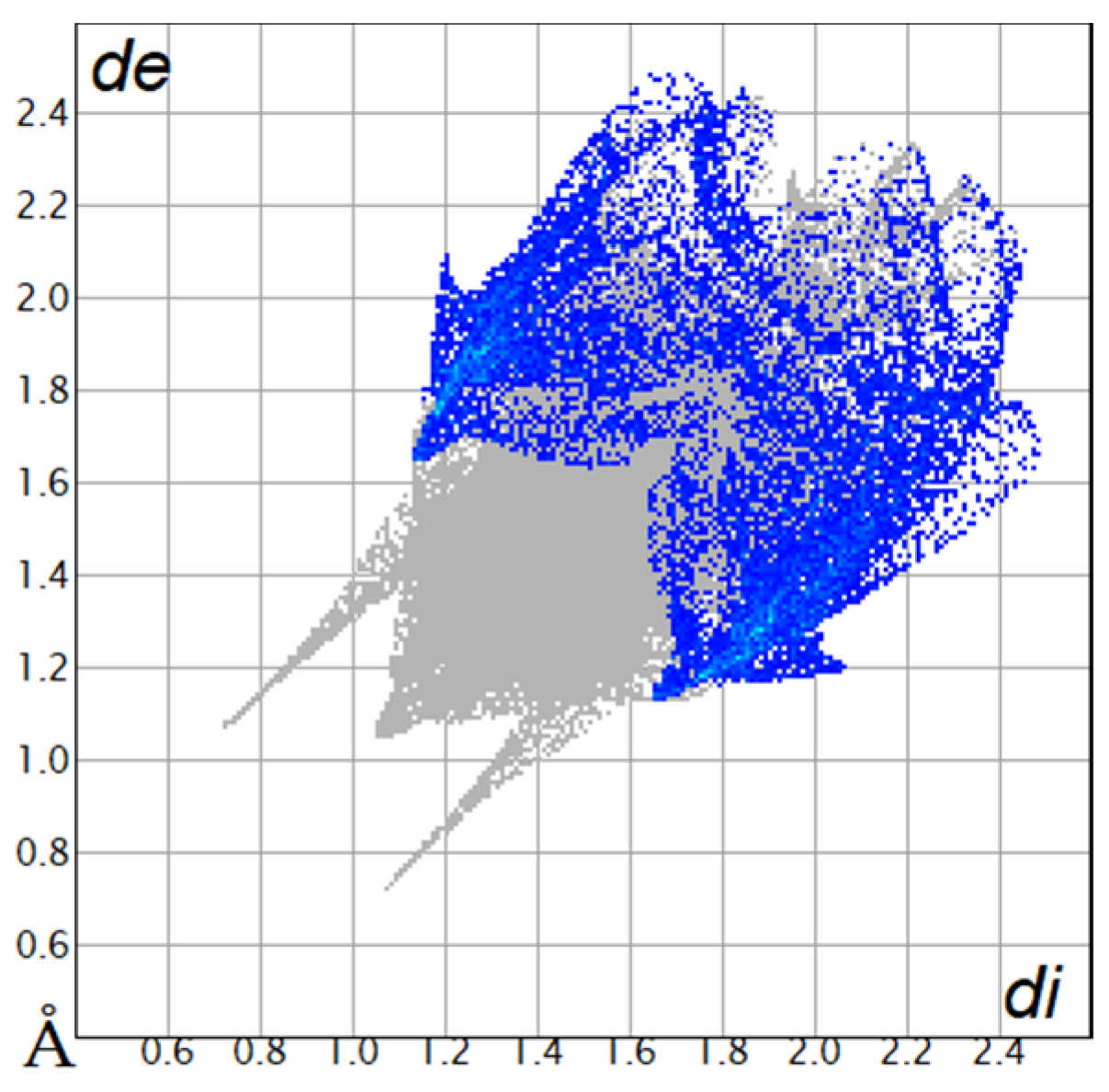
The Hirshfeld surfaces were plotted to visualize the van der Waals (VDW) distances and determine the interaction sites [17]. The surface was drawn using Crystal Explorer software and was based on our results from the X-ray studies in CIF format [18]. The plots are shown in Figure 4; the corresponding fingerprints were plotted and are shown in Table 4. The conspicuous red regions on the surface (close contacts) are due to O···H/H···O contacts accounting for 9.8% of all contacts in each compound; in fact, the two enantiomers had exactly the same fingerprints (Table 4) in which contacts H···H and C···H/H···C were 68.0 and 18.4%, respectively.
