
Molecular Mechanisms of Reversal of Multidrug Resistance in Breast Cancer by Inhibition of P-gp by Cytisine N-Isoflavones Derivatives Explored Through Network Pharmacology, Molecular Docking, and Molecular Dynamics

Abstract
1. Introduction
2. Results and Discussion
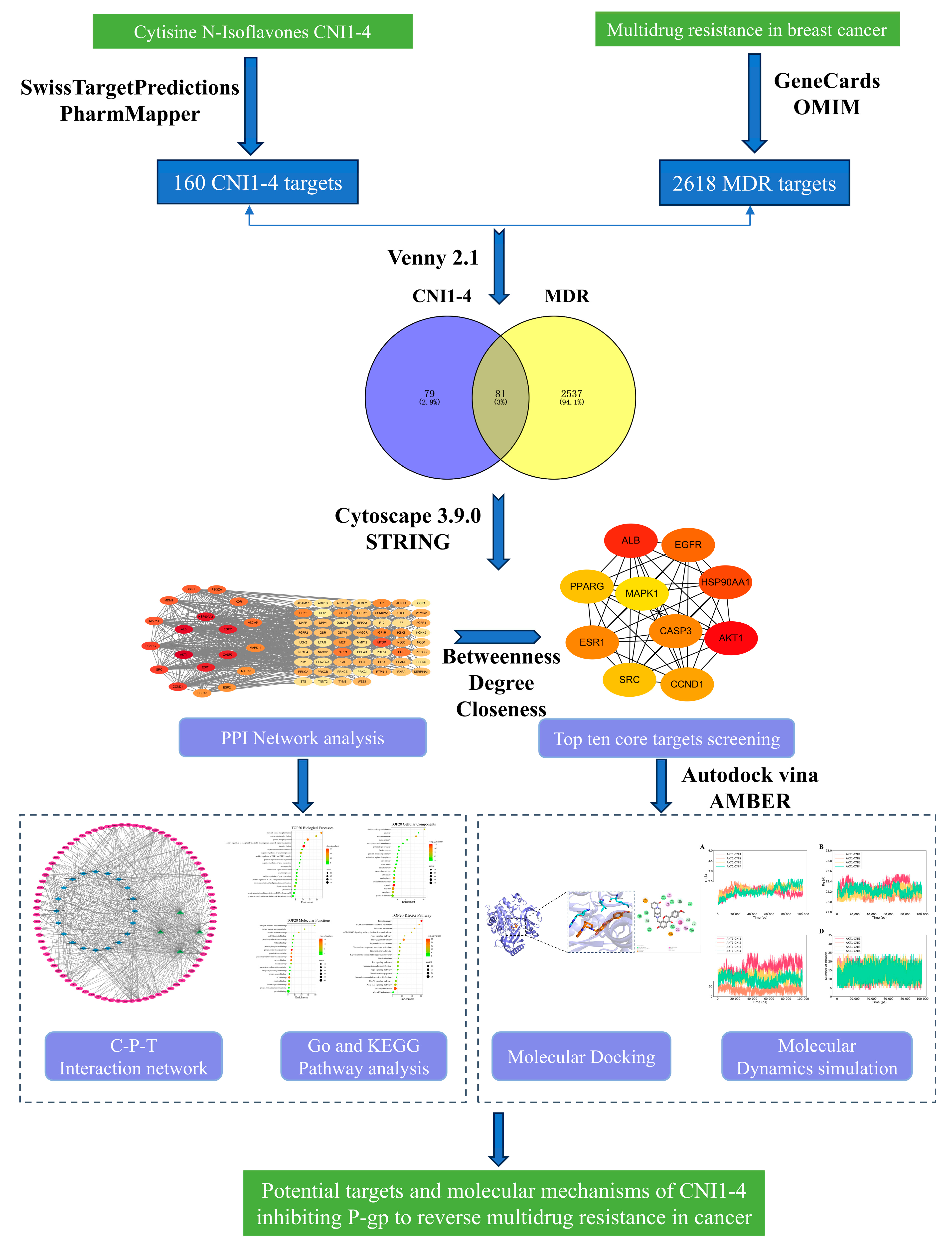
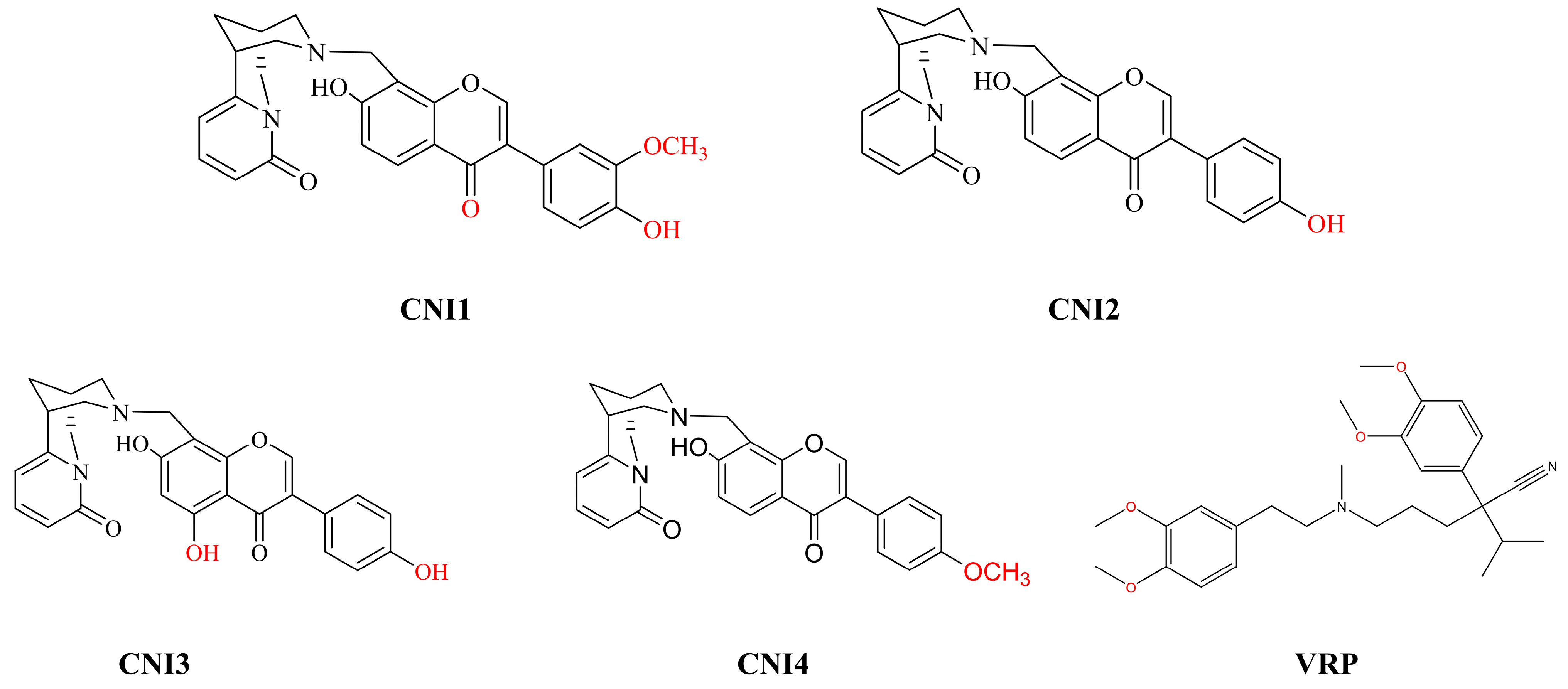
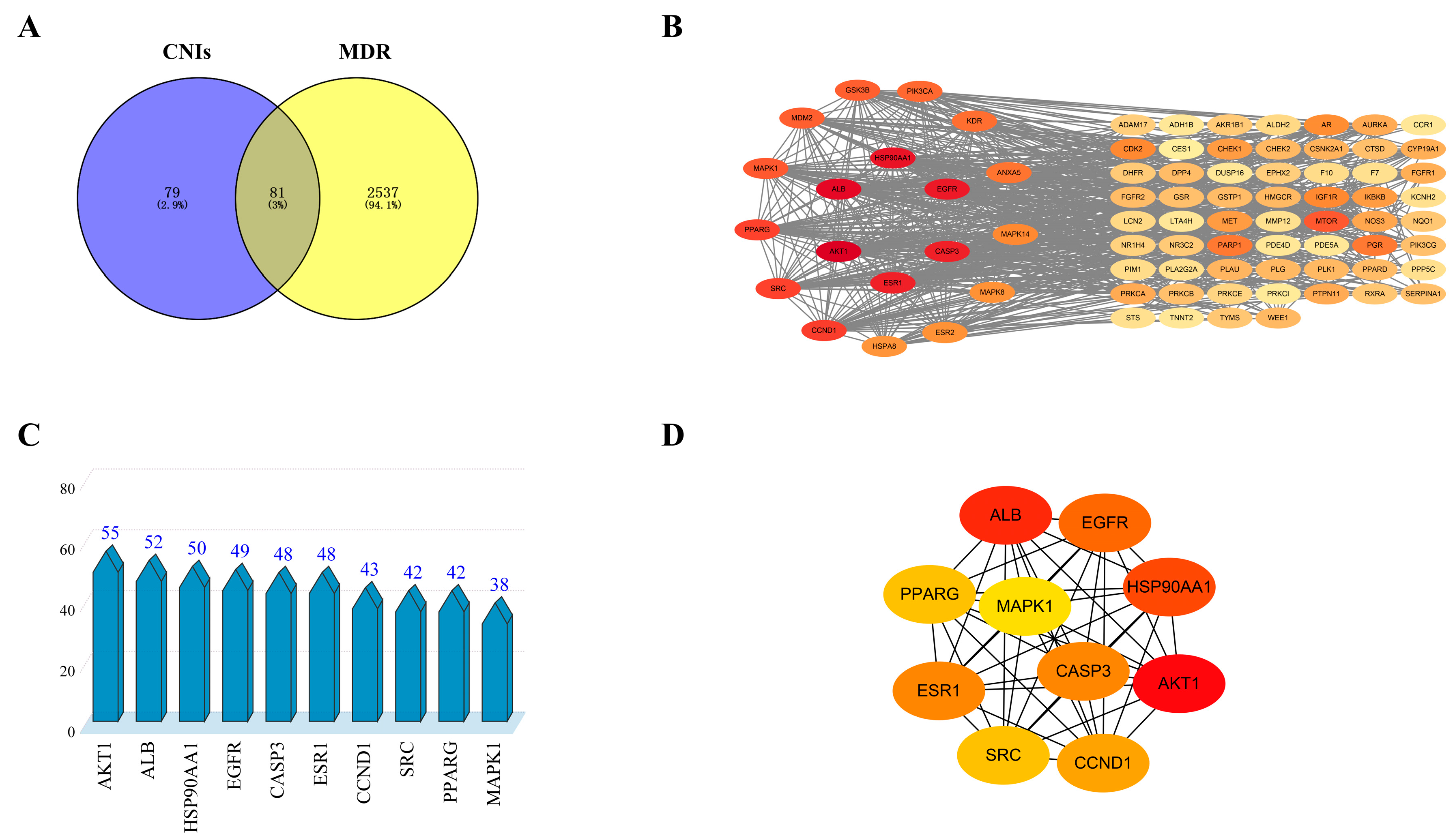
2.1. Prediction of Potential Targets for CNI1-4 to Reverse Multidrug Resistance in Breast Cancer
2.2. Construction of PPI Network
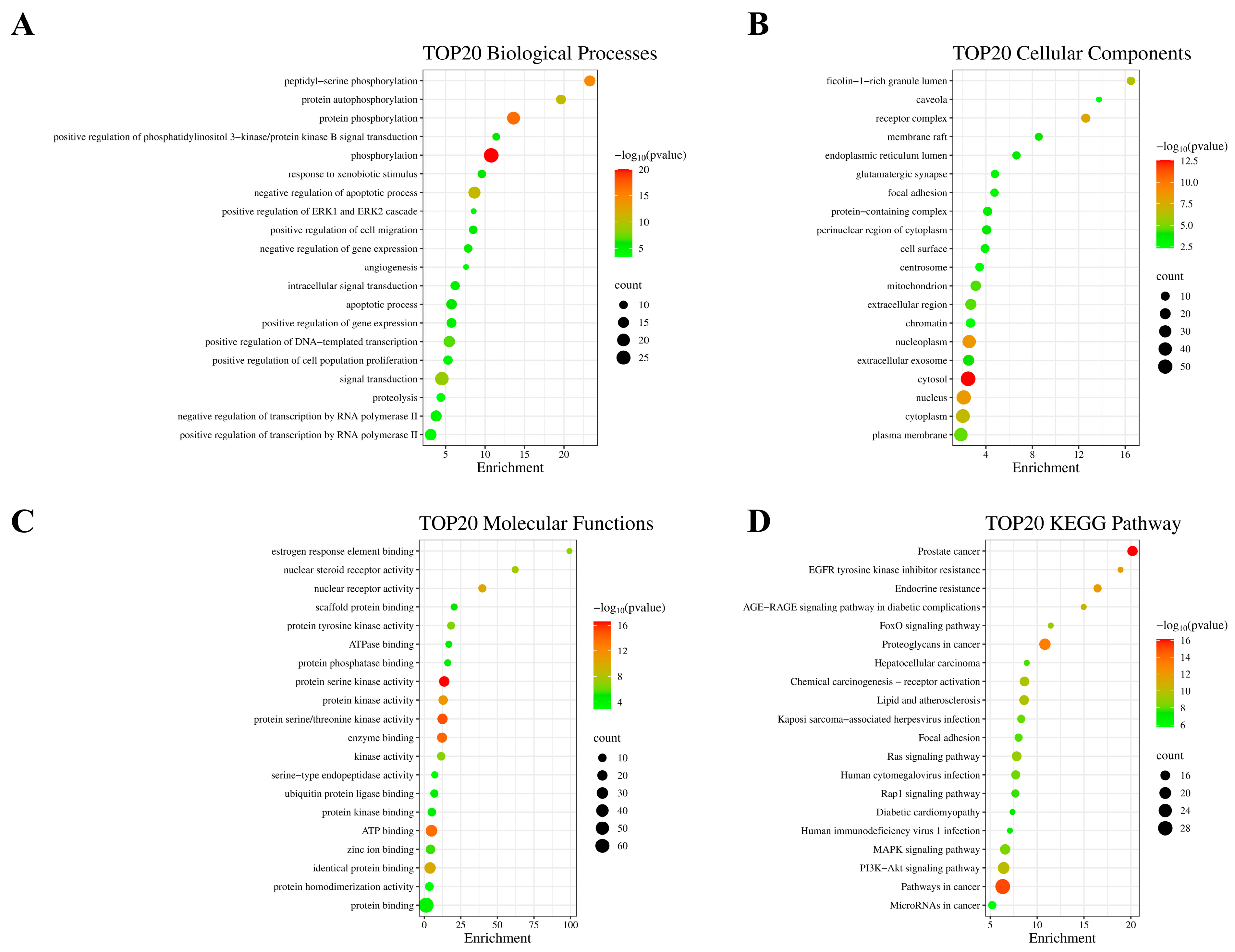
