
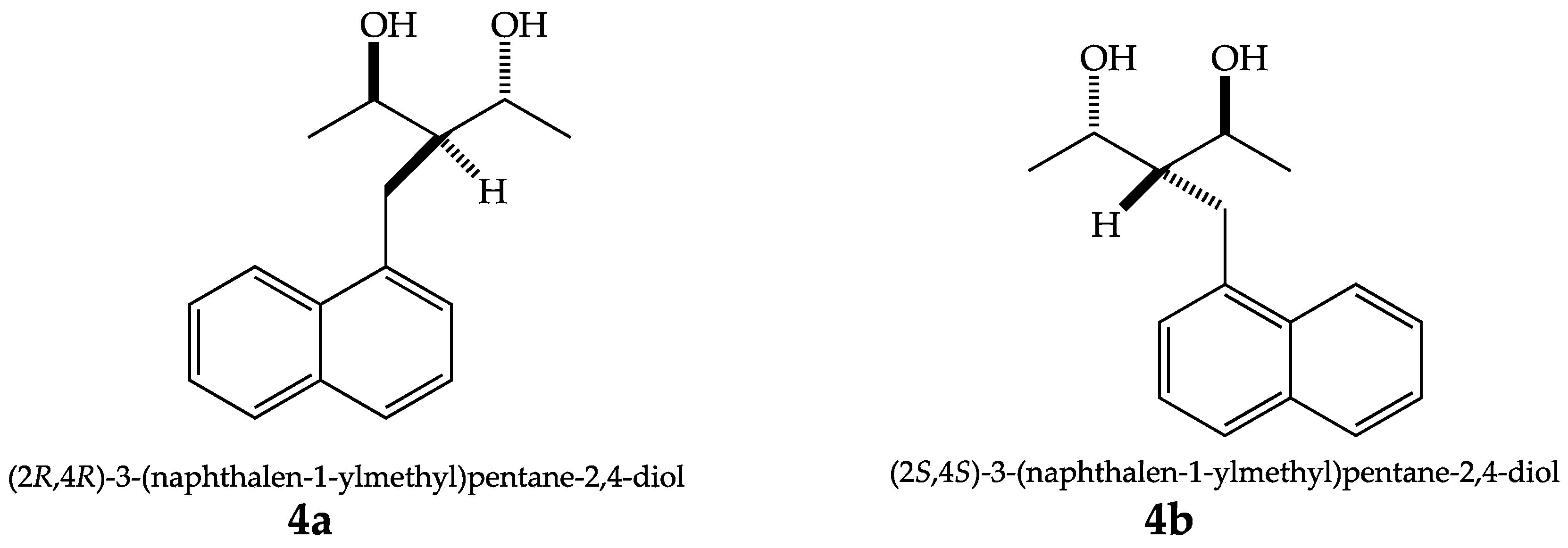
Spontaneous Enantiomeric Resolution of 1,3-Diols from the Naphtylidene Derivative of 2,4-Pentanedione

 ,
,  ,
,  , ,
, ,  and
and
Abstract
:
1. Introduction
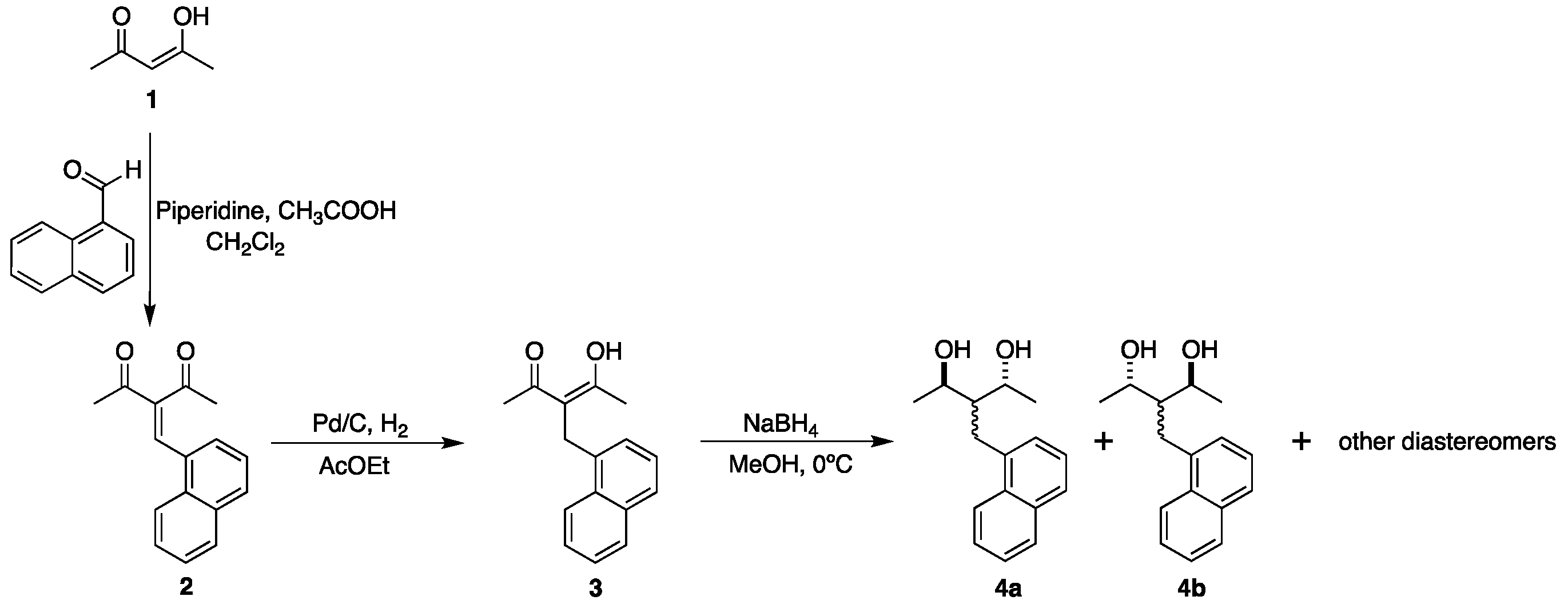
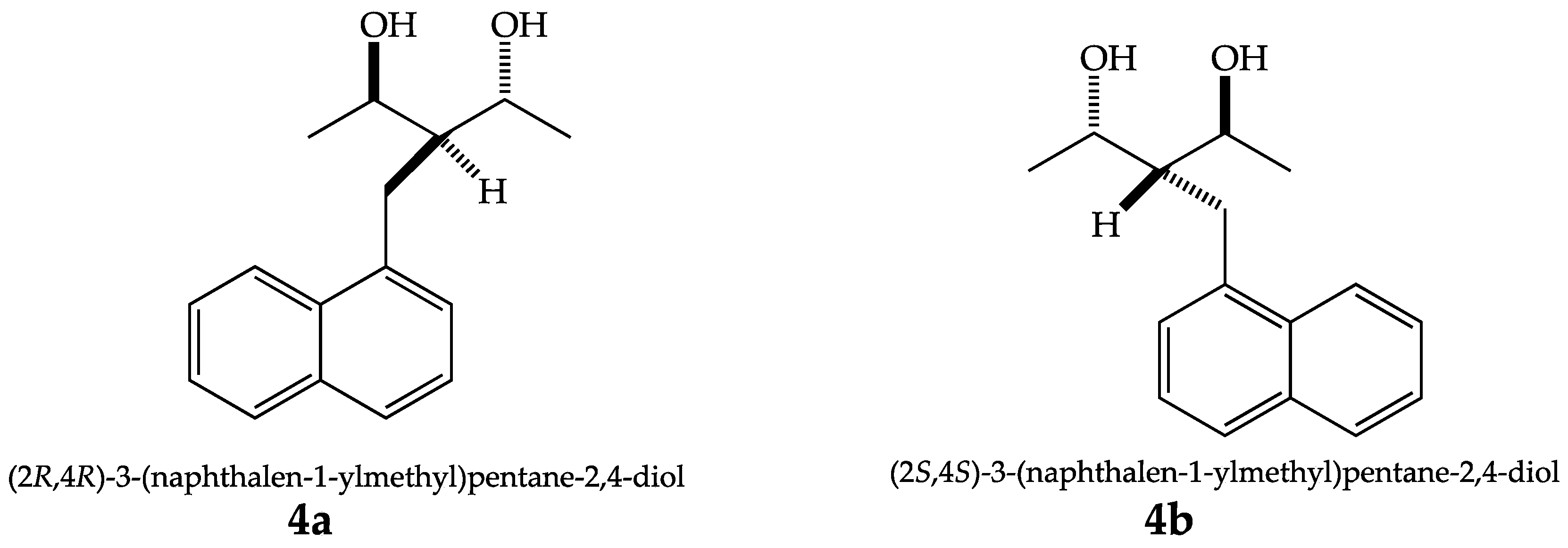
2. Results and Discussion
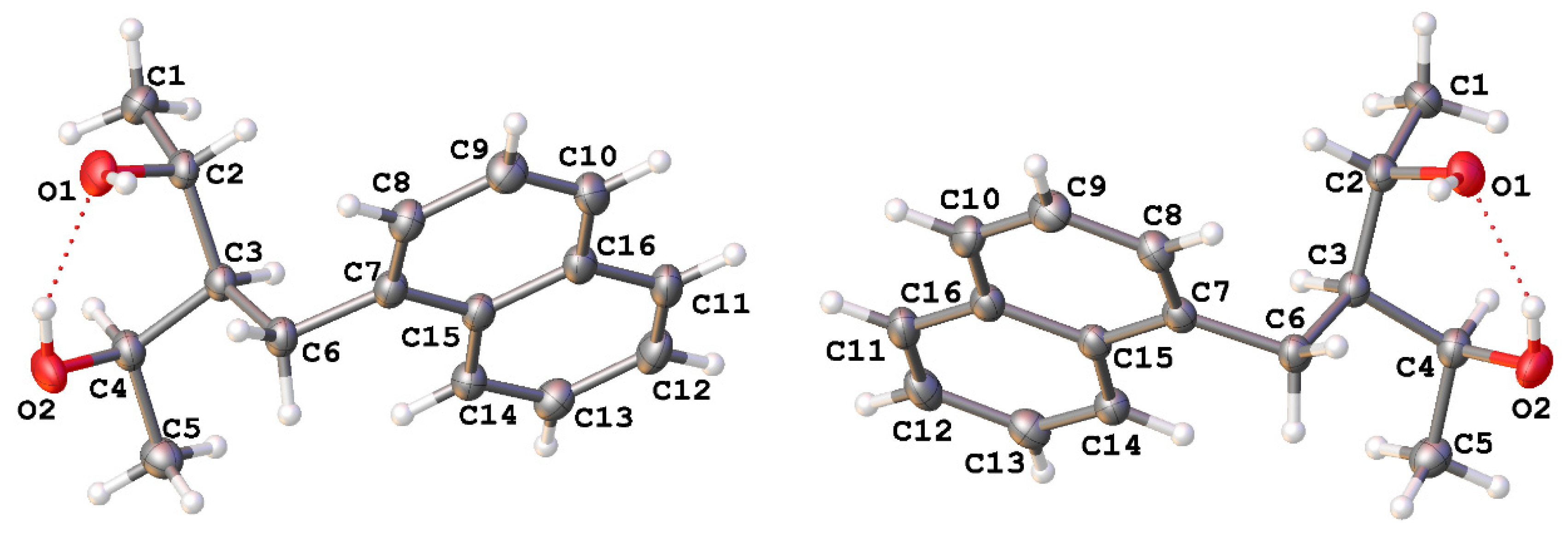
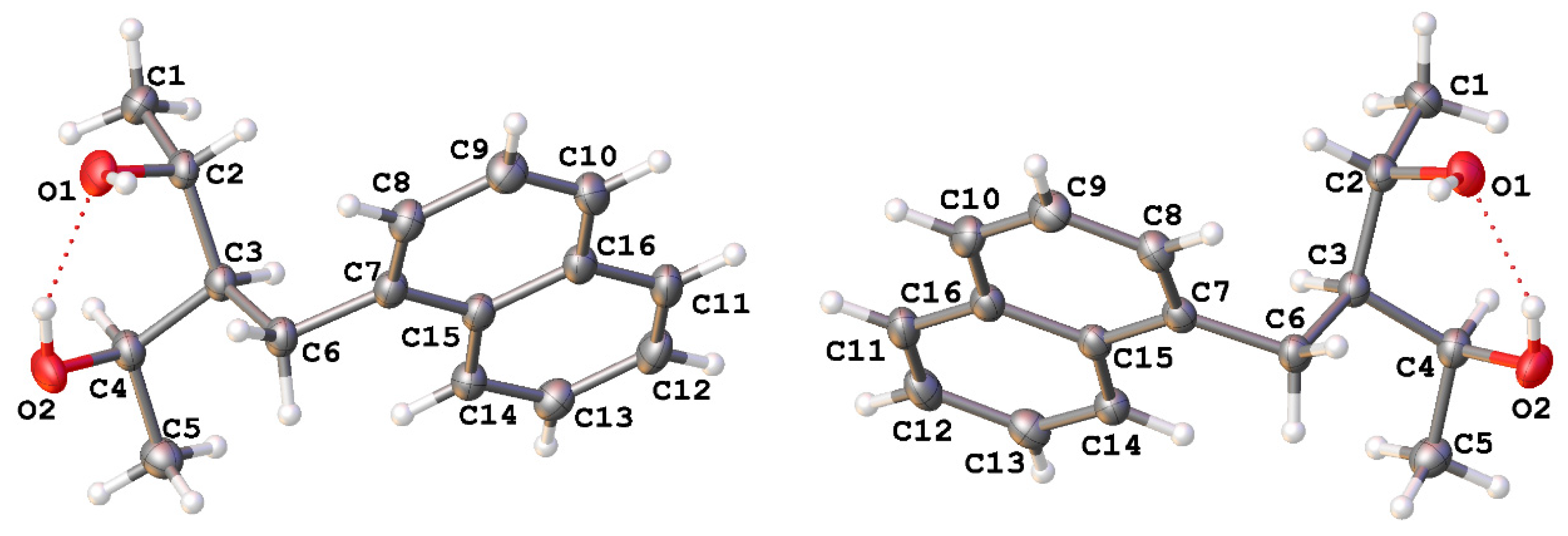
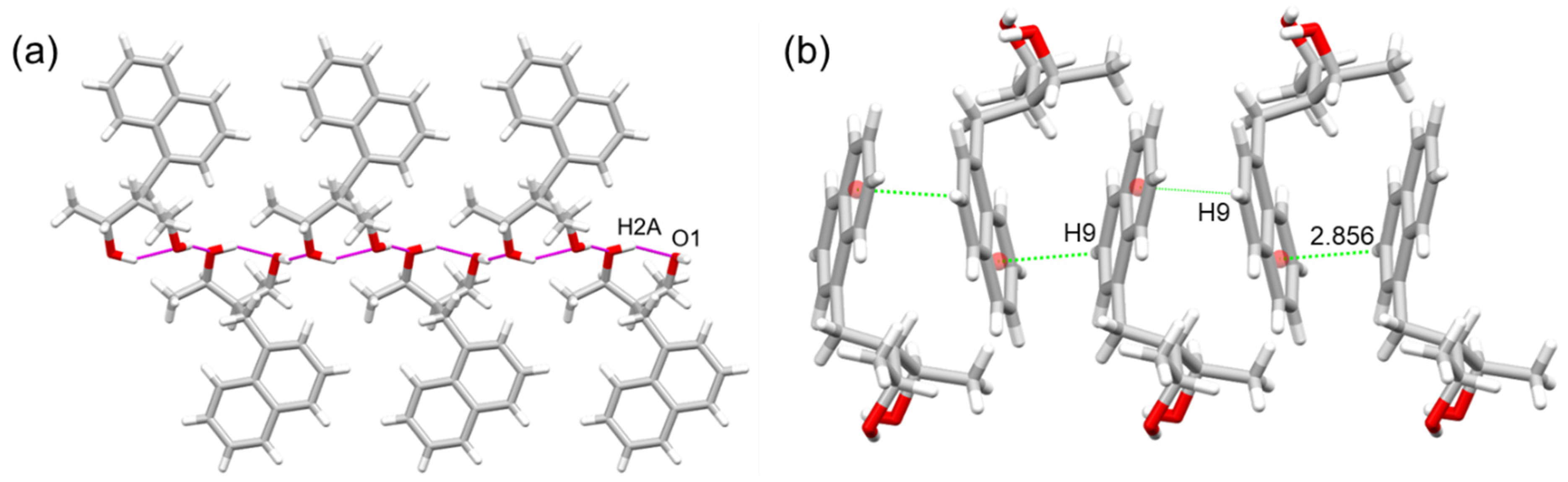
2.1. X-ray Diffraction
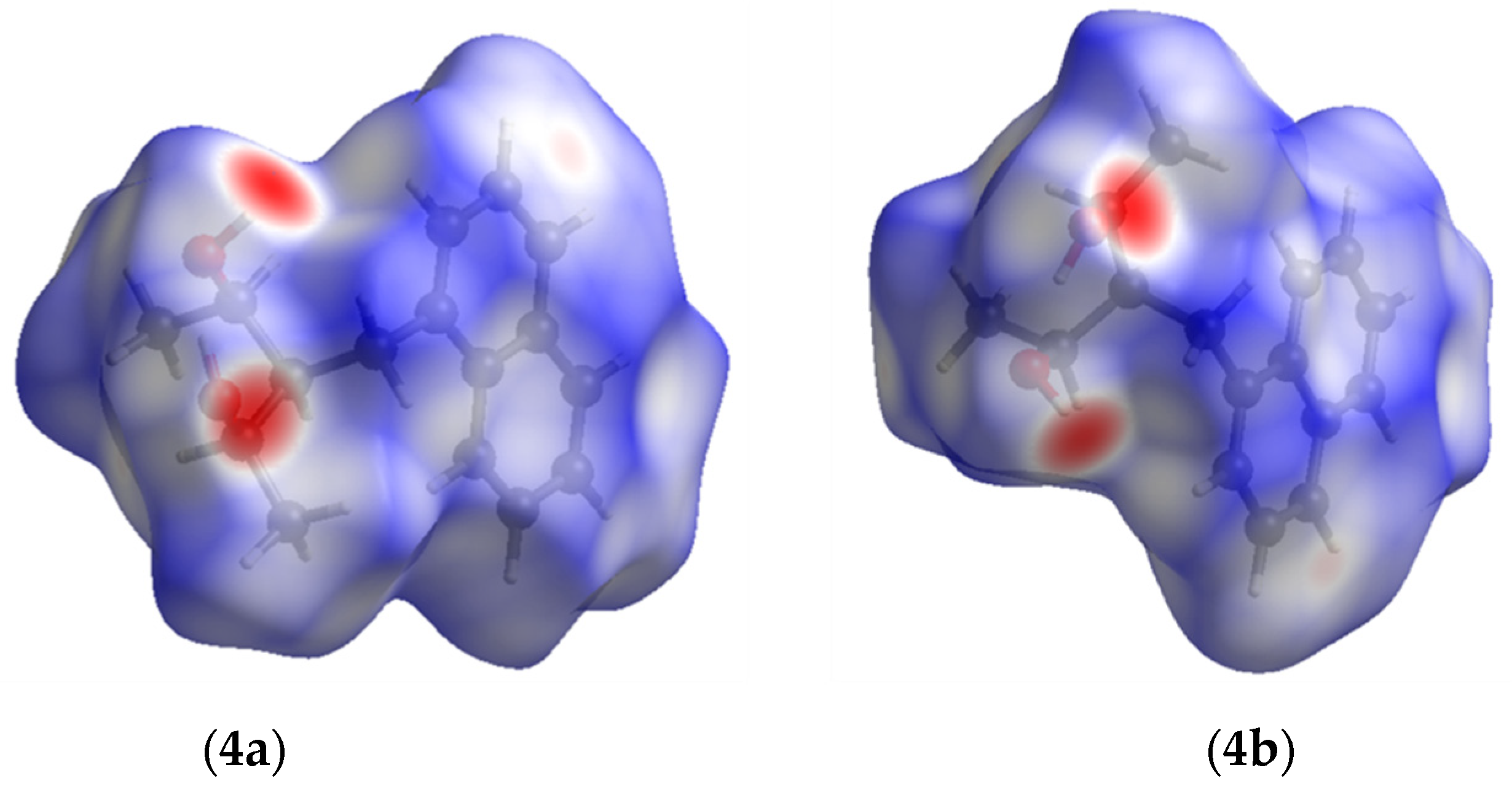
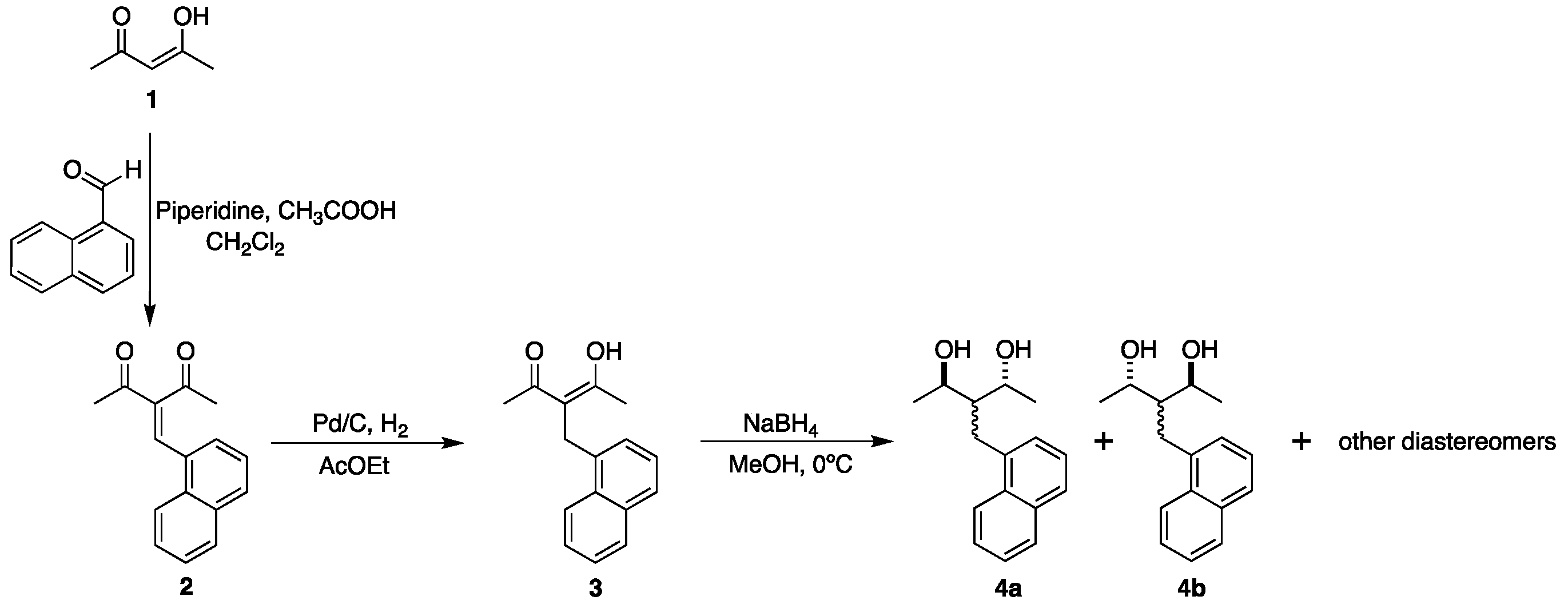
2.2. Hirshfeld Surface
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kel’in, A.V. Recent advances in the synthesis of 1,3-diketones. Curr. Org. Chem. 2003, 7, 1691–1711. [Google Scholar] [CrossRef]
- Shehzad, A.; Shahzad, R.; Lee, Y.S. Curcumin: A Potent Modulator of Multiple Enzymes in Multiple Cancers. Enzymes 2014, 36, 149–174. [Google Scholar] [PubMed]