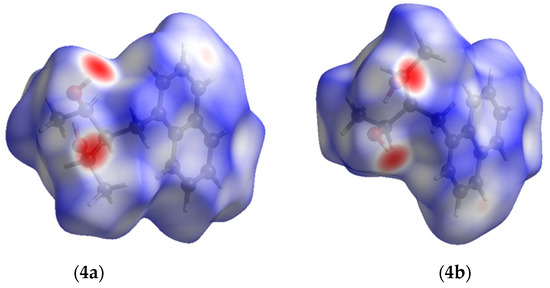
Figure 4.
Hirshfeld surface of compounds 4a and 4b.
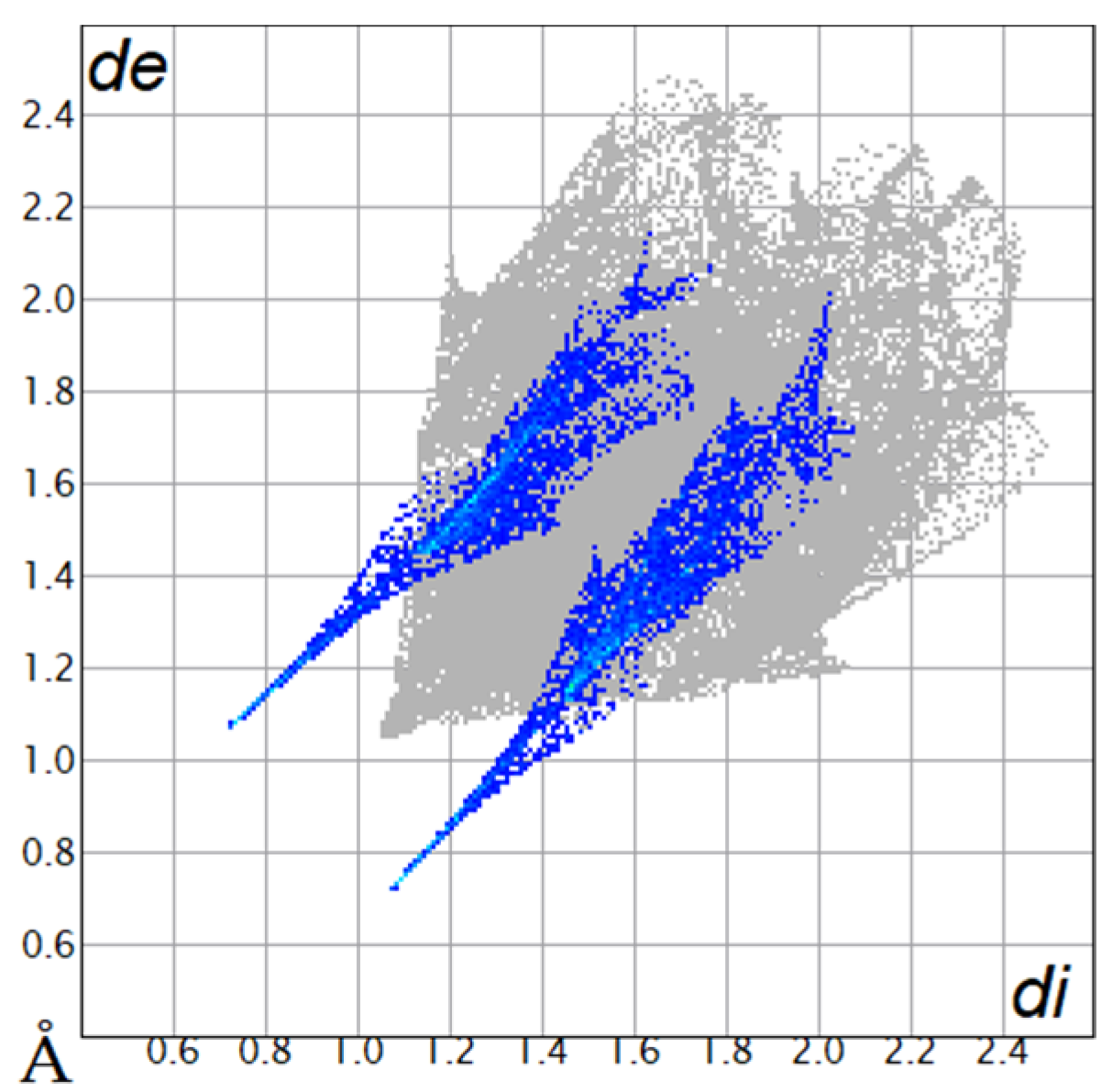
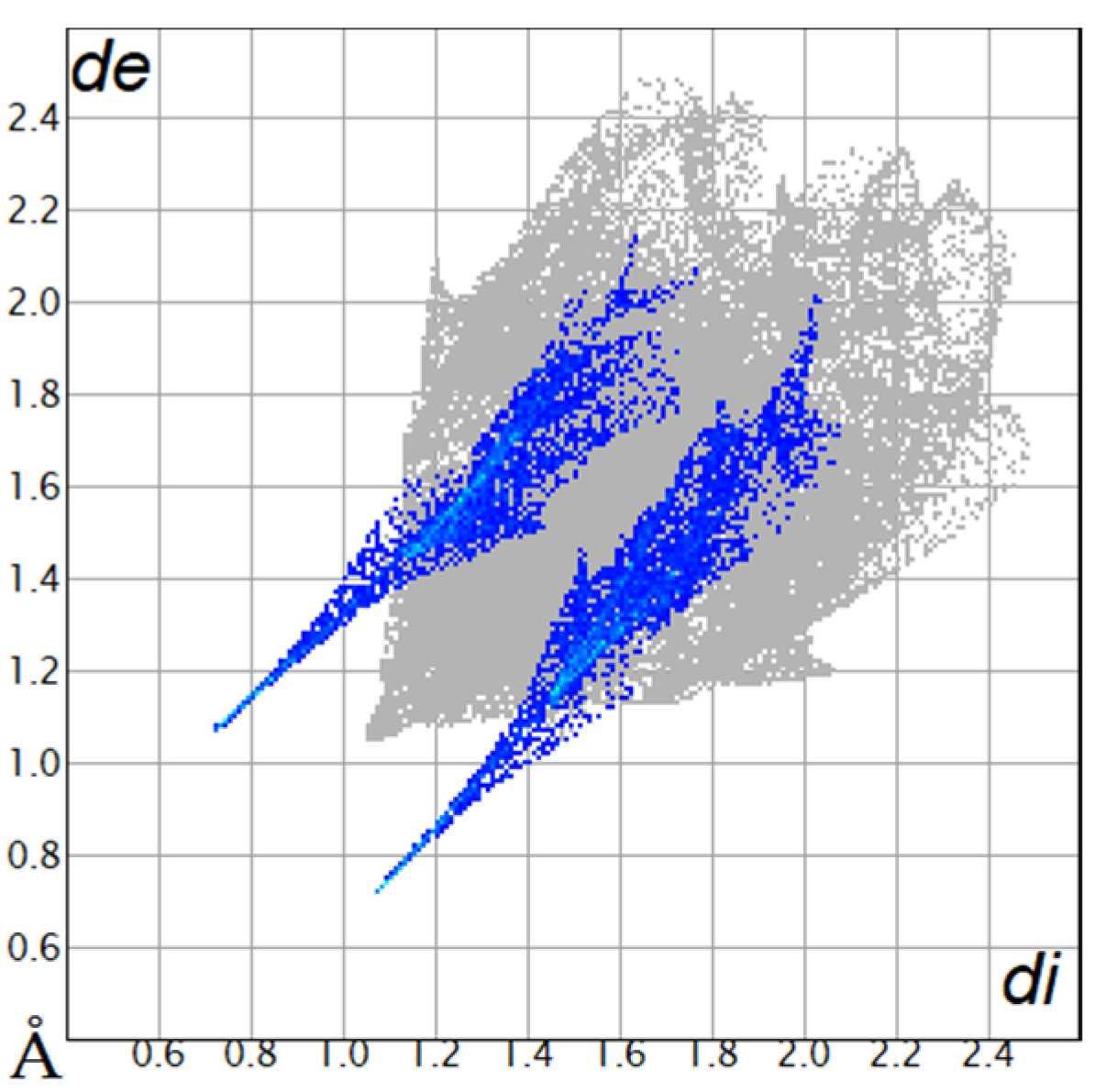
Table 4.
