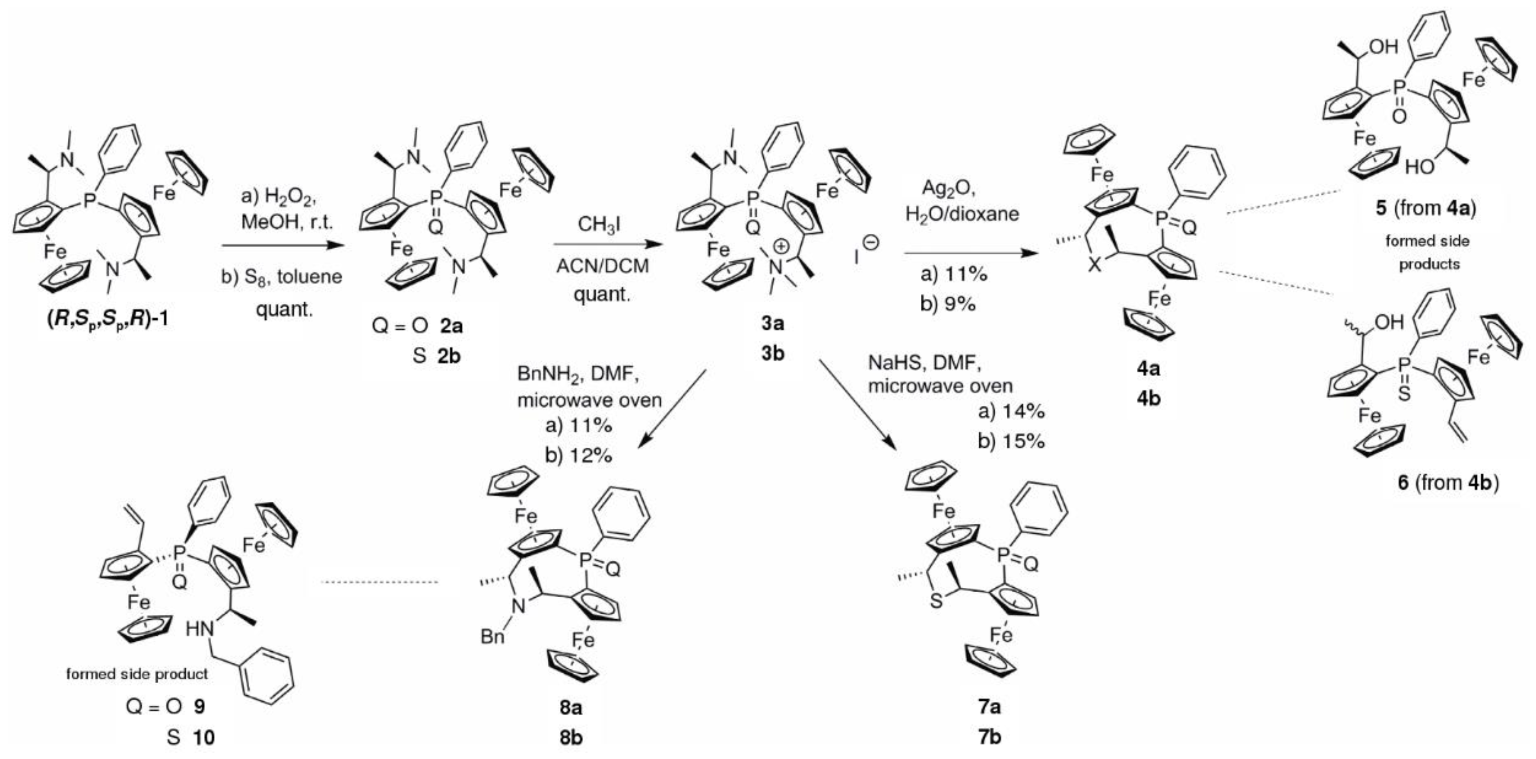
3.2. Synthesis
Bis{[(2Sp)-2-[(1R)-1-(dimethylamino)-ethyl]]ferrocenyl}phenyl phosphineoxide2a: [
19]
Diaminophosphine 1 (630 mg, 1.02 mmol) was dissolved in MeOH (8.0 mL) and cooled in an ice bath. Aqueous H2O2 (30%; 210 μL, 1.85 mmol, 1.82 equiv.) was added to the orange solution. The solution was warmed to r.t. and stirred for 40 min. The reaction mixture was quenched by adding 10% aqueous NaHSO3 solution followed by sufficient aqueous NaHCO3 solution until pH 7 was reached. The aqueous suspension was extracted using DCM (3 × 8 mL). The combined organic fractions were dried over MgSO4 and the solvent was removed under reduced pressure to yield diaminophosphineoxide 2a (647 mg, quant.) as an orange solid. The highly polar product was pure enough for the next step.