- Basile, V.; Ferrari, E.; Lazzari, S.; Belluti, S.; Pignedoli, F.; Imbriano, C. Curcumin derivatives: Molecular basis of their anti-cancer activity. Biochem. Pharmacol. 2009, 78, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.M.; Hunsaker, L.A.; Abcouwer, S.F.; Deck, L.M.; Jagt, D.L.V. Anti-oxidant activities of curcumin and related enones. Bioorg. Med. Chem. 2005, 13, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.A.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin-from Molecule to Biological Function. Angew. Chem. Int. Ed. 2012, 51, 5308–5332. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Tilekar, K.; Mehendale-Munj, S.; Mohan, R.; Ramaa, C. Synthesis and primary cytotoxicity evaluation of new 5-benzylidene-2,4-thiazolidinedione derivatives. Eur. J. Med. Chem. 2010, 45, 4539–4544. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Mahajan, N.; Taneja, S.C. Recent advances in the stereoselective synthesis of 1,3-diols using biocatalysts. Catal. Sci. Technol. 2013, 3, 2462–2480. [Google Scholar] [CrossRef]
- Hoyos, P.; Pace, V.; Alcántara, A.R. Dynamic Kinetic Resolution via Hydrolase-Metal Combo Catalysis in Stereoselective Synthesis of Bioactive Compounds. Adv. Synth. Catal. 2012, 354, 2585–2611. [Google Scholar] [CrossRef]
- Shao, N.; Monnier, V.; Charles, L.; Rodriguez, J.; Bressy, C.; Quintard, A. Multi-Catalytic Enantioselective Synthesis of 1,3-Diols Containing a Tetrasubstituted Fluorinated Stereocenter. Eur. J. Org. Chem. 2022, 2022, e202200031. [Google Scholar] [CrossRef]
- Bianchini, C.; Barbaro, P.; Scapacci, G.; Zanobini, F. In Situ and Reactor Study of the Enantioselective Hydrogenation of Acetylacetone by Ruthenium Catalysis with the New Chiral Diphosphine Ligand (R)-(R)-3-Benzyl-2,4-bis(diphenylphosphino)pentane. Organometallics 2000, 19, 2450–2461. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, A. Crystallization-Based Separation of Enantiomers. In Stereoselective Synthesis of Drugs and Natural Products; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Dalessandro, E.V.; Collin, H.P.; Valle, M.S.; Pliego, J.R. Mechanism