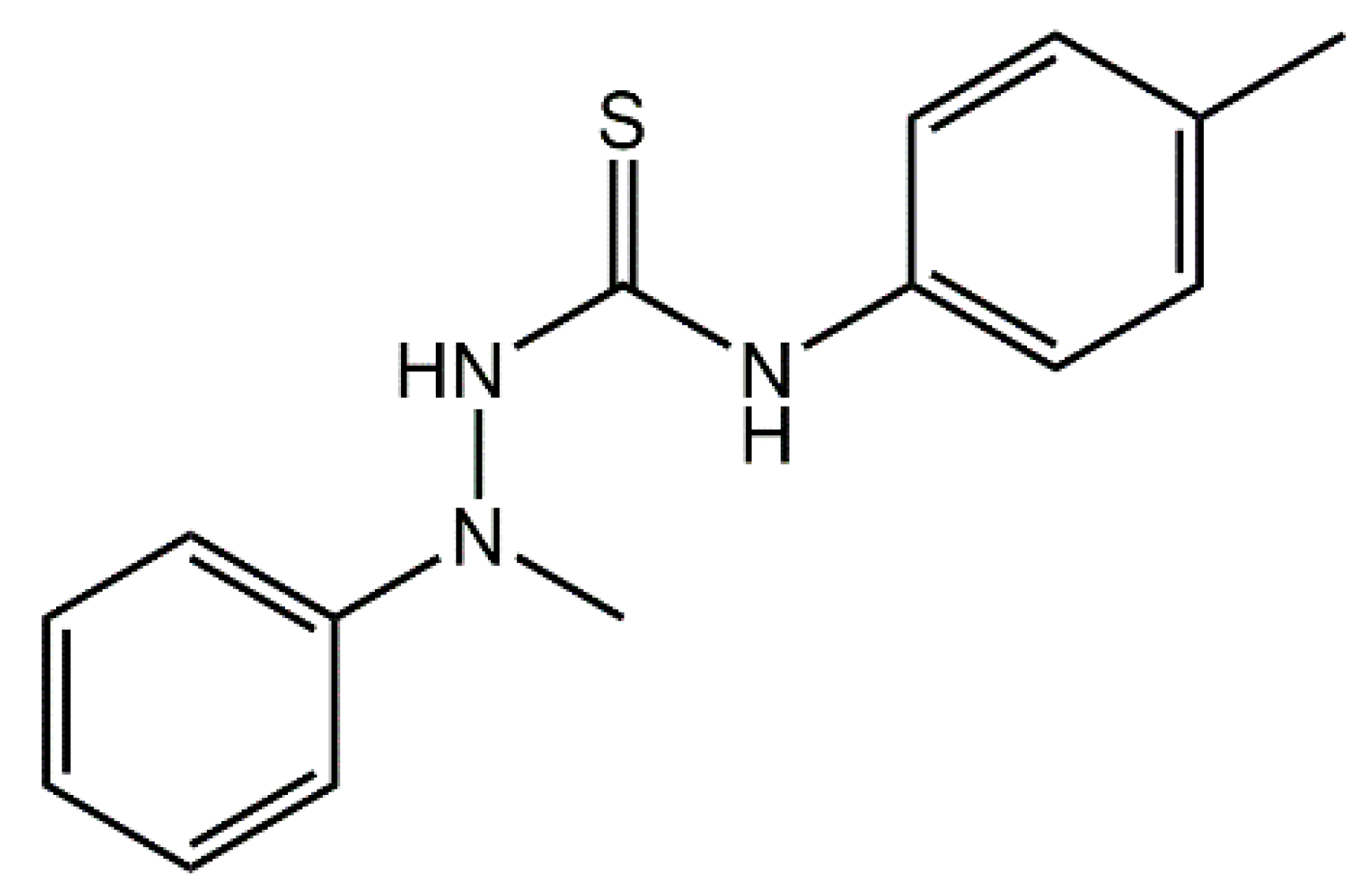
1. Introduction
Thiourea, H
2NC(=S)NH
2, is susceptible for to up to four substitutions to give a wide range of compounds, R
1(R
2)NC(=S)N(R
3)R
4 for R
1–4 = alkyl/aryl [
1]. These molecules can possess interesting properties and are well known for their ability to complex metal ions [
1]. Recent interest in thiourea derivatives revolves around their applications in dual hydrogen bonding catalysis for asymmetric synthesis [
2,
3] and for their potential biological potency [
4], including when complexing metals [
5]. Indeed, the original interest in the title compound 1-[
N-methyl-
N-(phenyl)amino]-3-(4-methylphenyl)thiourea (
1),
Scheme 1, related to the investigation of antiviral activity [
6]; since then there have been no other reports of (
1) in the literature. Herein, the synthesis, spectroscopic characterization, and X-ray crystal structure determination of the title compound, (
1), are described.