1H-NMR (600 MHz, CDCl3) δ = 7.80 (dd, J = 11.4, 8.0 Hz, 2H, C6H5); 7.43–7.35 (m, 3H, C6H5); 4.52 (q, J = 6.8 Hz, 1H, C2H4); 4.49 (m, 1H, C5H3); 4.44 (m, 1H, C5H3); 4.36 (m, 1H, C5H3); 4.33 (m, 1H, C5H3); 4.28 (m, 1H, C5H3); 4.25 (m, 1H, C5H3); 4.23 (s, 5H, C5H5); 3.73 (s, 5H, C5H5); 3.31–3.26 (m, 1H, C2H4); 2.26 (s, 6H, NCH3); 1.53 (s, 6H, NCH3); 1.47 (d, J = 6.8 Hz, 3H, C2H4); 1.21–1.15 (m, 3H, C2H4) ppm. 13C-NMR δ = 130.07 (d, JCP = 9.2 Hz, CH, C6H5); 129.44 (CH, C6H5); 127.20 (CH, C6H5); 89.90 (Cq, C5H3); 85.23 (Cq, C5H3); 75.14 (Cq, C5H3); 73.60 (CH, C5H3); 71.89 (CH, C5H3); 71.25 (Cq, C5H3); 71.72 (d, JCP = 14.9 Hz, CH, C5H3); 70.34 (CH, C5H5); 70.20 (CH, C5H5); 69.59 (d, JCP = 9.8 Hz, CH, C5H3); 69.32 (CH, C5H3); 67.91 (CH, C5H3); 55.17 (CH, C2H4); 40.97 (CH3, NCH3); 34.66 (CH, C2H4); 14.93 (CH3, NCH3); 13.05 (CH3, C2H4); 13.04 (CH3, C2H4) ppm; 1 Cq not observed. 31P-NMR δ = 29.59 (s) ppm. HRMS: m/z calculated for C34H41Fe2N2OP [M + H]+: 637.1734, found: 637.1726.
{[(2Sp)-2-[(1R)-1-(dimethylamino)-ethyl]]ferrocenyl}{[(2Sp)-2-[(1R)-1-(trimethylammonium)-ethyl]]ferrocenyl}phenyl phosphineoxide iodide (
3a): [
20]
Diaminophosphineoxide 2a (642 mg, 1.01 mmol) was dissolved in 15 mL of dry MeCN and 15 mL of dry DCM in a flame-dried Schlenk tube under Ar. MeI (375 μL, 6.00 mmol, 5.94 equiv.) was added to the solution and stirred for 2 h at r.t. Precipitation of ammonium iodide 3a was induced by adding Et2O (10 mL) yielding the salt as an orange powder (780 mg, quant.). HRMS: m/z calculated for C35H44Fe2IN2OP [M − I]+: 651.1885, found: 651.1868.
{[(2Sp)-2-[(1R)-1-(dimethylamino)-ethyl]]ferrocenyl}{[(2Sp)-2-[(1R)-1-(trimethylammonium)-ethyl]]ferrocenyl}phenyl phosphinesulfide iodide (3b):
Similar procedure as given for 3a yielded 3b from 2b in quantitative yield.
HRMS: m/z calculated for C35H44Fe2IN2PS [M − I]+: 667.1662, found: 667.1644.
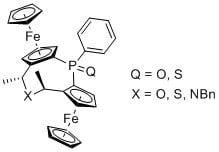
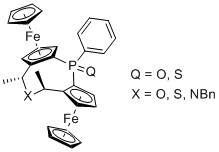
(2Sp,4R,6R,8Sp)-4,6-Dimethyl-1-phenyl-diferroceno-5-oxa-1-phosphineoxide (4a):
Ammonium iodide salt 3a (87 mg, 0.11 mmol) was dissolved in water (1.5 mL) and dioxane (1 mL). The solution was warmed to 50 °C and Ag2O (19 mg, 0.08 mmol, 1.5 equiv.) was added. The resulting suspension was stirred for 1 h. After complete consumption of the ammonium iodide salt 3a the mixture was cooled to r.t. and the solid material was removed by filtration. The solution was extracted with DCM (3 × 3 mL) and the combined organic fractions were washed with water (10 mL) and brine (10 mL) and dried (MgSO4). The solvent was removed under reduced pressure and the residue was purified by MPLC (SiO2, 75→100% EtOAc in heptane) yielding oxa cycle 4a (7 mg, 11%) as a glassy orange solid and dihydroxyphosphineoxide 5 (4 mg, 6%).