2.3. GO Functional Annotation Analysis and KEGG Enrichment Analysis
2.4. Construction of Compound–Target–Pathway Interaction Networks
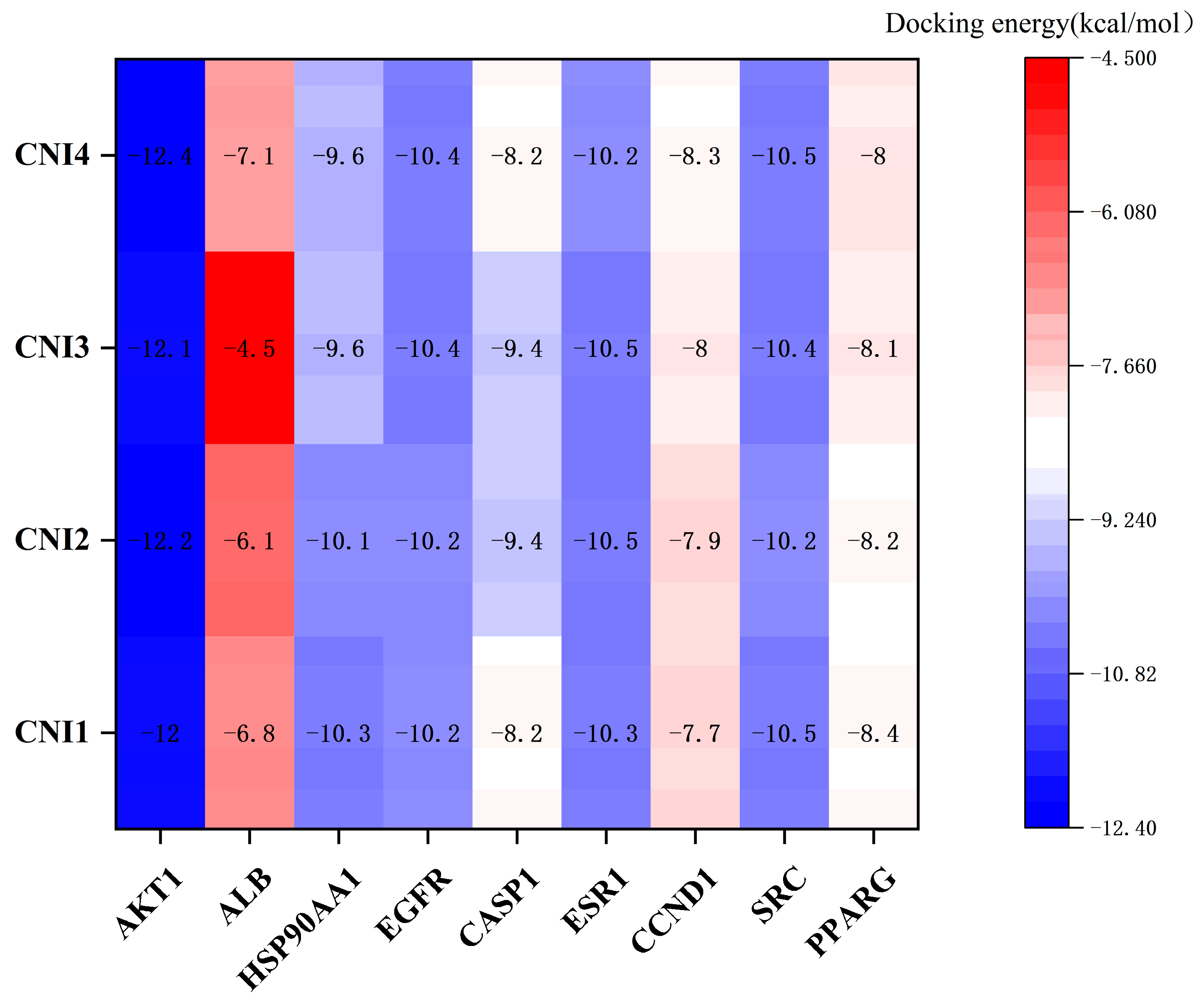
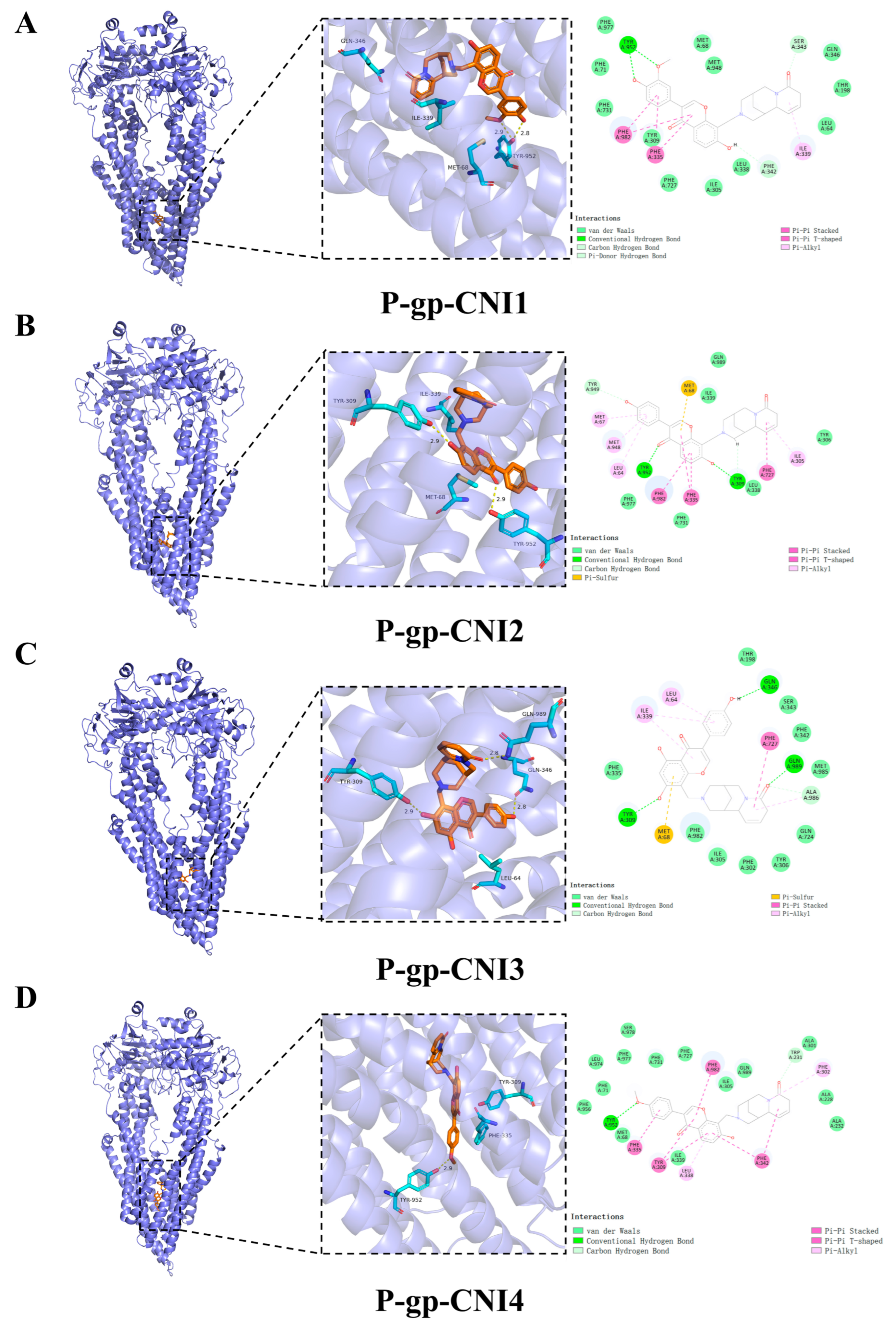
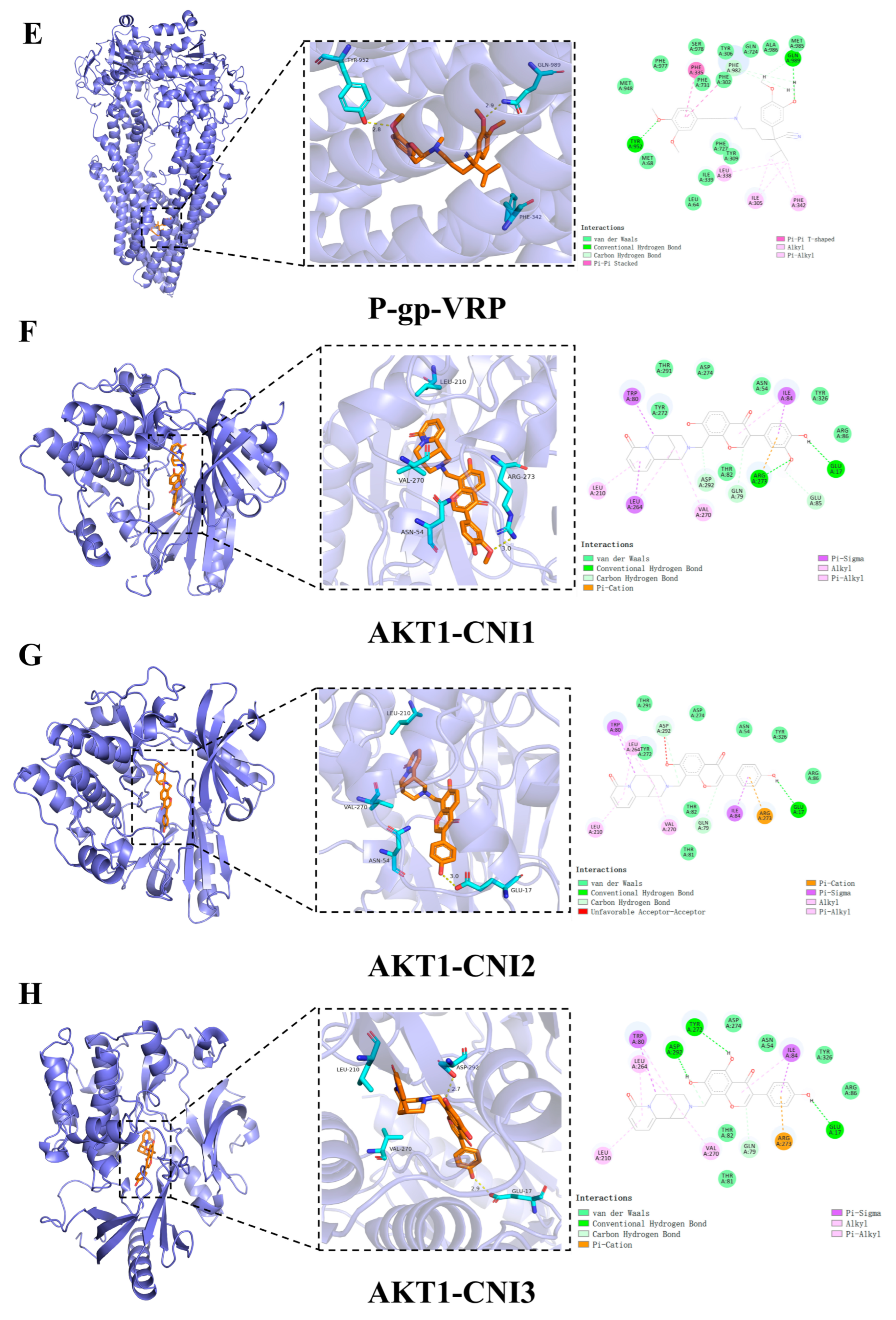
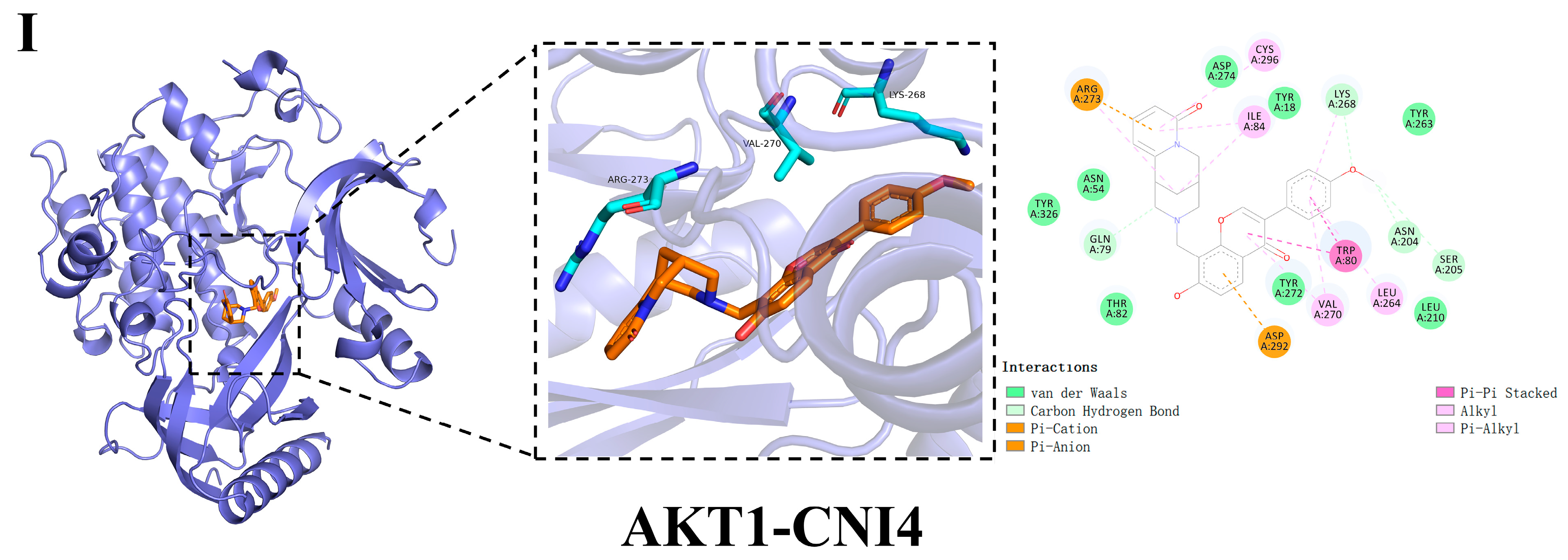
2.5. Molecular Docking
2.6. Molecular Dynamics Simulations
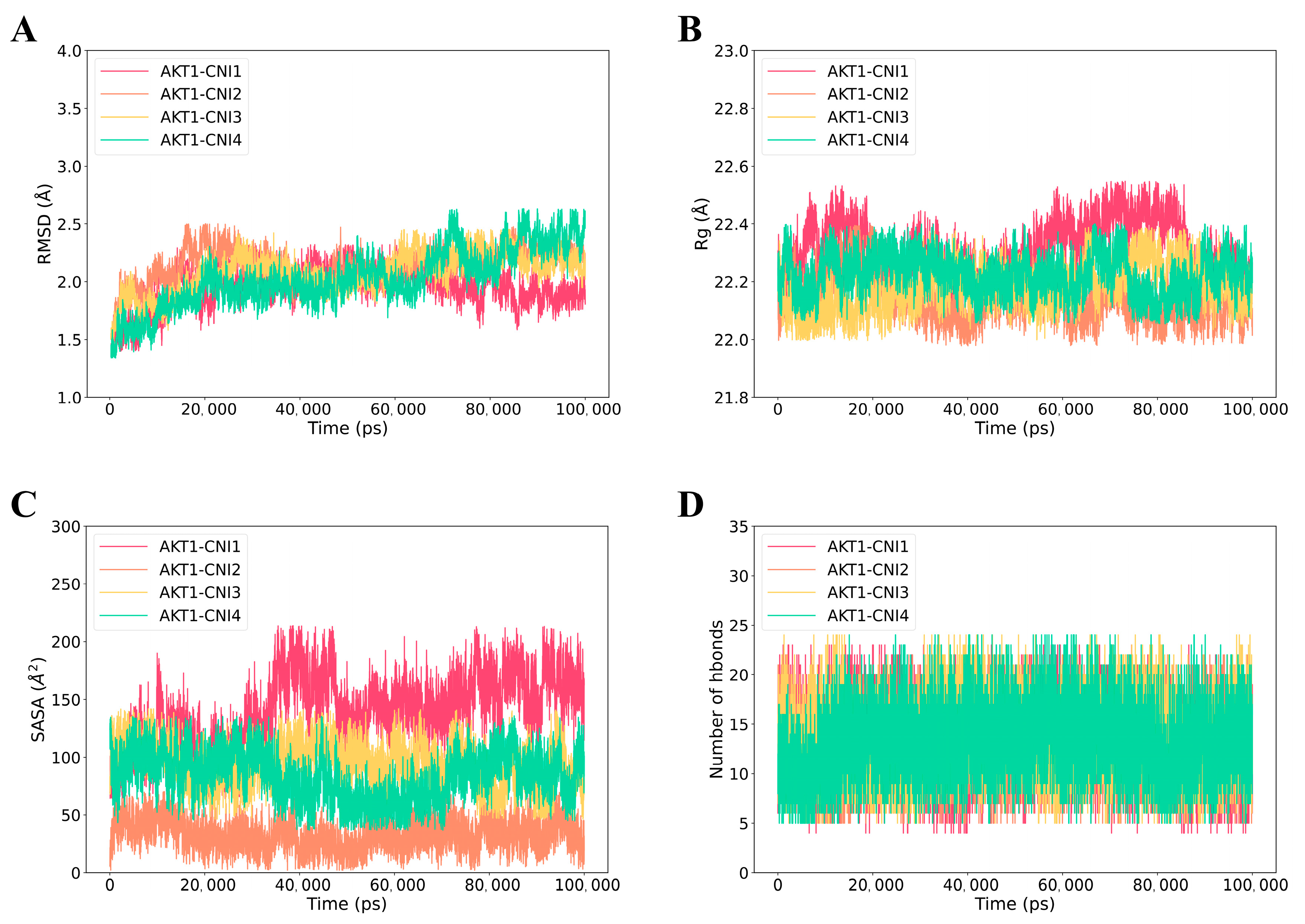
2.6.1. MD Simulation of AKT1-CNI1-4 Complexes
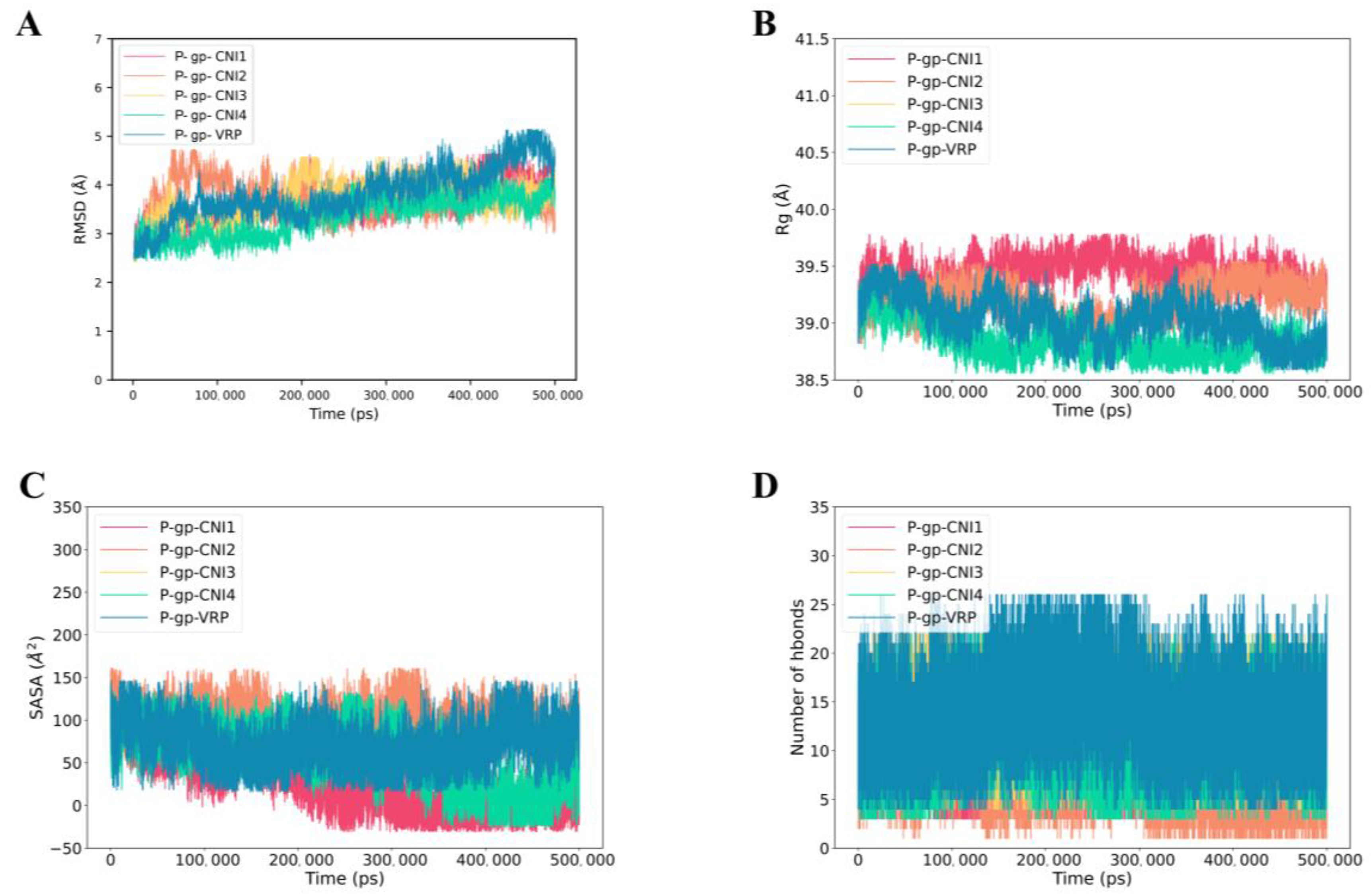
2.6.2. MD Simulation of P-gp-CNI1-4 and P-gp-VRP Complexes
2.7. Binding Free Energy MM-PBSA Calculations