Two-dimensional fingerprint plots of compounds 4a and 4b.
3. Materials and Methods
All chemicals were available commercially, and the solvents were purified using conventional methods before use. Melting points were determined in an Electrothermal Engineering IA9100X1 melting point apparatus and are reported uncorrected. IR absorption spectra were recorded in the range of 4000–230 cm−1 as KBr pellets on a BRUKER Tensor 27 spectrophotometer. The 1H, 13C, and 2D spectra were recorded in CDCl3 on a 500 MHz Bruker spectrometer using TMS as an internal reference. NMR spectra were processed with MestReC 12.0.0 software.
Electro Ionization Impact mode mass spectra were recorded in a JEOL, SX 102 A by JEOL, an AccuTOF JMS-T100LC using the Direct Analysis in Real Time (DART) mode, and an MStation JMS-700 in fast atom bombardment (FAB) mode. Single crystal X-ray diffraction analyses were performed on a Bruker D8 Venture κ-geometry diffractometer using CuKα radiation with a combination of ω- and φ-scans.
Data were corrected for absorption (semi-empirical from equivalents method) and polarization. Structures were solved using direct methods, and models were refined by the full-matrix least-squares method on F2 against all reflections. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters.
Synthesis of 3-(naphthalene-1-ylmethylene)pentane-2,4-dione (2): 1.5 mL of 2,4-pentanodione and 1.6 mL of 1-naphthaldehyde in 20 mL of dichloromethane (CH2Cl2) reacted with 0.05 mL of piperidine and 0.05 mL of glacial acetic acid (CH3COOH) in the presence of molecular sieves, which was removed by filtration once the reaction was completed. The reaction solvent was removed under reduced pressure and the product was extracted with a 1:1 mixture of water–dichloromethane. The organic layer was dried over anhydrous Na2SO4, the excess solvent was removed, and the sample was kept in a high vacuum. The product was purified by flash column chromatography using Hex-AcOEt-MeOH at 80:15:5 ratios. Yield: 53%., m.p. 66 °C. 1H NMR (500 MHz, CDCl3): δ(ppm) 8.29 (s, 1H), 7.99 (dd, J = 8.10, 0.92 Hz, 1H), 7.91 (m, 2H), 7.61 (ddd, J = 8.43, 6.89, 1.69 Hz, 1H), 7.58 (ddd, J = 8.17, 6.86, 1.49 Hz, 1H), 7.44 (m, 2H), 2.50 (s, 3H), 2.05 (s, 3H). FTIR (KBr, cm−1): 3055.05, 3004.01, 1692.04, 1659.04. EI+: (m/z): 238.
Synthesis of (Z)-4-hydroxy-3-(naphthalen-1-ylmethyl)pent-3-en-2-one (3): 516.3 mg of 1 and 10% mmol Pd/C-10% as catalysts were placed in 40 mL of AcOEt and allowed to react under hydrogen atmosphere at room temperature for 2 hours. The reaction was monitored by TLC and quenched by filtration using a sintered glass funnel with packed celite to retain the catalyst. Then, the filtrated solution was concentrated in a vacuum to give compound 3. Yield: 76%; m.p. 85°C. 1H NMR (500 MHz, CDCl3): δ(ppm) enol tautomer 16.95 (s, 1H), 8.13 (ddt, J = 8.43, 1.40, 0.82, 0.82 Hz, 1H), 7.91 (ddt, J = 8.04, 1.41, 0.63, 0.63 Hz, 1H), 7.76 (d, J = 7.74 Hz, 1H), 7.59 (ddd, J = 8.29, 6.76, 1.31 Hz, 1H), 7.54 (m, 1H), 7.41 (m, 1H), 7.18 (ddd, J = 7.12, 2.48, 1.17 Hz, 1H), 4.07 (s, 2H), 2.04 (s, 6H); ketone tautomer 7.96 (m, 0.82H), 7.87 (ddt, J = 8.00, 1.39, 0.62, 0.62 Hz, 0.83H), 7.74 (d, J = 7.60 Hz, 0.89H), 7.56 (m, 1H), 7.51 (ddd, J = 8.08, 6.80, 1.32 Hz, 0.82H), 7.28 (dd, J = 7.01, 1.15 Hz, 0.76H), 4.20 (t, J = 7.29, 7.29 Hz, 0.76H), 3.62 (d, J = 7.29 Hz, 1.57H), 2.10 (d, J = 0.39 Hz, 4.61H). FTIR (KBr, cm−1): 3049.58, 3012.45, 2919.42, 2860.93, 1699.61. EI+: (m/z): 240.