4a: 1H-NMR (600 MHz, CDCl3) δ = 8.02–7.95 (m, 2H, C6H5); 7.51–7.47 (m, 3H, C6H5); 5.13 (s, 1H, C5H3); 4.65 (q, J = 6.5 Hz, 1H, C2H4); 4.52 (s, 1H, C5H3); 4.45 (s, 1H, C5H3); 4.43 (s, 1H, C5H3); 4.38–4.36 (m, 1H, C5H3); 4.36–4.34 (m, 1H, C5H3); 4.24 (s, 5H, C5H5); 4.12 (q, J = 7.2 Hz, 1H, C2H4); 3.73 (s, 5H, C5H5); 1.53 (d, J = 6.5 Hz, 3H, C2H4); 1.51 (d, J = 6.4 Hz, 3H, C2H4) ppm. 13C-NMR δ = 138.11 (d, JCP = 112.4 Hz, Cq, C6H5); 131.56 (d, JCP = 10.0 Hz, CH, C6H5); 131.14 (d, JCP = 2.6 Hz, CH, C6H5); 127.79 (d, JCP = 12.3 Hz, CH, C6H5); 91.83 (d, JCP = 13.2 Hz, Cq, C5H3); 88.93 (d, JCP = 14.1 Hz, Cq, C5H3); 76.12 (d, JCP = 10.2 Hz, Cq, C5H3); 75.32 (Cq, C5H3); 74.74 (d, JCP = 10.6 Hz, CH, C5H3); 73.78 (d, JCP = 13.5 Hz, CH, C5H3); 70.44 (CH, C5H5); 70.17 (d, JCP = 11.1 Hz, CH, C5H3); 70.16 (CH, C5H5); 69.88 (d, JCP = 10.2 Hz, CH, C5H3); 69.15 (d, JCP = 9.8 Hz, CH, C5H3); 69.01 (d, JCP = 10.1, CH, C5H3); 68.31 (CH, C2H4); 66.13 (CH, C2H4); 20.93 (CH3, C2H4); 20.78 (CH3, C2H4) ppm. 31P-NMR δ = 31.13 (s) ppm. HRMS: m/z calculated for C30H29Fe2O2P [M + Na]+: 587.0502, found: 587.0507; [2M + Na]+ 1151.1106, found: 1151.1113.
5: 1H-NMR (600 MHz, CDCl3) δ = 7.95–7.90 (m, 2H, C6H5); 7.55–7.50 (m, 3H, C6H5); 5.93 (d, J = 5.4 Hz, 1H, OH); 5.22 (m, 1H, C5H3); 4.76 (m, 1H, C2H4); 4.57 (d, J = 5.4 Hz, 1H, C5H3); 4.56 (s, 1H, OH); 4.48 (pq, J = 2.4 Hz, 1H, C5H3); 4.44 (m, 1H, C5H3); 4.41 (m, 2H, C5H3, C2H4); 4.38 (pq, J = 2.4 Hz, 1H, C5H3); 4.17 (s, 5H, C5H5); 3.75 (s, 5H, C5H5); 1.64 (d, J = 6.7 Hz, 3H, C2H4); 1.21 (d, J = 6.7 Hz, 3H, C2H4) ppm. 13C-NMR δ = 136.54 (d, JCP = 110.2 Hz, Cq, C6H5); 131.55 (d, JCP = 2.9 Hz, CH, C6H5); 129.93 (d, JCP = 9.5 Hz, CH, C6H5); 128.45 (d, JCP = 12.1 Hz, CH, C6H5); 98.73 (d, JCP = 10.4 Hz, Cq, C5H3); 97.57 (d, JCP = 11.3 Hz, Cq, C5H3); 72.64 (Cq, C5H3); 71.85 (Cq, C5H3); 71.75 (d, JCP = 14.6 Hz, CH, C5H3); 70.97 (d, JCP = 15.0 Hz, CH, C5H3); 70.81 (d, JCP = 10.0 Hz, CH, C5H3); 70.46 (CH, C5H5); 70.45 (CH, C5H5); 70.27 (d, JCP = 11.0 Hz, CH, C5H3); 69.86 (d, JCP = 11.0 Hz, CH, C5H3); 69.64 (d, JCP = 9.8 Hz, CH, C5H3); 65.35 (CH, C2H4); 65.30 (CH, C2H4); 22.98 (CH3, C2H4); 21.95 (CH3), C2H4) ppm. 31P-NMR δ = 38.45 (s) ppm. HRMS: m/z calculated for C30H31Fe2O3P [M + Na]+: 605.0607, found: 605.0595; [2M + Na]+: 1187.1317, found: 1187.1295.
