1. Introduction
Functionalization of C–H bonds [
1,
2] has risen to be a powerful method for the transformation of inert C–H bonds into reactive ones. Accordingly, the bond activation science enables rapid access of functionalized molecules from simple non-reactive starting materials. In turn, C–H bond functionalization enables construction of complex molecules in an atom-economical and step-economical fashion. Site-selectivity remains a challenge in the environmentally benign strategy [
3,
4]. The problem has been tackled by development of monondentate [
5,
6] and bidentate directing groups [
7]. The Lewis-base-contained directing groups promote formation of cyclometallated complexes via chelation-assistance thus the setting stage for C–H bond functionalization. Specifically, relative thermodynamic stability favors the formation of five-membered chelates, allowing the formation of mono five-membered chelates (
1,
Figure 1) and double-five-membered chelates (
2,
Figure 1) by monodentate and bidentate directing groups, respectively (
Figure 1) [
8].
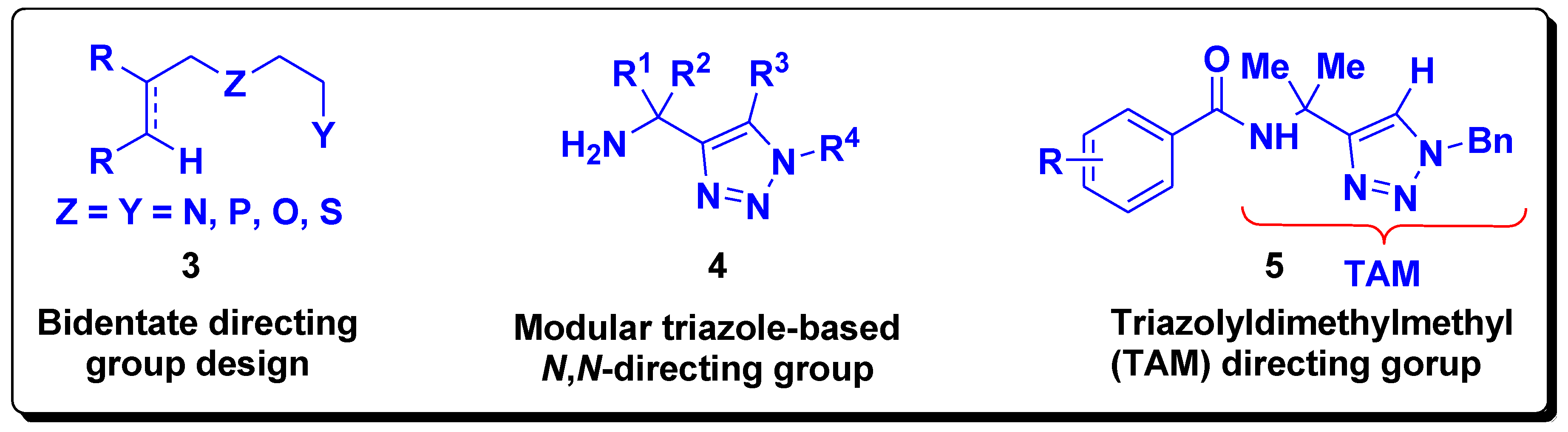
Formation of the thermodynamically stable five-membered chelates necessitates a two-carbon distance between the Lewis basic atom and the position to be functionalized and the same carbon distance among the two Lewis basic atoms in bidentate directing groups. Such structural requirements have triggered the design of bidentate directing groups (
3,
Figure 2) that led to modular triazole directing groups (
4,
Figure 2). Thus, Ackermann et al. have recently disclosed a novel design of a triazole-based
N,
N-bidentate (TAM) directing group (
5,
Figure 2) that enables different C–H bond functionalization reactions catalyzed by various transition metals [
9,
10].