2.7.1. Calculation of the Binding Free Energy of AKT1-CNI1-4
2.7.2. P-gp-CNI1-4 and P-gp-VRP Binding Free Energy Calculations
2.8. Discussion
3. Materials and Methods
3.1. Analysis of CNI1-4 Drug Targets Using Network Pharmacology
3.2. Network Pharmacology Screening for MDR Targets in Breast Cancer
3.3. Potential Targets for the Intersection of Active Compounds CNI1-4 with MDR in Breast Cancer
3.4. Construction of Protein–Protein Interaction (PPI) Networks
3.5. GO and KEGG Enrichment Analyses
3.6. Molecular Docking
3.7. Molecular Dynamics (MD) Simulations
3.8. Combined Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- Al-Malky, H.S.; Al Harthi, S.E.; Osman, A.-M.M. Major Obstacles to Doxorubicin Therapy: Cardiotoxicity and Drug Resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.; Kang, M.; Meadows, B.; Bakke, S.; Choyke, P.; Merino, M.; Goldspiel, B.; Chico, I.; Smith, T.; Chen, C.; et al. A Phase I Study of Infusional Vinblastine in Combination with the P-Glycoprotein Antagonist PSC 833 (Valspodar). Cancer 2001, 92, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Moiseenko, F.; Volkov, N.; Bogdanov, A.; Dubina, M.; Moiseyenko, V. Resistance