and free energy profile of base-catalyzed Knoevenagel condensation reaction. RSC Adv. 2016, 6, 57803–57810. [Google Scholar] [CrossRef]
- Alvarez-Ricardo, Y.F.; Sánchez-López, D.M.; Meza-Morales, W.E.; Obregón, M.A.; Arias-Olguín, I.I.; Nieto-Camacho, A.; Toscano, R.A.; Enríquez, R.G. Stereochemistry and Antioxidant Activity of 1,3-Diol Derivatives of Diacetylcurcumin-4H: A Joint NMR, X-Ray, and Biological Approach. ChemistrySelect 2020, 5, 1616–1622. [Google Scholar] [CrossRef]
- Evans, D.J.; Junk, P.C.; Smith, M.K. Intramolecular C–H⋯π interactions influence the conformation of N,N′-dibenzyl-4,13-diaza-18-crown-6 molecules. New J. Chem. 2002, 26, 1043–1048. [Google Scholar] [CrossRef]
- Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. CH/π hydrogen bonds in organic and organometallic chemistry. CrystEngComm 2009, 11, 1757–1788. [Google Scholar] [CrossRef]
- Anioła, M.; Dega-Szafran, Z.; Katrusiak, A.; Szafran, M. NH⋯O and OH⋯O interactions of glycine derivatives with squaric acid. New J. Chem. 2014, 38, 3556–3568. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 4a (CCDC 2189445) | 4b (CCDC 2189446) |
|---|---|---|
| Empirical formula | C16H20O2 | C16H20O2 |
| Formula weight | 244.32 | 244.32 |
| T(K) | 150(2) | 150(2) |
| Crystal system | Orthorombic | Orthorombic |
| Space group | P212121 | P212121 |
| a (Å) | 7.6041(6) | 7.6127(5) |
| b (Å) | 7.9171(6) | 7.9115(5) |
| c (Å) | 21.5553(17) | 21.5467(13) |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 90 | 90 |
| V (Å3) | 1297.68(18) | 1297.71(14) |
| Z | 4 | 4 |
| ρCalc (Mg m−3) | 1.251 | 1.251 |
| μ (mm−1) | 0.635 | 0.635 |
| F(000) | 528.0 | 528.0 |
| Crystal size (mm3) | 0.393 × 0.299 × 0.265 | 0.331 × 0.247 × 0.174 |
| Wavelength (Å) | 1.54178 | 1.54178 |
| 2θ range for data collection | 8.204 to 158.194° | 8.206 to 159.5° |
| Index ranges | −9 ≤ h ≤ 9, −10 ≤ k ≤ 9, −26 ≤ l ≤ 27 | −9 ≤ h ≤ 9, −10 ≤ k ≤ 10, −27 ≤ l ≤ 26 |
| Reflections collected | 29301 | 22941 |