Synthesis of (2S, 4S)- and (2R, 4R)-3-(naphthalene-1-ylmethyl) pentane-2,4-diols (4a, 4b): 533 mg of 3 in 40 mL of methanol (MeOH) were reacted with 224.9 mg of NaBH4 for 1h at 0 °C. After the reaction was complete, ice was added to the reaction mixture and 3% HCl was added until effervescence ceased, indicating neutralization of free hydride. The product was extracted in AcOEt–H2O at a 1:1 ratio. The organic phase was dried over anhydrous Na2SO4 and concentrated in a vacuum. Yield: 98%. The diastereomer mixture was separated on preparative layer chromatography using Hex-CH2Cl2-MeOH at 60:35:5 ratios. The constitution of the diastereomeric mixture was composed following the order of polarity (from least to most polar): 1,3-diol meso ca. 40%, racemate 4a/4b ca. 30%, and the hydroxyketone ca. 30%.
Compounds (4a, 4b): The racemic mixture crystallized from CH2Cl2 at room temperature and the independent single crystals of the enantiomers were obtained simultaneously. The X-ray diffraction results showed that a single enantiomer was found, which led to a search for the complementary one. The crystals were morphologically almost identical, with minimal perceptible differences in their crystalline habit. This led to the findings reported in the present work. The physical and spectroscopic data were as follows: m.p. 101 °C; 1H NMR (500 MHz, CDCl3): δ(ppm) 8.05 (dq, J = 8.59, 1.06, 0.93, 0.93 Hz, 1H), 7.86 (dd, J = 7.64, 1.82 Hz, 1H), 7.73 (dd, J = 7.45, 2.00 Hz, 1H), 7.51 (ddd, J = 8.45, 6.84, 1.72 Hz, 1H), 7.48 (ddd, J = 8.08, 6.79, 1.52 Hz, 1H), 7.39 (m, 2H), 4.36 (qd, J = 6.50, 6.50, 6.49, 1.83 Hz, 1H), 3.91 (qd, J = 6.46, 6.44, 6.44, 3.86 Hz, 1H), 3.30 (dd, J = 14.19, 9.74 Hz, 1H), 3.23 (dd, J = 14.22, 5.62 Hz, 1H), 1.83 (dddd, J = 9.61, 5.65, 3.85, 1.83 Hz, 1H), 1.46 (d, J = 6.49 Hz, 3H), 1.24 (d, J = 6.46 Hz, 3H). FTIR (film, cm−1): 3326.29, 3047.60, 2969.91, 2930.50, 2899.48. = −0.001 (c 1.5 mg/mL CHCl3). EI+: (m/z): calculated for C16H20O2 244.1463; found: 244.1464 [M+].
4. Conclusions
Suitable crystals of compound 4 were successfully crystallized and analyzed by single crystal X-ray diffraction analysis. The crystals of the two compounds were carefully separated by microscope optical inspection affording the two relative configurations, which were found to be the enantiomeric forms. Hirshfeld’s fingerprints showed that two enantiomers had the same fingerprints.
Supplementary Materials
The following supporting information can be downloaded. Supplementary data for compounds 4a and 4b have been deposited at the Cambridge Crystallographic Data Centre. Copies of this information are available free of charge on request from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (Fax: +44-1223-336033); e-mail: deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk, quoting the deposition numbers CCDC 2189445-2189446.
