2-(4-Heptyloxyphenyl)benzothiazole

Abstract
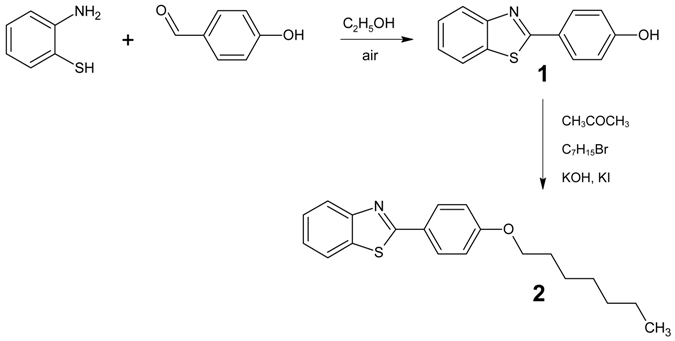
:
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Acknowledgements
References and Notes
- Funahashi, M.; Hanna, J.I. Jpn. J. Appl. Phys 1996, 35, L703.
- Funahashi, M.; Hanna, J.I. Phys. Rev. Lett. 1997, 78, 2184.
- Funahashi, M.; Hanna, J.I. Mol. Cryst. Liq. Cryst. 1997, 304, 429.
- Prajapati, A.K.; Bonde, N.L. J. Chem. Sci. 2006, 118, 203.
- Ha, S.T.; Koh, T.M.; Yeap, G.Y.; Lin, H.C.; Boey, P.L.; Yip, F.W.; Ong, S.T.; Ong, L.K. Mol. Cryst. Liq. Cryst. 2009, in press.
- Ha, S.T.; Koh, T.M.; Yeap, G.Y.; Lin, H.C.; Beh, J.K.; Win, Y.F.; Boey, P.L. Chin. Chem. Lett. 2009, in press.
- Tokunaga, K.; Yamashita, T.; Hanna, J. JP 2006248948, 2006.
- Koyanagi, T.; Komatsu, M. PCT Int. Appl. WO 2001003232, 2001.
- Hanna, J.; Funabashi, M.; Akata, M. JP 09059266, 1997.
- Hanna, J.; Funabashi, M.; Akada, M.; Ando, M.; Kosaka, Y. EP 763532, 1997.
- Ha, S.T.; Ong, L.K.; Wong, J.P.W.; Win, Y.F.; Koh, T.M. Molbank 2009, 1, M598.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ha, S.-T.; Koh, T.-M.; Ong, S.-T.; Lee, T.-L.; Sivasothy, Y. 2-(4-Heptyloxyphenyl)benzothiazole. Molbank 2009, 2009, M610. https://doi.org/10.3390/M610
Ha S-T, Koh T-M, Ong S-T, Lee T-L, Sivasothy Y. 2-(4-Heptyloxyphenyl)benzothiazole. Molbank. 2009; 2009(3):M610. https://doi.org/10.3390/M610
Chicago/Turabian StyleHa, Sie-Tiong, Teck-Ming Koh, Siew-Teng Ong, Teck-Leong Lee, and Yasodha Sivasothy. 2009. "2-(4-Heptyloxyphenyl)benzothiazole" Molbank 2009, no. 3: M610. https://doi.org/10.3390/M610
APA StyleHa, S.-T., Koh, T.-M., Ong, S.-T., Lee, T.-L., & Sivasothy, Y. (2009). 2-(4-Heptyloxyphenyl)benzothiazole. Molbank, 2009(3), M610. https://doi.org/10.3390/M610