(2Sp,4R,6R,8Sp)-4,6-Dimethyl-1-phenyl-diferroceno-5-oxa-1-phosphinesulfide (4b):
Similar procedure as given for 4a yielded cycle 4b from 3b in 9% yield and 6% of eliminated side product 6.
4b: 1H-NMR (600 MHz, CDCl3) δ = 8.13–8.07 (m, 2H, C6H5); 7.45–7.38 (m, 3H, C6H5); 5.44 (m, 1H, C5H3); 4.61 (m, 1H, C5H3); 4.59–4.55 (m, 2H, C2H4); 4.47 (m, 1H, C5H3); 4.45–4.39 (m, 2H, C5H3); 4.37 (m, 1H, C5H3); 4.16 (s, 5H, C5H5); 4.00 (s, 5H, C5H5); 1.45 (d, J = 6.4 Hz, 3H, C2H4); 1.41 (d, J = 6.7 Hz, 3H, C2H4) ppm. 13C-NMR δ = 137.31 (d, JCP = 90.0 Hz, Cq, C6H5); 132.51 (d, JCP = 11.2 Hz, CH, C6H5); 130.82 (d, JCP = 3.0 Hz, CH, C6H5); 127.33 (d, JCP = 12.7 Hz, CH, C6H5); 92.84 (d, JCP = 9.9 Hz, Cq, C5H3); 89.73 (d, JCP = 10.2 Hz, Cq, C5H3); 77.97 (Cq, C5H3); 77.42 (d, JCP = 15.5 Hz, CH, C5H3); 76.15 (Cq, C5H3); 75.04 (d, JCP = 15.2 Hz, CH, C5H3); 70.82 (d, JCP = 8.8 Hz, CH, C5H3); 70.81 (CH, C5H5); 70.41 (CH, C5H5); 69.58 (d, JCP = 11.7 Hz, CH, C5H3); 69.41 (d, JCP = 11.5 Hz, CH, C5H3); 69.33 (d, JCP = 9.0 Hz, CH, C5H3); 65.79 (2 CH, C2H4); 21.22 (CH3, C2H4); 20.56 (CH3, C2H4) ppm. 31P-NMR δ = 43.82 (s) ppm. HRMS: m/z calculated for C30H29Fe2OPS [M + Na]+: 603.0273, found: 603.0279; [2M + Na]+ 1183.0649, found: 1183.0662.
6: 1H-NMR (600 MHz, CDCl3) δ = 8.10 (dd, J = 17.6, 10.8 Hz, 1H, C2H3); 7.87–7.81 (m, 2H, C6H5); 7.51–7.42 (m, 3H, C6H5); 5.49 (dd, J = 17.6, 1.6 Hz; 1H, C2H3); 5.23–5.17 (m, 1H, C5H3); 5.20 (dd, J = 10.8, 1.7 Hz, 1H, C2H3); 4.88 (m, 1H, C5H3); 4.49 (m, 1H, C5H3); 4.34 (s, 5H, C5H5); 4.33 (m, 1H, C5H3); 4.24 (m, 1H, C5H3); 4.17 (s, 5H, C5H5); 3.77 (m, 1H, C5H3); 3.71 (m, 1H, OH); 2.41 (d, J = 5.3 Hz; 1H, C2H4); 1.26 (d, J = 6.6 Hz, 3H, C2H4) ppm. 13C-NMR δ = 135.30 (d, JCP = 87.1 Hz, Cq, C6H5); 134.25 (CH, C6H5); 132.10 (d, JCP = 10.3 Hz, CH, C6H5); 131.38 (d, JCP = 2.8 Hz, CH, C6H5); 127.96 (d, JCP = 12.1 Hz, CH, C2H3); 111.74 (CH2, C2H3); 94.90 (d, JCP = 12.3 Hz, Cq, C5H3); 88.43 (d, JCP = 11.8 Hz, Cq, C5H3); 78.56 (d, JCP = 95.4 Hz, Cq, C5H3); 75.05 (d, JCP = 12.0 Hz, CH, C5H3); 74.97 (d, JCP = 12.6 Hz, CH, C5H3); 73.48 (d, JCP = 96.0 Hz, Cq, C5H3); 71.22 (CH, C5H5); 71.00 (d, JCP = 9.7 Hz, CH, C5H3); 70.72 (CH, C5H5); 70.10 (d, JCP = 10.4 Hz, CH, C5H3); 68.38 (d, JCP = 9.1 Hz, CH, C5H3); 68.04 (d, JCP = 10.5 Hz, CH, C5H3); 64.38 (CH, C2H4); 21.91 (CH3, C2H4) ppm. 31P-NMR δ = 40.51 (s) ppm. HRMS: m/z calculated for C30H29Fe2OPS [M]+ 580.0376, found: 580.0360; [M + Na]+ 603.0273, found: 603.0273; [M + K]+ 619.0013, found: 619.0018.