2. Results and Discussion
The reaction of 1-methyl-1-phenylhydrazine and 4-tolyl isothiocyanate yielded the title compound (
1) in good yield (71%). The compound was characterized by spectroscopy; see the
Supplementary Materials for original spectra. The
1H spectrum showed the expected resonances and integration. The downfield shift for the NH proton proximate to the C=S and N(Me)Ph groups cf. NH(4-tolyl) is ascribed to the proximity to the nitrogen bound lone pair of electrons. The most prominent feature of the
13C{
1H} spectrum was the downfield resonance at
δ 180.2 ascribed to N
2C(=S). In the IR spectrum, characteristic bands of ν(N–H) [3266 (m)], ν(C–N) [1485 (vs)], and ν(C=S) [1256 (vs)] were observed. In the UV spectrum, two bands were observed at λ
abs 242 nm and 267 nm and are ascribed to π‒π* and n‒π* transitions, respectively.
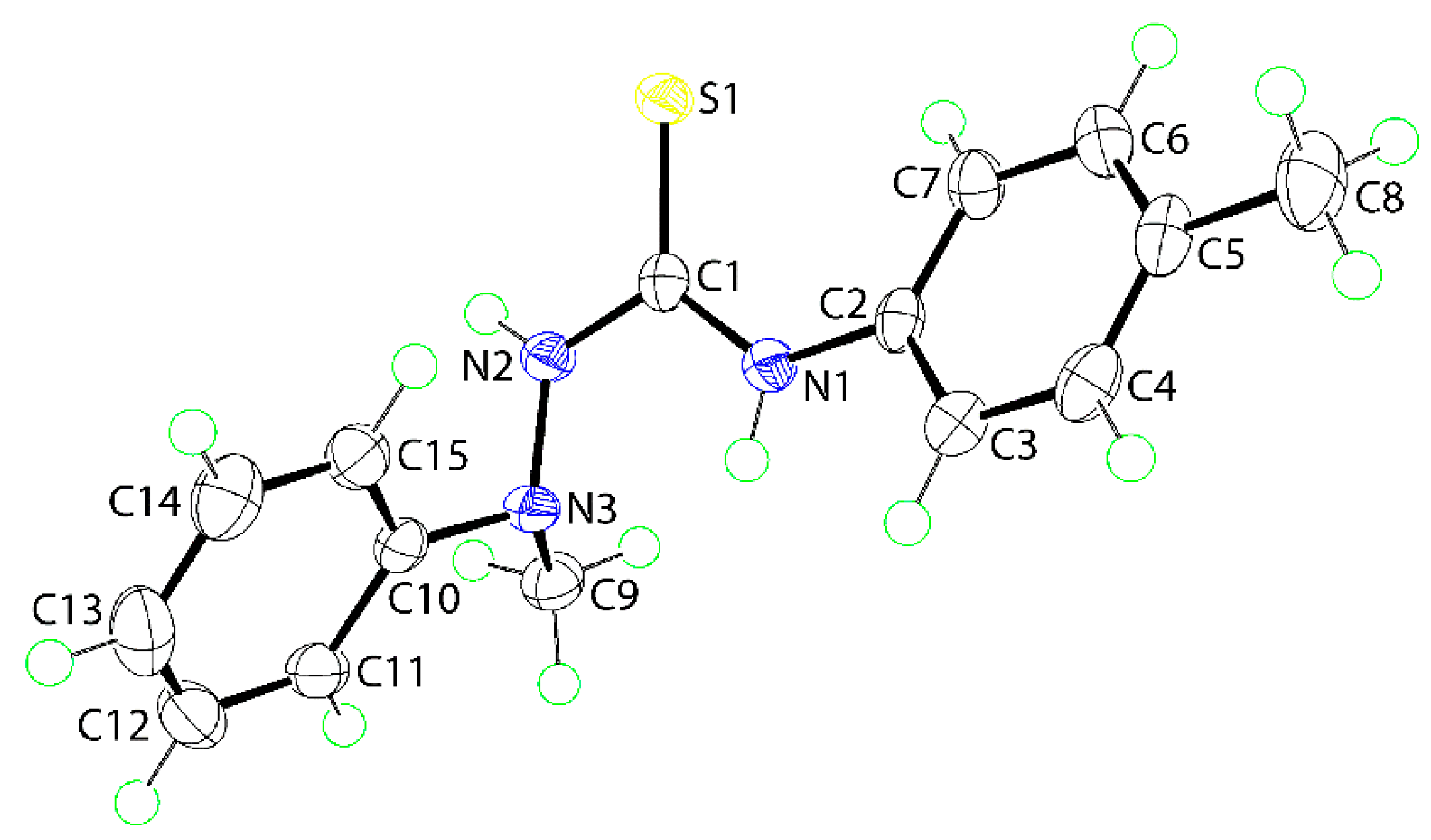
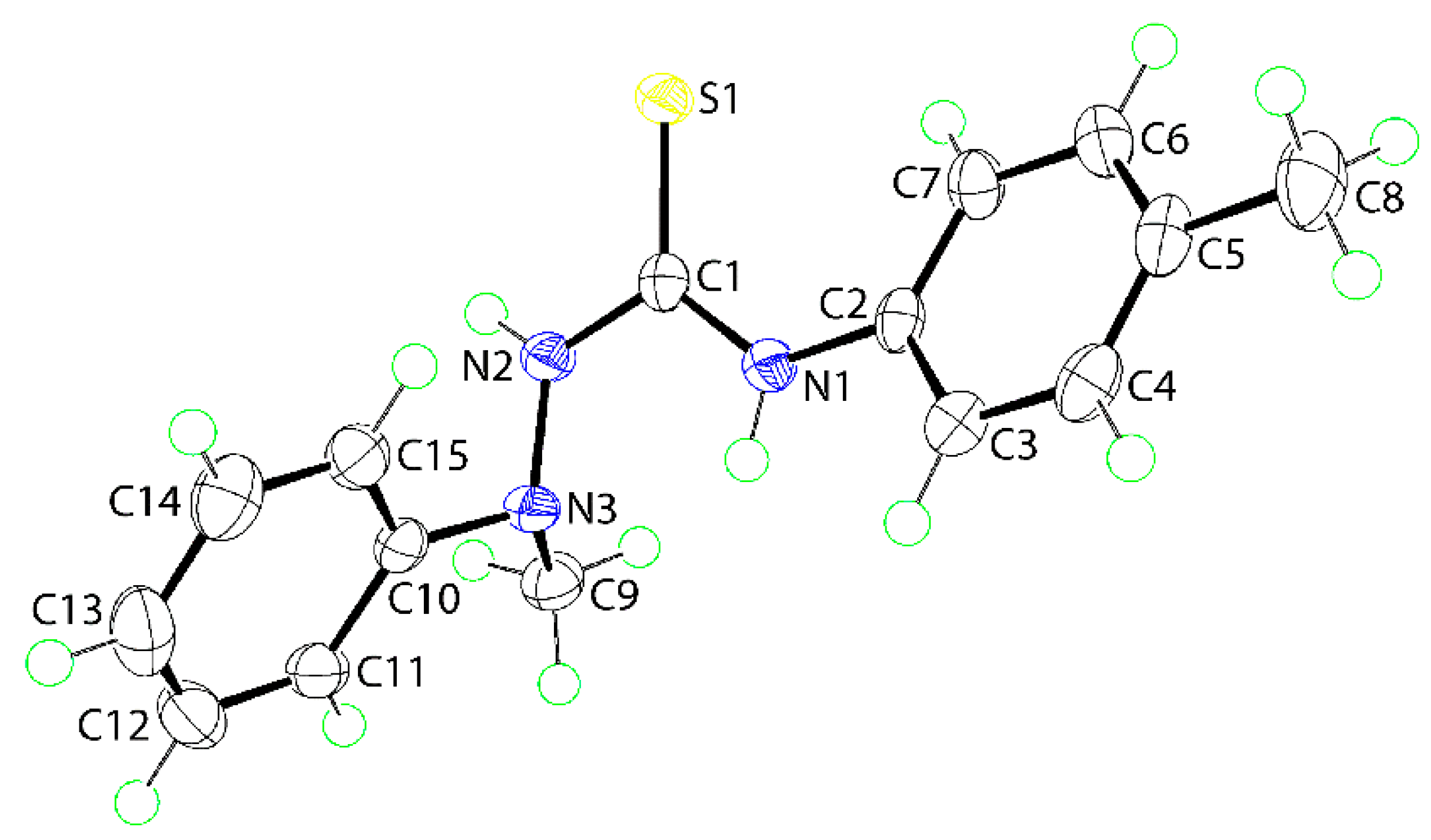
The molecular structure of (
1) was also determined by X-ray crystallographic methods and is shown in