Mechanisms to Drug Therapy in Breast Cancer and Other Solid Tumors: An Opinion. F1000Research 2017, 6, 288. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Chen, Z.-S.; Ambudkar, S.V. Tyrosine Kinase Inhibitors as Modulators of ABC Transporter-Mediated Drug Resistance. Drug Resist. Updat. 2012, 15, 70–80. [Google Scholar] [CrossRef]
- de Klerk, D.J.; Honeywell, R.J.; Jansen, G.; Peters, G.J. Transporter and Lysosomal Mediated (Multi)Drug Resistance to Tyrosine Kinase Inhibitors and Potential Strategies to Overcome Resistance. Cancers 2018, 10, 503. [Google Scholar] [CrossRef]
- Amawi, H.; Sim, H.-M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC Transporter-Mediated Multidrug-Resistant Cancer. Adv. Exp. Med. Biol. 2019, 1141, 549–580. [Google Scholar] [CrossRef]
- Dong, J.; Yuan, L.; Hu, C.; Cheng, X.; Qin, J.-J. Strategies to Overcome Cancer Multidrug Resistance (MDR) through Targeting P-Glycoprotein (ABCB1): An Updated Review. Pharmacol. Ther. 2023, 249, 108488. [Google Scholar] [CrossRef]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remião, F. Modulation of