Author Contributions
Conceptualization, R.G.E., D.M.-M. and Y.A.-R.; methodology, Y.A.-R., D.S.-L., W.M.-M. and M.A.O.-M.; writing—review and editing, R.G.E. and D.M.-M.; validation X-rays: R.A.T.; molecular and supramolecular analysis: M.R.Z.-O., J.M.G.-A. and A.A.-C.; project administration and funding acquisition: R.G.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by PAPIIT (DGAPA, UNAM, IN208516, IN210520, and IT200720) and CONACYT (CB 252524, A1-S-033933, and FOINS-307152).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Scholarships from CONACYT to YAR (No. 576706) and WMM (No. 576707) are gratefully acknowledged. We are indebted to María del Rocío Patiño Maya from Instituto de Química, UNAM (FT-IR and optical rotation).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kel’in, A.V. Recent advances in the synthesis of 1,3-diketones. Curr. Org. Chem. 2003, 7, 1691–1711. [Google Scholar] [CrossRef]
- Shehzad, A.; Shahzad, R.; Lee, Y.S. Curcumin: A Potent Modulator of Multiple Enzymes in Multiple Cancers. Enzymes 2014, 36, 149–174. [Google Scholar] [PubMed]
- Basile, V.; Ferrari, E.; Lazzari, S.; Belluti, S.; Pignedoli, F.; Imbriano, C. Curcumin derivatives: Molecular basis of their anti-cancer activity. Biochem. Pharmacol. 2009, 78, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.M.; Hunsaker, L.A.; Abcouwer, S.F.; Deck, L.M.; Jagt, D.L.V. Anti-oxidant activities of curcumin and related enones. Bioorg. Med. Chem. 2005, 13, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.A.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin-from Molecule to Biological Function. Angew. Chem. Int. Ed. 2012, 51, 5308–5332. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Tilekar, K.; Mehendale-Munj, S.; Mohan, R.; Ramaa, C. Synthesis and primary cytotoxicity evaluation of new 5-benzylidene-2,4-thiazolidinedione derivatives. Eur. J. Med. Chem. 2010, 45, 4539–4544. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Mahajan, N.; Taneja, S.C. Recent advances in the stereoselective synthesis of 1,3-diols using biocatalysts. Catal. Sci. Technol. 2013, 3, 2462–2480. [Google Scholar] [CrossRef]
- Hoyos, P.; Pace, V.; Alcántara, A.R. Dynamic Kinetic Resolution via Hydrolase-Metal Combo Catalysis in Stereoselective Synthesis of Bioactive Compounds. Adv. Synth. Catal. 2012, 354, 2585–2611. [Google Scholar] [CrossRef]
- Shao, N.; Monnier, V.; Charles, L.; Rodriguez, J.; Bressy, C.; Quintard, A. Multi-Catalytic Enantioselective Synthesis of 1,3-Diols Containing a Tetrasubstituted Fluorinated Stereocenter. Eur. J. Org. Chem. 2022, 2022, e202200031. [Google Scholar] [CrossRef]
- Bianchini, C.; Barbaro, P.; Scapacci, G.; Zanobini, F. In Situ and Reactor Study of the Enantioselective Hydrogenation of Acetylacetone by Ruthenium Catalysis with the New Chiral Diphosphine Ligand (R)-(R)-3-Benzyl-2,4-bis(diphenylphosphino)pentane. Organometallics 2000, 19, 2450–2461. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, A. Crystallization-Based Separation of Enantiomers. In Stereoselective Synthesis of Drugs and Natural Products; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Dalessandro, E.V.; Collin, H.P.; Valle, M.S.; Pliego, J.R. Mechanism and free energy profile of base-catalyzed Knoevenagel condensation reaction. RSC Adv. 2016, 6, 57803–57810. [Google Scholar] [CrossRef]
- Alvarez-Ricardo, Y.F.; Sánchez-López, D.M.; Meza-Morales, W.E.; Obregón, M.A.; Arias-Olguín, I.I.; Nieto-Camacho, A.; Toscano, R.A.; Enríquez, R.G. Stereochemistry and Antioxidant Activity of 1,3-Diol Derivatives of Diacetylcurcumin-4H: A Joint NMR, X-Ray, and Biological Approach. ChemistrySelect 2020, 5, 1616–1622. [Google Scholar] [CrossRef]
- Evans, D.J.; Junk, P.C.; Smith, M.K. Intramolecular C–H⋯π interactions influence the conformation of N,N′-dibenzyl-4,13-diaza-18-crown-6 molecules. New J. Chem. 2002, 26, 1043–1048. [Google Scholar] [CrossRef]
- Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. CH/π hydrogen bonds in organic and organometallic chemistry. CrystEngComm 2009, 11, 1757–1788. [Google Scholar] [CrossRef]
- Anioła, M.; Dega-Szafran, Z.; Katrusiak, A.; Szafran, M. NH⋯O and OH⋯O interactions of glycine derivatives with squaric acid. New J. Chem. 2014, 38, 3556–3568. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).