Figure 1. The five atoms of the central chromophore, that is, S1, C1, and N1–N3, are effectively co-planar as evidenced in the root mean square deviation of 0.0223 Å; the maximum deviation from the least-squares plane was 0.0337(8) Å for the N2 atom. The appended C2–tolyl and C10–phenyl rings form dihedral angles of 25.42(6) and 81.18(3)° with the central residue, respectively, indicating skewed and orthogonal relationships. The dihedral angle between the terminal rings is 74.05(4)° which suggests that, to a first approximation, the molecule has the shape of the letter L. The C1–N1, N2 bond lengths are similar [1.3491(16) cf. 1.3458(16) Å] but, the S1–C1–N1 angle [127.50(10)°] is significantly wider than the S1–C1–N2 angle [117.36(9)°], presumably to minimize steric repulsion. The thioamide–NH atoms adopt an anti-disposition. An intramolecular thioamide-N1-H⋯N3 hydrogen bond is noted [N1–H1n⋯N3: H1n⋯N3 = 2.200(14) Å, N1⋯N3 = 2.6684(15) Å with angle at H1n = 113.5(11)°] that closes an S(5) loop.
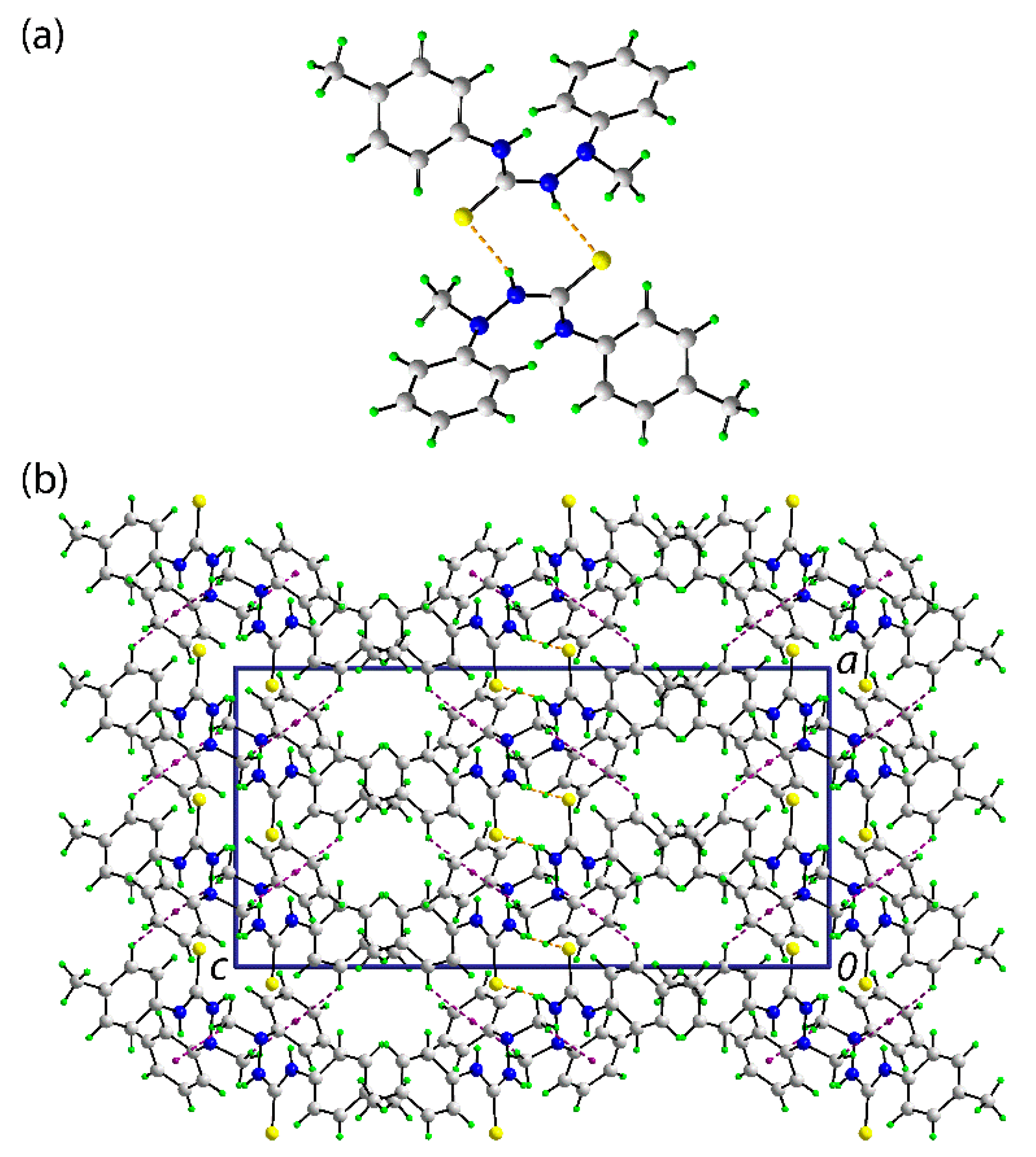
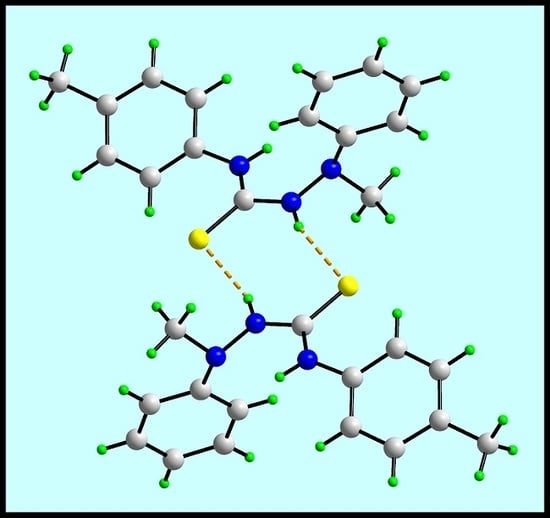