In order to mimic the modular triazole design
4 (
Figure 2) and, in turn, mimic TAM group
5 (
Figure 2), we developed a design of the
N,
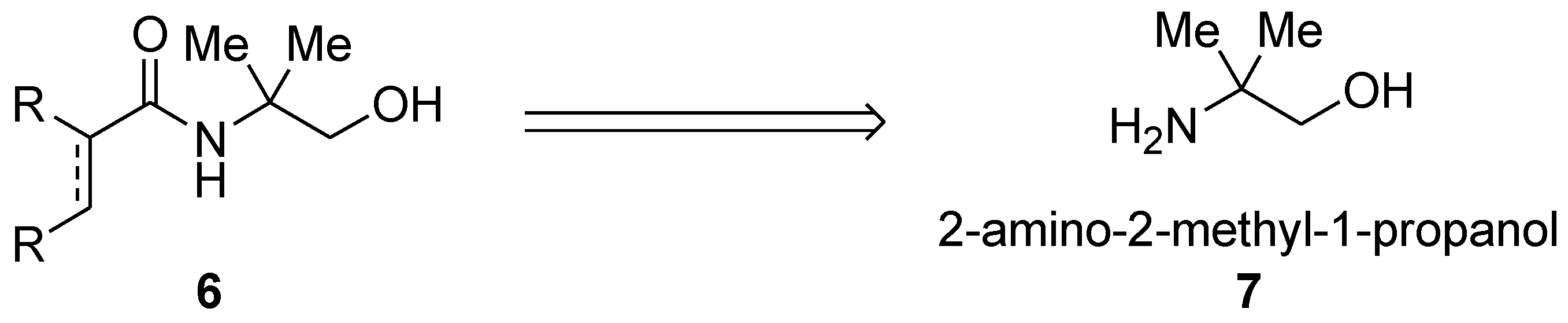
O-bidentate directing groups based on 2-aminoethanol. The basis was the presence of the two-carbon distance between the N and O Lewis basic atoms thus enabling the design-based formation of the five-membered and double five-membered cyclometallated complexes. Thus, amides containing hydroxymethyl(dimethylmethyl) were envisaged (
6,
Scheme 1) which would have as a retrosynthetic precursor 2-amino-2-methyl-1-propanol
7 (
Scheme 1).
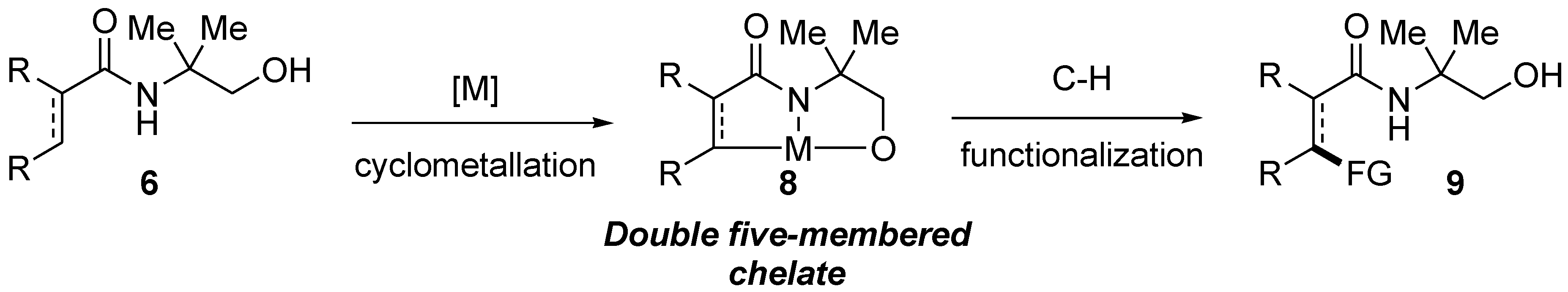
Based on the strategic design considerations above and as part of our research program on the development of design-based bidentate directing groups, [
11] we initiated synthesis of amides containing 2-amino-2-methyl-1-propanol as a potential
N,
O-bidentate directing group. The aim would ultimately be to test such amides in metal-catalyzed C–H bond functionalization reactions. Based on the possession of N and O Lewis basic atoms strategically separated by two carbons in the amides, it was projected that amides containing 2-amino-2-methyl-1-propanol (
6,
Scheme 2) could potentially allow formation of the corresponding double-five-membered chelate (
8,
Scheme 2) which sets the stage for the ultimate C–H bond cleavage leading to functionalized amide
9 (
Scheme 2). Therefore, amides containing 2-amino-2-methyl-1-propanol could potentially function as a bidentate directing group in C–H bond functionalization reactions.