| Independent reflections | 2783 [Rint = 0.0354, Rsigma = 0.0155]] | 2797 [Rint = 0.0325, Rsigma = 0.0179] |
| Data/restraints/parameters | 2783/0/171 | 2797/0/171 |
| Goodness of fit (GOF) on F2 | 1.075 | 1.064 |
| Final R indices [I≥2σ(I)] | R1 = 0.0301, wR2 = 0.0811 | R1 = 0.0315, wR2 = 0.0797 |
| Final R indices [all data] | R1 = 0.0305, wR2 = 0.0815 | R1 = 0.0318, wR2 = 0.0801 |
| Largest difference peak/hole (e Å-3) | 0.22 and −0.17 | 0.24 and −0.16 |
| Flack parameter | −0.02(5) | −0.01(6) |
| Contact | Distance H···A (Å) | Distance D···A (Å) | Angle D-H···A (°) | Symmetry Code |
|---|---|---|---|---|
| C9H9···Cg * | 2.856 | 3.473 | 123.65 | −1/2 + x, 1.5 − y, 1 − z |
| C11···H5B-C5 | 2.891 | 3.662 | 136.24 | −1/2 + x, 1/2 − y, 1 − z |
| O2···H1-O1 | 1.909 | 2.777 | 165.23 | 1 − x, −1/2 + y, 1.5 − z |
| Contact | Distance H···A (Å) | Distance D···A (Å) | Angle D-H···A (°) | Symmetry Code |
|---|---|---|---|---|
| C9H9···Cg * | 2.855 | 3.475 | 123.89 | −1/2 + x, 1/2 − y, 1 − z |