In the crystal of (
1), the most prominent feature of the molecular packing is the formation of thioamide-N1–H⋯S1(thione) hydrogen bonds. These lead to the formation of a supramolecular dimer via a centrosymmetric, eight-membered {⋯HNCS}
2 synthon, as shown in
Figure 2a; geometric details characterizing intermolecular interactions discussed herein are given in the figure caption. The connections between the dimers are of the type phenyl- and methyl–C–H⋯
π(phenyl) whereby the C10–C15 phenyl ring effectively functions as a bridge between the dimers. The result is a supramolecular layer in the
bc-plane. Layers stack along the
a-axis with no directional interactions between them. A view of the unit cell contents is shown in
Figure 2b.
There is a closely related structure in the literature where the N-bound phenyl group of (
1) is substituted by a methyl group and the original 4-tolyl ring is now a phenyl ring, that is, Me
2NN(H)C(=S)N(H)Ph (
2) [
7]. Molecule (
2) adopts essentially the same conformation as just described for (1) but, some systematic changes in the key bond lengths are evident. Thus, while the C1=S1 [1.6813(18) Å] bond length in (
2) is experimentally equivalent to that in (
1), the C1–N1 [1.330(2) Å] and C1–N2 [1.330(3) Å] bond lengths in (
2) are shorter. The reduction in the specified bond lengths is consistent with there being reduced delocalization of
π-electron density over the ring as a result of replacing phenyl in (
1) with electron donating methyl in (
2), and 4-tolyl with electron-withdrawing phenyl. The molecular packing of (
2) features the same eight-membered {⋯HNCS}
2 synthon seen in the crystal of (
1).
In conclusion, an X-ray crystallographic analysis on (1) confirms the molecule to exist as the thioamide tautomer, to have an anti-disposition of the thioamide–N–H atoms and an intramolecular N1–H⋯N3 hydrogen bond. Overall, the conformation of the molecule resembles that of the letter L. The presence of thioamide–N–H⋯S(thione) hydrogen bonds in the molecular packing leads to the formation of eight-membered {⋯HNCS}2 synthons.