In the present communication, a synthesis of the title compound,
N-(2-hydroxy-1,1-dimethylethyl)-3-methylbenzamide is reported. The importance of the compound lies in its potential reactivity as a
N,
O-bidentate directing group in metal-catalyzed C–H bond functionalization reactions. The literature reports studies involving a similar synthesis which suggests the possible formation of the title compound as a possible reaction intermediate or byproduct [
12,
13]. However, the compound was not isolated, its formation was not confirmed, and neither its characterization by spectroscopic methods nor structure determination have been reported. Li et al. [
12] reported Pd-catalyzed C–H fluorination of arenes using oxazolines as removable directing groups. The reported synthesis could have involved the title compound as a possible reaction intermediate or a byproduct. However, no spectroscopic characterization was reported. In addition, a similar oxazoline synthesis was reported by Swenton et al. [
13]. The reported synthesis could have involved the title compound as a possible reaction intermediate or byproduct. However, yet again, the title compound was not isolated and, thus, spectroscopic information or characterization have not been reported.
We wish to report herein a standard synthesis of the title compound,
N-(2-hydroxy-1,1-dimethylethyl)-3-methylbenzamide (
11), from the corresponding benzoyl chloride or benzoic acid. The compound was full characterized by various spectroscopic methods, elemental analysis, and its structure was determined and confirmed by X-ray analysis (Please see the
Supporting Information for spectra and other
Supporting Materials).
2. Results and Discussion
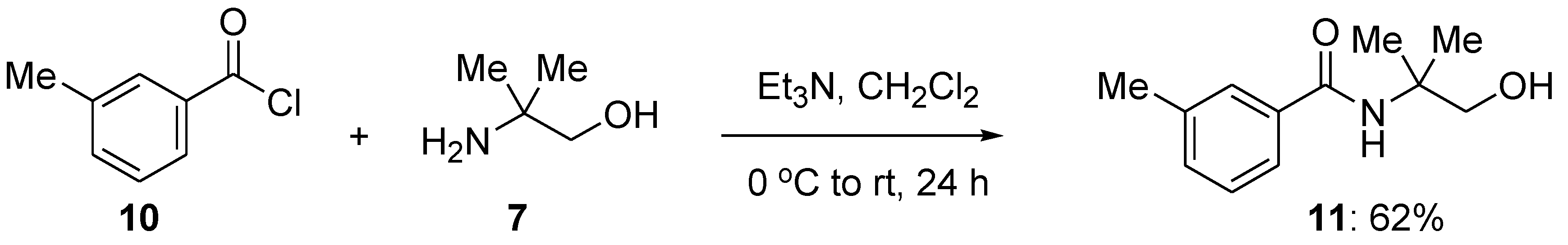
Toward investigating 2-amino-2-methyl-1-propanol as a potential directing group in metal-catalyzed C–H functionalization, 3-methylbenzamide containing the requisite amino alcohol indicated above, the title compound,
N-(2-hydroxy-1,1-dimethylethyl)-3-methylbenzamide (
11), was synthesized. Thus, 3-methylbenzolyl chloride (
10,
Scheme 3) was reacted with 2-amino-2-methyl-1-propanol (
7,
Scheme 3) under standard amide formation reaction conditions to give the corresponding amide in isolated 62% yield.
For comparison purposes, the target compound (
11) was also obtained using a carboxylic acid-amine coupling method. Thus the 3-methylbenzoic acid (
12,
Scheme 4) was treated with the coupling agent; DCC (dicyclohexylcarbodiimide) and 2-amino-2-methyl-1-propanol (
7) in the presence of DMAP (4-
N,
N-dimethylaminopyridine) to give the desired title compound,
N-(2-hydroxy-1,1-dimethylethyl)-3-methylbenzamide (
11), in 11% yield.