(2Sp,4R,6R,8Sp)-4,6-Dimethyl-1-phenyl-diferroceno-5-sulfa-1-phosphineoxide (7a):
Ammonium iodide salt 3a (75 mg, 0.10 mmol) and NaHS·H2O (7.5 mg, 0.10 mmol, 1.0 equiv.) were dissolved in 8 mL of DMF and heated for 8 min at 100 °C in a microwave oven. The mixture was cooled to r.t., 10 mL of DCM were added and the organic layer was washed four times with 50 mL of water each, once with 20 mL of brine and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography (Al2O3; 0→100% EtOAc in heptane) yielding 14% of the intended sulfidephosphineoxide diferrocenyl cycle 7a (8 mg) as a glassy orange solid.
1H-NMR(700 MHz, CDCl3) δ = 7.92–7.85 (m, 2H, C6H5); 7.52–7.48 (m, 3H, C6H5); 5.30 (s, 1H, C5H3); 5.11 (s, 1H, C5H3); 4.52 (s, 1H, C5H3); 4.37 (s, 1H, C5H3); 4.36 (s, 1H, C5H3); 4.35 (s, 5H, C5H5); 4.24 (s, 1H, C5H3); 3.83 (s, 5H, C5H5); 3.74 (q, J = 6.9 Hz, 1H, C2H4); 3.21 (q, J = 7.1 Hz, 1H, C2H4); 1.58 (d, J = 7.2 Hz, 3H, C2H4); 1.51 (d, J = 7.0 Hz, 3H, C2H4) ppm. 13C-NMR δ = 138.59 (d, JCP = 111.7 Hz, Cq, C6H5); 131.81 (d, JCP = 9.8 Hz, CH, C6H5); 131.17 (d, JCP = 2.7 Hz, CH, C6H5); 127.66 (d, JCP = 12.1 Hz, CH, C6H5); 96.88 (d, JCP = 13.2 Hz, Cq, C5H3); 92.56 (d, JCP = 13.8 Hz, Cq, C5H3); 74.83 (d, JCP = 37.5 Hz, Cq, C5H3); 74.20 (d, JCP = 42.9 Hz, Cq, C5H3); 73.50 (d, JCP = 10.7 Hz, CH, C5H3); 73.32 (d, JCP = 13.8 Hz, CH, C5H3); 70.36 (CH, C5H5); 70.32 (d, JCP = 11.8 Hz, CH, C5H3); 70.11 (d, JCP = 10.1 Hz, CH, C5H3); 69.95 (CH, C5H5); 67.45 (d, JCP = 9.6 Hz, CH, C5H3); 67.43 (d, JCP = 10.0 Hz, CH, C5H3); 36.62 (CH, C2H4); 35.71 (CH, C2H4); 20.28 (CH3, C2H4); 20.20 (CH3, C2H4) ppm. 31P-NMR δ = 29.47 (s) ppm. HRMS: m/z calculated for C30H29Fe2OPS [M]+: 580.0376, found: 580.0369.
(2Sp,4R,6R,8Sp)-4,6-Dimethyl-1-phenyl-diferroceno-5-sulfa-1-phosphinesulfide (7b):
Similar procedure as given for 7a yielded cycle 7b from 3b in 15% yield.