3. Materials and Methods
3.1. General Information
All standard chemicals and solvents were sourced from Merck (Darmstadt, Germany) and used without further purification. The melting point was determined on a Biobase automatic melting point apparatus MP450 (Biobase Group, Jinan, Shandong Province, China). The IR spectrum was measured on a Bruker Vertex 70v FTIR (Billerica, MA, USA) spectrophotometer from 4000 to 80 cm−1. Elemental analyses were performed on a Leco TruSpec Micro CHN Elemental Analyzer (Saint Joseph, MI, USA). 1H and 13C{1H} NMR spectra were recorded in CDCl3 solution on a Bruker Ascend 400 MHz NMR (Billerica, MA, USA) spectrometer with chemical shifts relative to tetramethylsilane. The optical absorption spectra were obtained from an acetonitrile solution of 1 × 10−5 M in the range 200–800 nm on a Shimadzu UV-3600 plus UV/VIS/NIR (Shimadzu Corporation, Kyoto, Japan) spectrophotometer.
3.2. Synthesis and Characterization of (1)
1-Methyl-1-phenylhydrazine (0.01 mol, 1.18 mL) was added dropwise to 4-tolyl isothiocyanate (0.01 mol, 1.49 g) in acetone (15 mL). The resulting mixture was stirred for 2 h and then left for slow evaporation under ambient conditions. The solid obtained was recrystallized in the solvent mixture 1:1
v/
v chloroform/acetonitrile. Colorless crystals were formed after two weeks. Yield: 1.93 g, 71%. M. pt: 159.5–161.7 °C; Lit. 165 °C [
6]. Anal. Calc. for C
15H
17N
3S: C, 66.39; H, 6.31; N, 15.48%. Found: C, 66.34; H, 6.26; N, 15.35%. IR (cm
−1): 3266 (m)
ν(N-H), 3138 (m)
ν(C–H), 1485 (vs)
ν(C–N), 1256 (vs)
ν(C=S).
1H NMR (CDCl
3):
δ 8.78 (s, br, 1H, Ph–NH), 7.69 (s, br, 1H, N–NH), 7.41 (d, 2H, C
6H
4-3,
3JHH = 8.26 Hz), 7.36–7.32 (m, 2H, C
6H
5-3), 7.15 (d, 2H, C
6H
4-2,
3JHH = 8.13 Hz), 7.05–7.00 (m, 3H, C
6H
5-2,4), 3.21 (s, 3H, NCH
3), 2.32 (s, 3H, aryl–CH
3).
13C{
1H} NMR (CDCl
3):
δ 180.2 (Cq), 148.9 (C
6H
5, C1), 136.1 (C
6H
4, C1), 135.0 (C
6H
4, C4), 129.5 (C
6H
4, C3, C5), 129.3 (C
6H
4, C2, C6), 124.7 (C
6H
5, C3, C5), 122.4 (C
6H
5, C4), 114.8 (C
6H
5, C2, C6), 41.8 (NCH
3), 21.0 (C
6H
4–CH
3). UV (acetonitrile; nm, L·cm
−1·mol
−1): λ
abs = 267, ε = 26,700; λ
abs = 242, ε = 22,500.
3.3. Crystallography
Intensity data for (1) were measured at T = 100(2) K on a XtaLAB Synergy Dual AtlasS2 (Rigaku Polska SP. Z O O, Wrocław, Poland) diffractometer fitted with Cu Kα radiation (
λ = 1.54184 Å) using
ω-scans in the
θmax range 3.8°–67.1°. Data reduction, including absorption correction, was accomplished with CrysAlis Pro [
8]. Of the 18,012 reflections measured, 2550 were unique (
Rint = 0.030), and of these, 2356 data satisfied the
I ≥ 2
σ(
I) criterion of observability. The structure was solved by direct methods [
9] and refined (anisotropic displacement parameters and C-bound H atoms in the riding model approximation) on
F2 [
10]. The thioamide–N–H atoms were located from a difference map and refined with a N–H restraint of 0.88 ± 0.01 Å. A weighting scheme of the form
w = 1/[
σ2(
Fo2) + (0.039
P)
2 + 1.297
P] was introduced, where
P = (
Fo2 + 2
Fc2)/3). Based on the refinement of 180 parameters, the final values of
R and
wR (all data) were 0.028 and 0.077, respectively. The molecular structure diagram was generated with ORTEP for Windows [
11] and the packing diagram using DIAMOND [
12].
Crystal data for C15H17N3S (1): M = 271.38, orthorhombic, Pbca, a = 10.50030(10) Å, b = 11.70230(10) Å, c = 23.2486(3) Å, V = 2856.73(5) Å3, Z = 8, Dx = 1.262 g cm−3, F(000) = 1152 and μ = 1.918 mm−1. CCDC deposition number: 1898244.



{kind=link}
{kind=link}
{kind=link}
{kind=link}