The product was characterized by various spectroscopic methods;
1H NMR,
13C NMR, IR, GC-MS, and its elemental composition was confirmed by elemental analysis. In addition, the crystal structure of the compound was determined and confirmed by X-ray analysis (
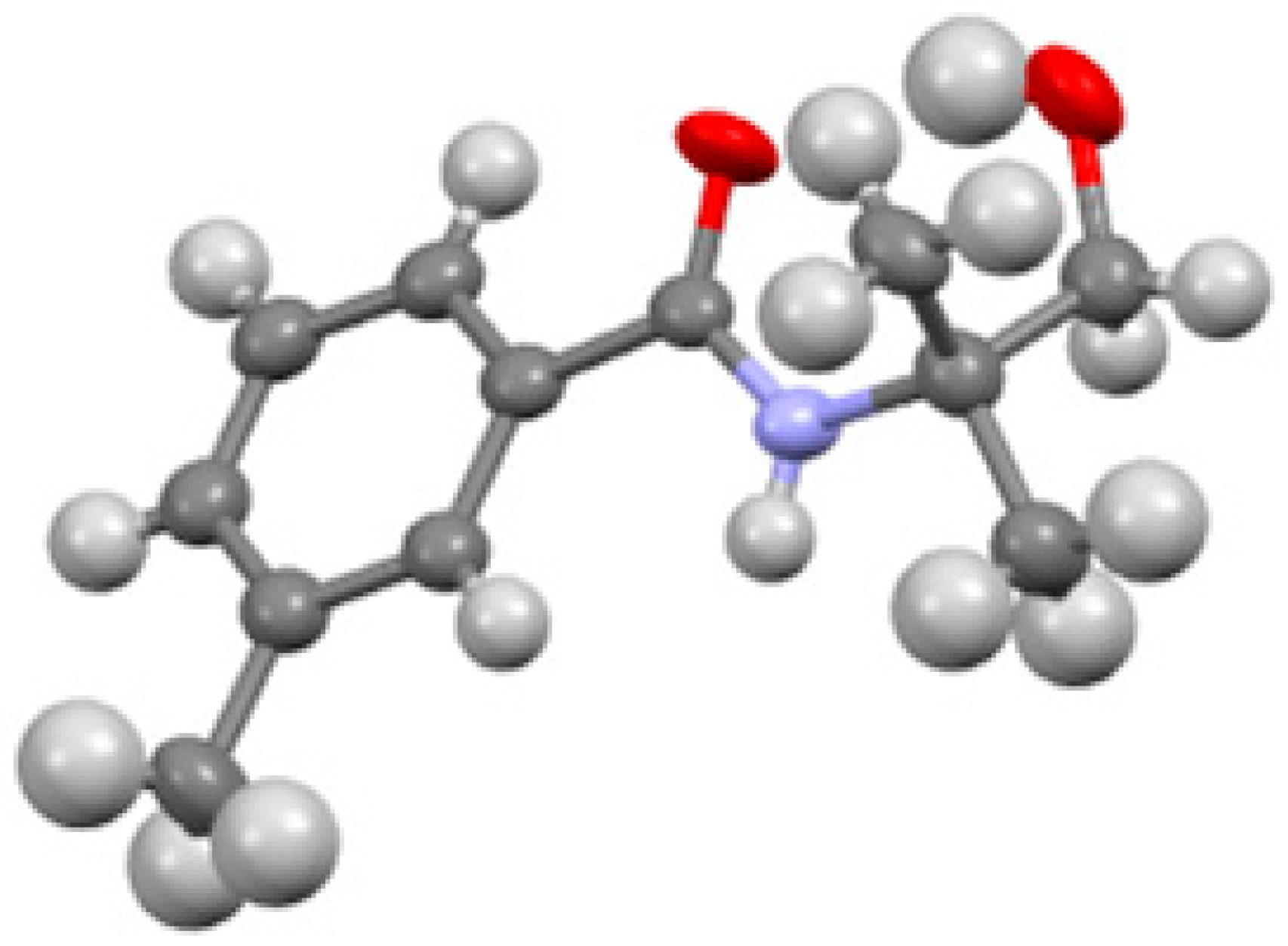
Figure 3). CCDC 1965367 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via
http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
Formation of amides was recently reported by Machetti et al. [
14] to proceed from methyl esters and amines. For comparison purposes, the title amide compound (
11) was attempted to be prepared using the reported ester amidation reaction. Thus, methyl 3-methylbenzoate was treated with an equimolar amount of 2-amino-2-methyl-1-propanol (
7) at 60 °C for 24 h. The reaction resulted in formation of only a very small amount of the desired amide product as detected by thin-layer chromatography (TLC) and
1H NMR.