1H-NMR (600 MHz, CDCl3) δ = 7.95–7.90 (m, 2H, C6H5); 7.47–7.43 (m, 1H, C6H5); 7.43–7.38 (m, 2H, C6H5); 5.38 (m, 1H, C5H3); 4.64 (q, J = 2.2 Hz, 1H, C5H3); 4.50 (m, 1H, C5H3); 4.42–4.40 (m, 1H, C5H3); 4.40–4.38 (m, 2H, C5H3); 4.28 (s, 5H, C5H5); 4.20 (s, 5H, C5H5); 3.63 (q, J = 6.8 Hz, 1H, C2H4); 2.96 (q, J = 7.3 Hz, 1H, C2H4); 1.54 (d, J = 7.3 Hz, 3H, C2H4); 1.31 (d, J = 6.8 Hz, 3H, C2H4) ppm. 13C-NMR δ = 137.96 (d, JCP = 88.8 Hz, Cq, C6H5); 132.57 (d, JCP = 11.0 Hz, CH, C6H5); 131.13 (d, JCP = 3.1 Hz, CH, C6H5); 127.45 (d, JCP = 12.5 Hz, CH, C6H5); 96.94 (d, JCP = 11.0 Hz, Cq, C5H3); 92.90 (d, JCP = 10.6 Hz, Cq, C5H3); 75.32 (d, JCP = 15.6 Hz, CH, C5H3); 75.02 (Cq, C5H3); 74.42 (Cq, C5H3); 73.75 (d, JCP = 15.0 Hz, CH, C5H3); 70.91 (CH, C5H5); 70.29 (CH, C5H5); 70.22 (d, JCP = 11.5 Hz, CH, C5H3); 70.05 (d, JCP = 11.5 Hz, CH, C5H3); 68.58 (d, JCP = 8.6 Hz, CH, C5H3); 68.34 (d, JCP = 9.1 Hz, CH, C5H3); 37.27 (CH, C2H4); 35.39 (CH, C2H4); 20.82 (CH3, C2H4); 20.79 (CH3, C2H4) ppm. 31P-NMR δ = 42.92 (s) ppm. HRMS: m/z calculated for C30H29Fe2PS2 [M]+: 596.0147, found: 596.0141.
(2Sp,4R,6R,8Sp)-4,6-Dimethyl-5-benzyl-1-phenyl-diferroceno-5-amino-1-phosphineoxide (8a):
Ammonium iodide salt 3a (77 mg, 0.10 mmol) and BnNH2 (11 μL, 0.10 mmol, 1.0 equiv.) were dissolved in 8 mL of DMF and heated for 8 min at 100 °C in a microwave oven. The reaction mixture was cooled to r.t., 10 mL of DCM were added and the organic solution was washed four times with 50 mL of water each, once with 20 mL of brine and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography (Al2O3; 0→100% EtOAc in heptane) yielding 11% of the intended benzylic aminephosphineoxide diferroceno cycle 8a (7 mg) as a glassy orange solid as well as 8% of the sec. amine elimination product 9 (5 mg).
8a: 1H-NMR (600 MHz, CDCl3) δ = 8.11–8.06 (m, 2H, C6H5); 7.57–7.50 (m, 3H, C6H5); 7.41 (d, J = 7.4 Hz, 2H, C7H7); 7.28–7.24 (m, 2H, C7H7); 7.17 (pt, J = 7.3 Hz, 1H, C7H7); 4.80 (m, 1H, C5H3); 4.47 (m, 1H, C5H3); 4.39 (m, 2H, C5H3); 4.35 (m, 1H, C5H3); 4.29 (m, 1H, C5H3); 4.24 (s, 5H, C5H5); 4.04 (q, J = 6.8 Hz, 1H, C2H4); 3.94 (d, J = 16.4 Hz, 1H, C7H7); 3.90 (s, 5H, C5H5); 3.73 (q, J = 6.6 Hz, 1H, C2H4); 3.39 (d, J = 16.3 Hz, 1H, C7H7); 1.26 (d, J = 6.5 Hz, 3H, C2H4); 1.25 (d, J = 6.9 Hz, 3H, C2H4) ppm. 13C-NMR δ = 143.69 (Cq, C7H7); 137.02 (d, JCP = 110.2 Hz, Cq, C6H5); 132.35 (d, JCP = 9.38 Hz, CH, C6H5); 131.20 (d, JCP = 2.7 Hz, CH, C6H5); 127.82 (CH, C7H7); 127.63 (d, JCP = 5.5 Hz, CH, C6H5); 127.54 (d, JCP = 6.6, CH, C7H7); 126.01 (CH, C7H7); 93.20 (d, JCP = 13.2 Hz, Cq, C5H3); 93.06 (d, JCP = 13.6 Hz, Cq, C5H3); 75.38 (d, JCP = 22.2 Hz, Cq, C5H3); 74.62 (d, JCP = 18.2 Hz, Cq, C5H3); 73.65 (d, JCP = 11.4 Hz, CH, C5H3); 72.64 (d, JCP = 13.9 Hz, CH, C5H3); 70.45 (CH, C5H5); 70.39 (d, JCP = 10.6 Hz, CH, C5H3); 70.17 (CH, C5H5); 69.81 (d, JCP = 11.2 Hz, CH, C5H3); 69.58 (d, JCP = 9.6 Hz, 2 CH, C5H3); 54.66 (CH, C2H4); 53.55 (CH, C2H4); 53.12 (CH2, C7H7); 21.44 (CH3, C2H4); 20.52 (CH3, C2H4) ppm. 31P-NMR δ = 29.63 (s) ppm. HRMS: m/z calculated for C37H36Fe2NOP [M + H]+: 654.1306, found: 654.1306; [M + Na]+: 676.1126, found: 676.1129.