| C11···H5B-C5 | 2.888 | 3.660 | 136.26 | −1/2 + x, 1.5 − y, 1 − z |
| O2···H1-O1 | 1.918 | 2.778 | 166.76 | 1 − x, −1/2 + y, ½ − z |
| Contacts | 4a | 4b |
|---|---|---|
| All contacts 100% | 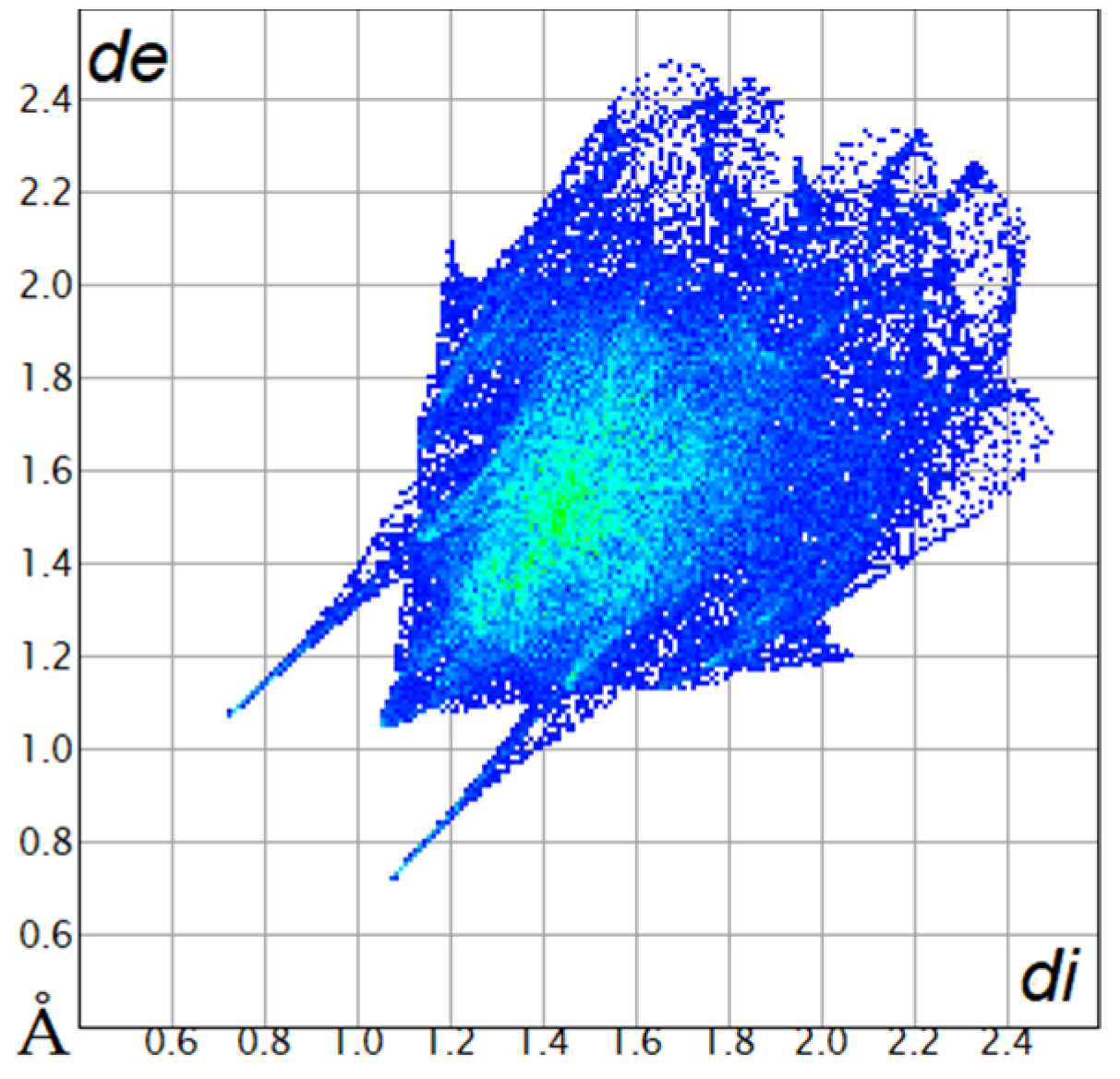 | 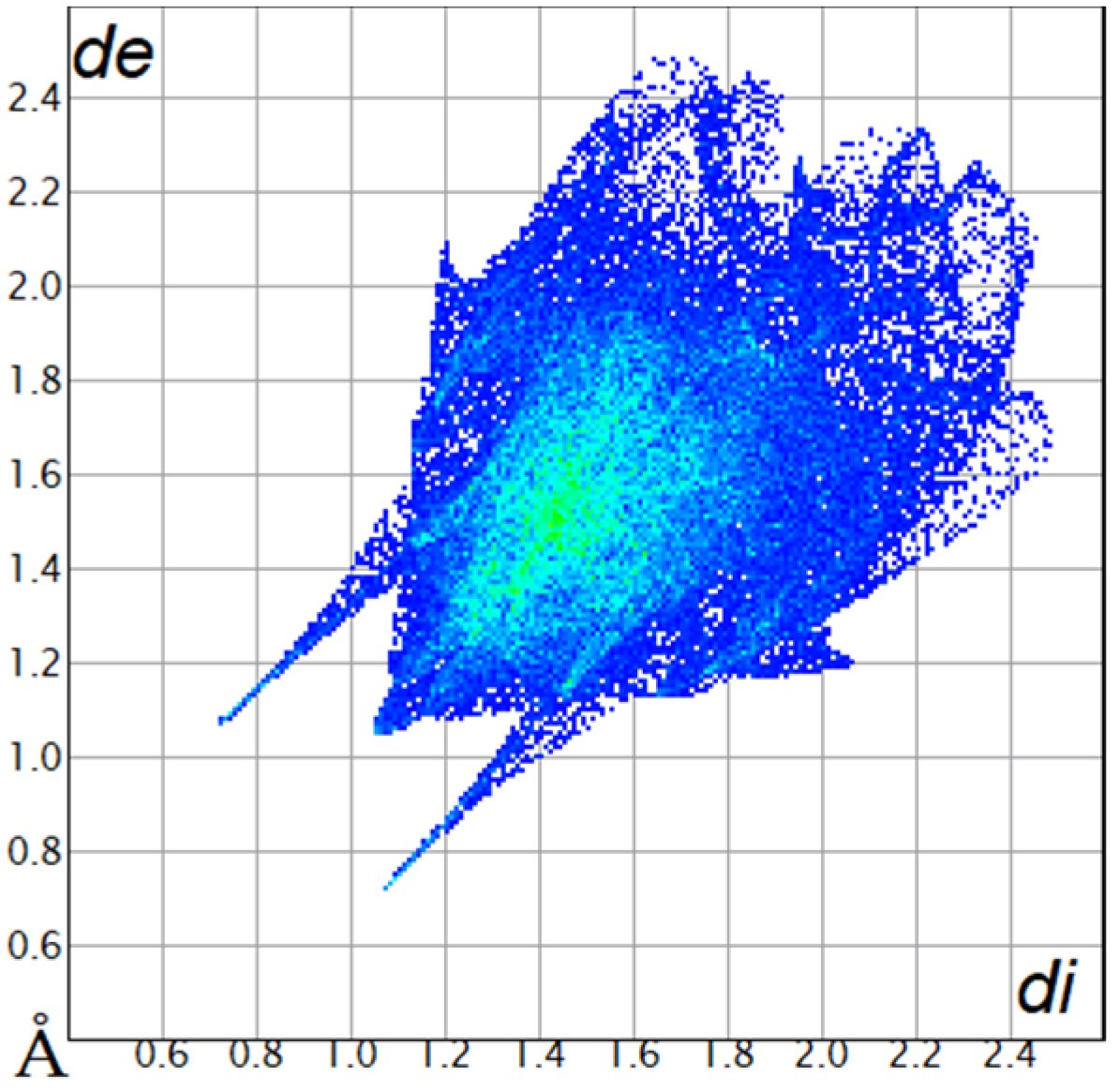 |
| H···H68.0% |  |  |
| O···H/H···O9.8% |  | 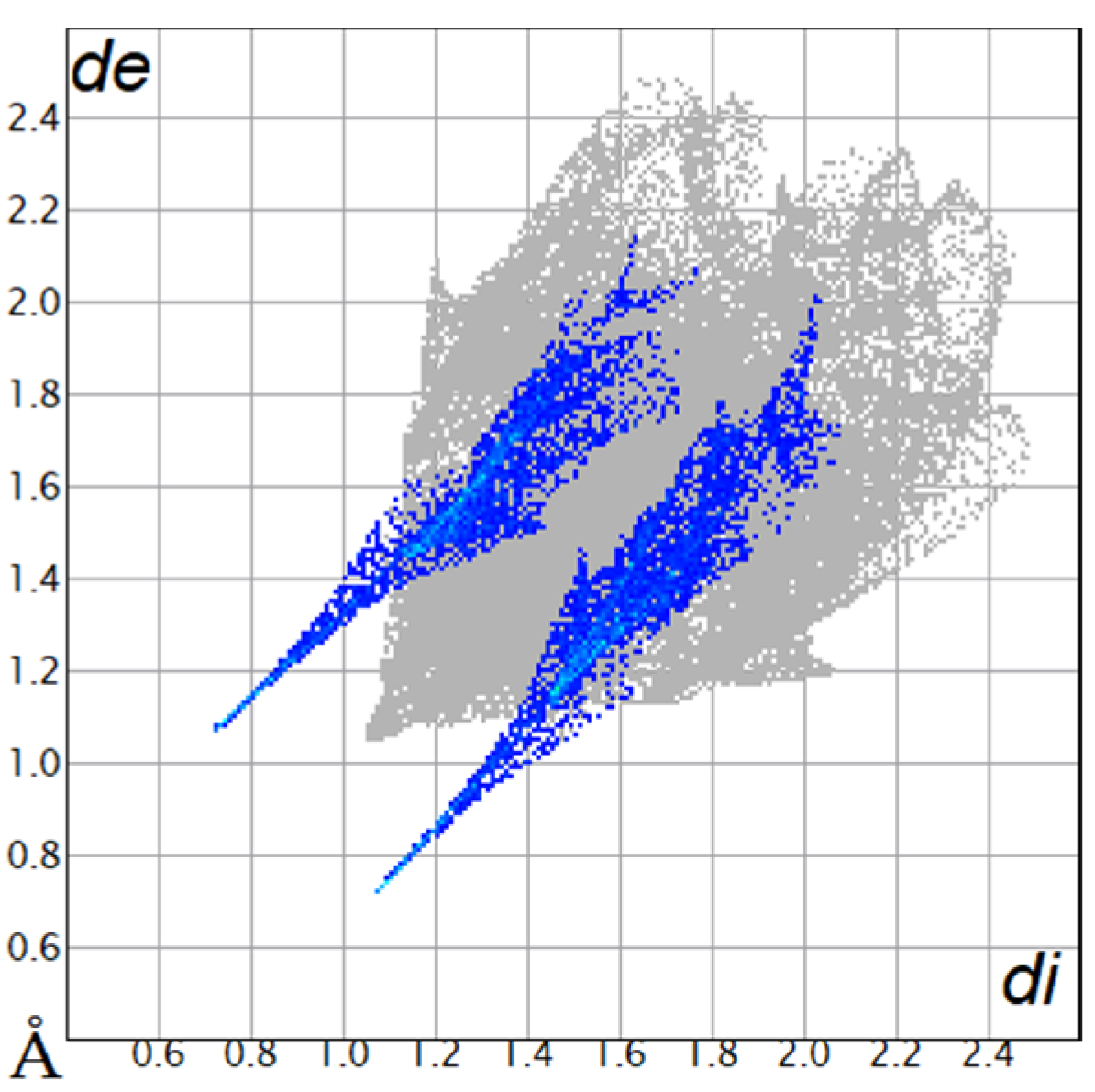 |
| C···H/H···C18.4% | 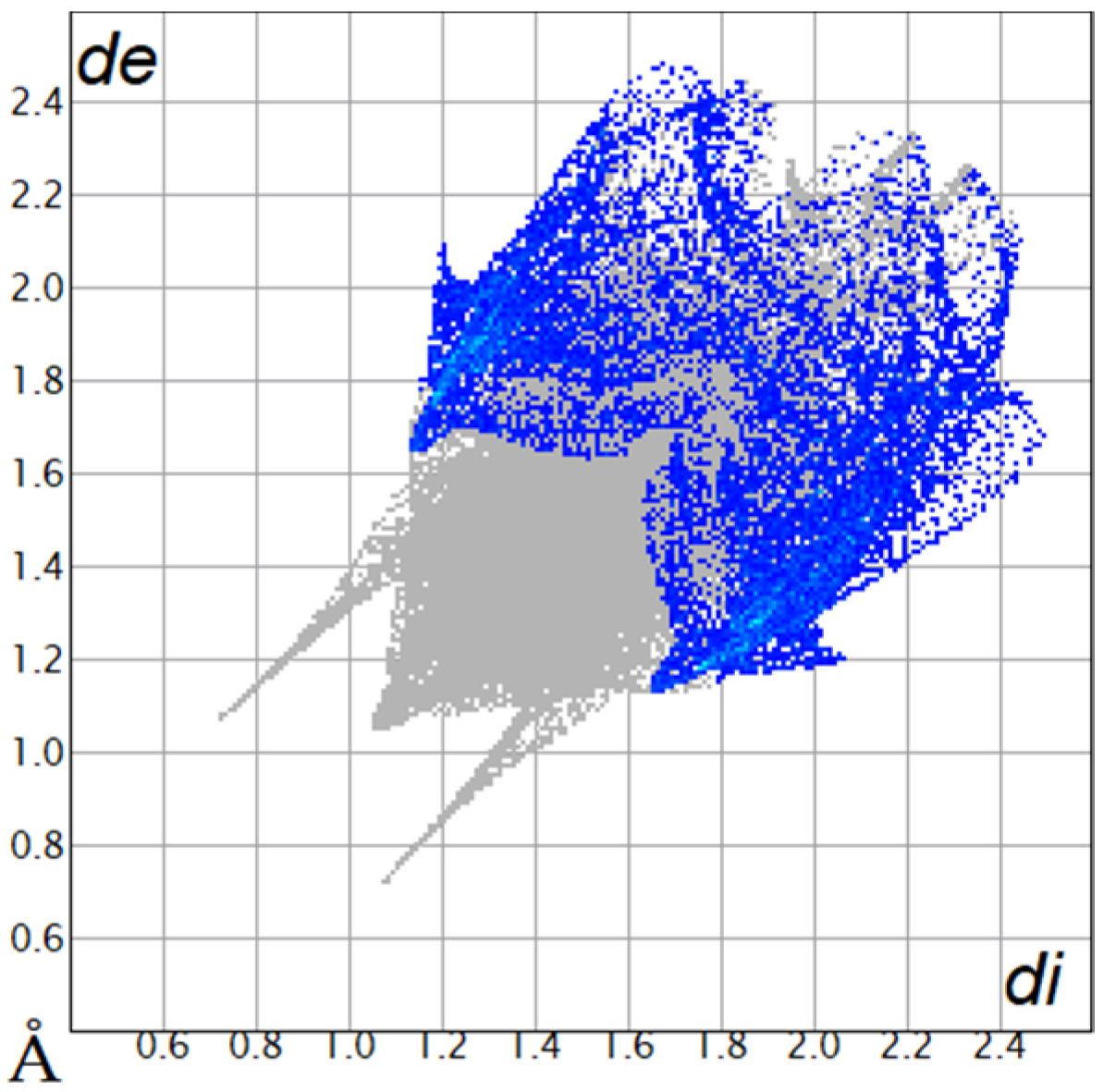 | 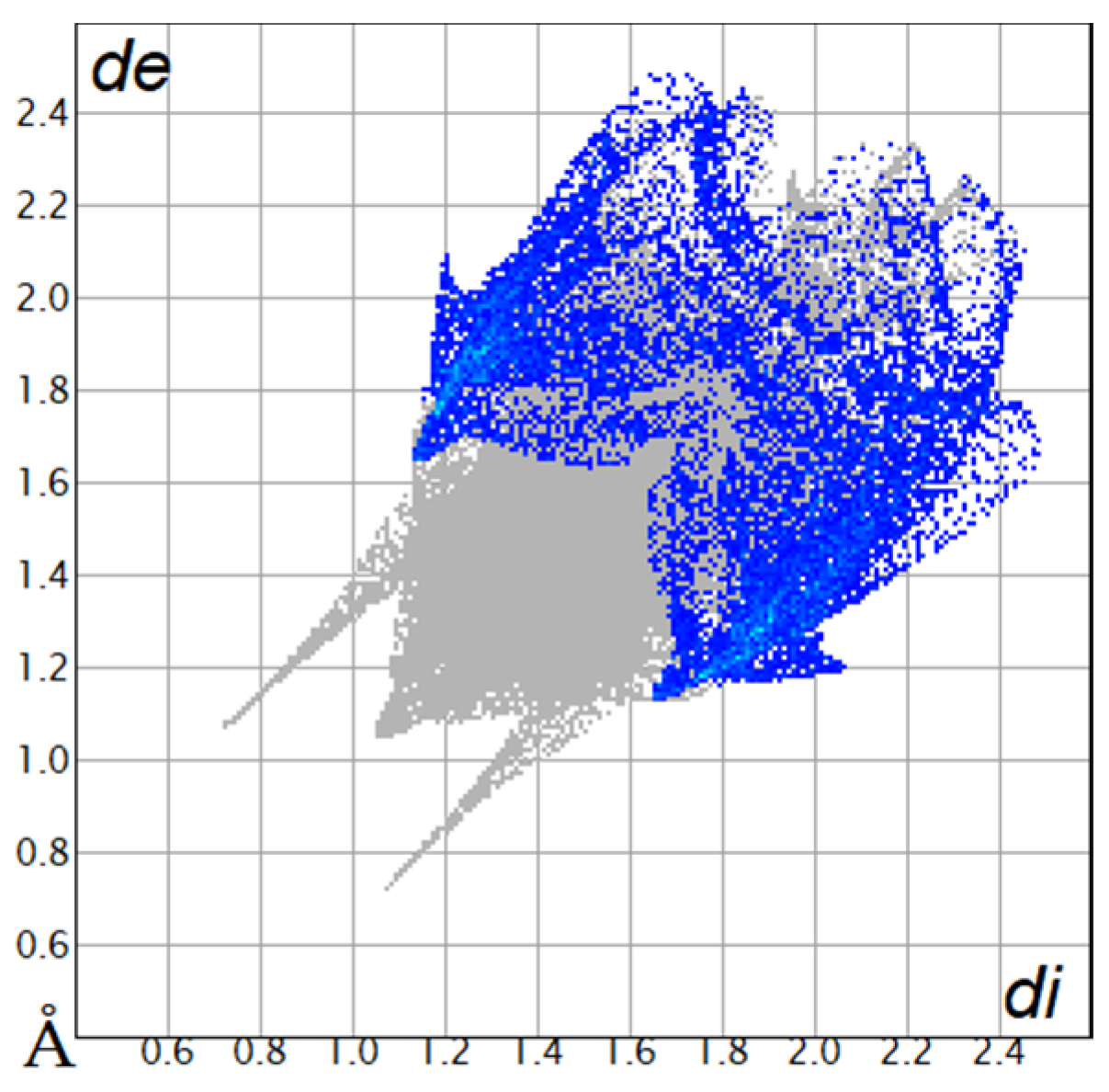 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez-Ricardo, Y.; Sánchez-López, D.; Meza-Morales, W.; Obregón-Mendoza, M.A.; Arenaza-Corona, A.; Germán-Acacio, J.M.; Toscano, R.A.; Zermeño-Ortega, M.R.; Morales-Morales, D.; Enríquez, R.G. Spontaneous Enantiomeric Resolution of 1,3-Diols from the Naphtylidene Derivative of 2,4-Pentanedione. Molbank 2022, 2022, M1457. https://doi.org/10.3390/M1457