3. Materials and Methods
3.1. General Methods
All chemicals, reagents, and solvents were purchased from chemical companies (Sigma–Aldrich Chemie GmbH, Taufkirchen, Germany) and were used as received without prior purification. Reactions that required dry conditions were performed in an inert atmosphere with Ar gas. Syringes and needles for the transfer of reagents were oven dried and cooled in a desiccator over silica gel before use. The reaction’s progress was monitored by thin-layer chromatography (TLC) on glass plates pre-coated with Merck silica gel. Thhe TLC plates were examined under a UV lamplight (UVGL-58 Handheld 254/365 nm). Büchi-USA rotary evaporators were used to evaporate solvents using appropriate temperatures. Flash column chromatography was performed using silica gel (Kieselgel) (70–230) mesh as an adsorbent. The purified products were characterized using analyses NMR (1H NMR, 13C NMR), IR, mass spectra, and melting points. Melting points were recorded on a GallenKamp-MPd350.bm2.5 melting point apparatus (Gallenkamp, Kent, UK). Attenuated total-reflectance IR spectra were recorded on pure samples on Agilent Technologies Cary 630 FTIR (Agilent, Santa Clara, CA, USA). 1H NMR spectra were recorded in CDCl3 on JEOL ECX-400 spectrometers (JEOL Ltd., Tokyo, Japan). 1H NMR chemical shifts (δ) were assigned in parts per million (ppm) downfield using an internal standard trimethyl silane (TMS) and were referenced to CDCl3, δ = 7.24. Abbreviations s, d, t, q, quin, sept, and m refer to singlet, doublet, triplet, quartet, quintet, septet, and multiplet, respectively. Chemical shifts in 13C spectra (175 MHz) were quoted in ppm and referenced to the central line of the CDCl3 triplet, δ C 77.0. Coupling constants (J) were recorded in hertz (Hz). The GC-MS spectra were obtained using an Agilent mass spectrometer (Agilent, Santa Clara, CA, USA). Elemental analysis was performed using an EuroEA Elemental Analyzer (configuration CHN (EuroVector Instruments & Software, Milano, Italy) with a calibration type of K-factor. Single-crystal X-ray structure determinations were performed at room temperature on a Stoe IPS II diffractometer using monochromatic Mo−Kα radiation (λ = 0.71073 Å). A multi-scan absorption correction was applied. The data reduction, including an empirical absorption correction using spherical harmonics, was implemented in LANA. The crystal structures were solved by direct methods using the online version of WinGX and then refined by full-matrix least-squares (SHELXL2014) on F2. The non-hydrogen atoms were refined anisotropically. All of the hydrogen atoms were positioned geometrically in idealized positions and refined with the riding model approximation with Uiso(H) = 1.2 or 1.5 Ueq(C). The molecular graphics program MERCURY from the Cambridge Structural Database (CSD) package was used.