9: 1H-NMR (600 MHz, CDCl3) δ = 7.80–7.69 (m, 3H, C6H5); 7.55–7.34 (m, 2H, C6H5); 7.33–7.27 (m, 3H, C7H7); 7.16–7.10 (m, 2H, C7H7); 6.84–6.80 (m, 1H, C2H3); 5.46 (dd, J = 17.7, 1.6 Hz, 1H, C2H3); 5.16 (dd, J = 10.8, 10.6 Hz, 1H, C2H3); 4.86 (m, 1H, C5H3); 4.49 (m, 1H, C2H4); 4.41 (pq, J = 2.4 Hz, 1H, C5H3); 4.39 (pt, J = 2.0 Hz, 1H, C5H3); 4.36 (m, 1H, C5H3); 4.27 (s, 5H, C5H5); 4.24 (s, 1H, C5H3); 3.93 (s, 5H, C5H5); 3.89 (d, J = 4.3 Hz, 1H, C5H3); 3.20 (s, 2H, C7H7); 1.43 (d, J = 6.6, 3H, C2H4) ppm. 13C-NMR δ = 133.52 (Cq, C6H5); 131.28 (CH, C6H5); 130.29 (CH, C6H5); 128.38 (CH, C6H5); 128.26 (CH, C7H7); 128.15 (CH, C7H7); 127.95 (CH, C7H7); 111.43 (CH2, C2H3); 90.13 (Cq, C2H3); 86.08 (Cq, C5H3); 71.65 (CH, C5H3); 71.26 (CH, C5H3); 71.17 (CH, C5H3); 70.81 (CH, C5H3); 70.73 (CH, C5H5); 70.31 (CH, C5H5); 70.28 (CH, C5H3); 69.58 (CH, C5H3); 67.25 (CH, C2H4); 53.15 (CH2, C7H7); 20.56 (CH3, C2H4) ppm; 4 Cq not observed. 31P-NMR δ = 30.54 (s) ppm. HRMS: m/z calculated for C37H36Fe2NOP [M + H]+: 654.1306, found: 654.1314.
(2Sp,4R,6R,8Sp)-4,6-Dimethyl-5-benzyl-1-phenyl-diferroceno-5-amino-1-phosphinesulfide (8b):
Similar procedure as given for 8a yielded cycle 8b from 3b in 12% yield and side product 10 in 13% yield.