Alvarez-Ricardo Y, Sánchez-López D, Meza-Morales W, Obregón-Mendoza MA, Arenaza-Corona A, Germán-Acacio JM, Toscano RA, Zermeño-Ortega MR, Morales-Morales D, Enríquez RG. Spontaneous Enantiomeric Resolution of 1,3-Diols from the Naphtylidene Derivative of 2,4-Pentanedione. Molbank. 2022; 2022(4):M1457. https://doi.org/10.3390/M1457
Chicago/Turabian StyleAlvarez-Ricardo, Yair, Dylan Sánchez-López, William Meza-Morales, Marco A. Obregón-Mendoza, Antonino Arenaza-Corona, Juan M. Germán-Acacio, Rubén A. Toscano, Miriam R. Zermeño-Ortega, David Morales-Morales, and Raúl G. Enríquez. 2022. "Spontaneous Enantiomeric Resolution of 1,3-Diols from the Naphtylidene Derivative of 2,4-Pentanedione" Molbank 2022, no. 4: M1457. https://doi.org/10.3390/M1457
APA StyleAlvarez-Ricardo, Y., Sánchez-López, D., Meza-Morales, W., Obregón-Mendoza, M. A., Arenaza-Corona, A., Germán-Acacio, J. M., Toscano, R. A., Zermeño-Ortega, M. R., Morales-Morales, D., & Enríquez, R. G. (2022). Spontaneous Enantiomeric Resolution of 1,3-Diols from the Naphtylidene Derivative of 2,4-Pentanedione. Molbank, 2022(4), M1457. https://doi.org/10.3390/M1457