3.2. Synthesis of N-(2-Hydroxy-1,1-dimethylethyl)-3-methylbenzamide (11)
3-Methylbenzoyl chloride (10) (1.30 mL, 9.86 mmol) was added dropwise under an atmosphere of N2 into a cold (0 °C ice-water bath) solution of 2-amino-2-methylpropan-1-ol (7) (0.50 mL, 5.2 mmol), in CH2Cl2 (20 mL). Triethylamine (Et3N (1.40 mL, 10.0 mmol) was then added to the 0 °C mixture under N2. The mixture was stirred for 1 h at 0 °C, allowed to warm up to room temperature, and then stirred for an additional 23 h. To the reaction mixture, an aqueous saturated NaHCO3 solution (20 mL) was added. The mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried over anhydrous MgSO4 and filtered. The solvents were evaporated under reduced pressure, and the crude residue was purified by flash chromatography (SiO2) using hexane:Et2O (5:1), giving the title compound 11 (0.66 g, 62%) as white crystals after recrystallization from CH2Cl2:hexane; Rf = 0.12 (Pet. Ether/EtOAc, 1:1) and mp = 81–83 °C. 1H NMR (400 MHz, CDCl3): δ 7.52 (s, 1H), 7.49 (d, J = 2.3 Hz, 1 H), 7.48 (d, J = 1.9 Hz, 1 H), 7.28 Hz (dd, J = 3.8, 3.3 Hz, 1 H), 7.25 (s, 1 H), 6.24 (s, 1 H), 3.66 (s, 2 H), 2.37 (s, 3 H), 1.39 (s, 6 H). 13C NMR (176 MHz, CDCl3): δ 168.8, 138.6, 134.8, 132.5, 128.6, 127.7, 123.9, 70.8, 56.5, 24.8, 24.4. IR (film): ν max/cm−1: 3340, 3198, 2924, 1627, 1535, 1548, 1481. Elemental analysis calculated: C (69.54), H (8.27), N (6.76), found (average of two runs): C (69.33), H (8.17), N (6.53).
3.3. Refinement Details of the X-ray Structure
Single-crystal X-ray structure determinations were performed at room temperature on a Stoe IPS II diffractometer using monochromatic Mo−K
α radiation (λ = 0.71073 Å). A multiscan absorption correction was applied. The data reduction, including an empirical absorption correction using spherical harmonics, implemented in LANA. The crystal structures were solved by direct methods using the online version of WinGX [
15] and then refined by full-matrix least-squares (SHELXL2014) on F
2 [
16,
17]. The nonhydrogen atoms were refined anisotropically. All of the hydrogen atoms were positioned geometrically in idealized positions and refined with the riding model approximation, with Uiso(H) = 1.2 or 1.5 Ueq(C). The molecular graphics the program MERCURY from the CSD package was used [
18]. The structure was solved by direct methods. The data used for the refinement are up to a resolution of 2-theta = 37.7 degrees as the intensity of the data dropped rapidly after this point. The flack parameter (x = −0.901) was refined by full-matrix least squares (i.e., using the TWIN/BASF commands in the
SHELXL.ins file) [
19]. Hydrogen atoms were included in calculated positions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
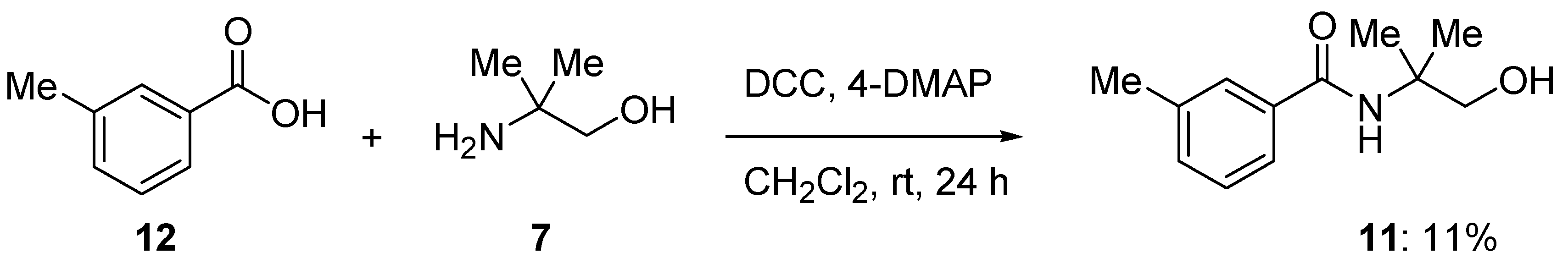{kind=link}