8b: 1H-NMR (600 MHz, CDCl3) δ = 7.91 (ddd, J = 13.6, 7.8, 1.5 Hz, 2H, C6H5); 7.47–7.41 (m, 3H, C6H5); 7.31 (d, J = 7.5 Hz, 2H, C7H7); 7.28–7.24 (m, 2H, C7H7); 7.17 (pt, J = 7.2 Hz, 1H, C7H7); 5.36 (m, 1H, C5H3); 4.65 (dd, J = 4.3, 2.4 Hz, 1H, C5H3); 4.57 (m, 1H, C5H3); 4.48 (m, 1H, C5H3); 4.46 (m, 1H, C5H3); 4.38 (m, 1H, C5H3); 4.21 (s, 5H, C5H5); 4.14 (s, 5H, C5H5); 3.98 (s, 1H, C7H7); 3.73 (q, J = 6.9 Hz, 1H, C2H4); 3.46 (q, J = 6.3 Hz, 1H, C2H4); 3.19 (d, J = 16.9 Hz, 1H, C7H7); 1.19 (d, J = 7.0 Hz, 3H, C2H4); 1.09 (d, J = 6.4 Hz, 3H, C2H4) ppm. 13C–NMR δ = 144.18 (Cq, C7H7); 138.58 (d, JCP = 89.2 Hz, Cq, C6H5); 132.79 (d, JCP = 11.3 Hz, CH, C6H5); 130.88 (d, JCP = 3.0 Hz, CH, C6H5); 127.88 (CH, C7H7); 127.32 (d, JCP = 12.4 Hz, CH, C6H5); 127.00 (CH, C7H7); 126.05 (CH, C7H7); 93.39 (d, JCP = 11.1 Hz, Cq, C5H3); 92.56 (d, JCP = 12.1 Hz, Cq, C5H3); 75.55 (Cq, C5H3); 75.47 (d, JCP = 15.6 Hz, CH, C5H3); 74.93 (Cq, C5H3); 73.93 (d, JCP = 15.0 Hz, CH, C5H3); 71.41 (d, JCP = 9.7 Hz, CH, C5H3); 71.01 (CH, C5H5); 70.35 (CH, C5H5); 70.07 (d, JCP = 11.5 Hz, CH, C5H3); 69.96 (d, JCP = 9.1 Hz, CH, C5H3); 69.85 (d, JCP = 11.4 Hz, CH, C5H3); 55.79 (CH, C2H4); 53.18 (CH2, C7H7); 52.14 (CH, C2H4); 22.12 (CH3, C2H4); 21.35 (CH3, C2H4) ppm. 31P-NMR δ = 43.41 (s) ppm. HRMS: m/z calculated for C37H36Fe2NPS [M + H]+: 670.1078, found: 670.1068.
10: M.p.: 179–180 °C (decomposition). 1H-NMR (600 MHz, CDCl3) δ = 7.82 (dd, J = 12.7, 6.5 Hz, 2H, C6H5); 7.29–7.23 (m, 3H, C6H5); 7.13–7.08 (m, 3H, C7H7); 6.80–6.76 (m, 3H, C7H7, C2H3); 5.47 (dd, J = 17.7, 1.6 Hz, 1H, C2H3); 5.17 (dd, J = 10.8, 1.6 Hz, 1H, C2H3); 4.86 (m, 1H, C5H3); 4.62 (q, J = 6.7 Hz, 1H, C2H4); 4.56 (m, 1H, C5H3); 4.33 (s, 5H, C5H5); 4.30 (m, 1H, C5H3); 4.24 (dd, J = 4.1, 2.6 Hz, 1H, C5H3); 4.12 (s, 5H, C5H5); 3.77 (m, 1H, C5H3); 3.67 (m, 1H, C5H3); 3.21 (dd, J = 23.6, 12.8 Hz, 2H, C7H7); 1.45 (d, J = 6.6 Hz, 3H, C2H4) ppm. 13C-NMR δ = 140.46 (Cq, C7H7); 135.15 (d, JCP = 86.3 Hz, Cq, C6H5); 134.47 (CH, C7H7); 131.80 (d, JCP = 10.2 Hz, CH, C6H5); 131.14 (d, JCP = 2.9 Hz, CH, C6H5); 127.85 (CH, C7H7); 127.82 (d, JCP = 11.4 Hz, CH, C6H5); 127.78 (CH, C7H7); 126.14 (CH, C2H3); 111.48 (CH2, C2H3); 95.12 (d, JCP = 12.7 Hz, Cq, C5H3); 88.55 (d, JCP = 12.0 Hz, Cq, C5H3); 77.91 (d, JCP = 95.5 Hz, Cq, C5H3); 75.03 (d, JCP = 12.2 Hz, CH, C5H3); 74.42 (d, JCP = 12.1 Hz, CH, C5H3); 73.98 (d, JCP = 95.4 Hz, Cq, C5H3); 71.15 (CH, C5H5); 71.10 (d, JCP = 10.1 Hz, CH, C5H3); 70.67 (CH, C5H5); 69.97 (d, JCP = 10.2 Hz, CH, C5H3); 68.22 (d, JCP = 9.1 Hz, CH, C5H3); 67.94 (d, JCP = 10.6 Hz, CH, C5H3); 50.71 (CH2, C7H7); 50.08 (CH, C2H4); 19.53 (CH3, C2H4) ppm. 31P-NMR δ = 39.55 (s) ppm. HRMS: m/z calculated for C37H36Fe2NPS [M + H]+: 670.1078, found: 670.1075.



{kind=link}
{kind=link}