P-Glycoprotein Efflux Pump: Induction and Activation as a Therapeutic Strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef]
- Maliepaard, M.; van Gastelen, M.A.; de Jong, L.A.; Pluim, D.; van Waardenburg, R.C.; Ruevekamp-Helmers, M.C.; Floot, B.G.; Schellens, J.H. Overexpression of the BCRP/MXR/ABCP Gene in a Topotecan-Selected Ovarian Tumor Cell Line. Cancer Res. 1999, 59, 4559–4563. [Google Scholar] [PubMed]
- Jonker, J.W.; Smit, J.W.; Brinkhuis, R.F.; Maliepaard, M.; Beijnen, J.H.; Schellens, J.H.; Schinkel, A.H. Role of Breast Cancer Resistance Protein in the Bioavailability and Fetal Penetration of Topotecan. J. Natl. Cancer Inst. 2000, 92, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, S.L.A.; Van Der Kolk, D.M.; De Bont, E.S.J.M.; Vellenga, E.; Kamps, W.A.; De Vries, E.G.E. Breast Cancer Resistance Protein (BCRP) in Acute Leukemia. Leuk. Lymphoma 2004, 45, 649–654. [Google Scholar] [CrossRef]
- Miyake, M.M.; Nocera, A.; Miyake, M.M. P-Glycoprotein and Chronic Rhinosinusitis. World J. Otorhinolaryngol.-Head Neck Surg. 2018, 4, 169–174. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Ashby, C.R.; Assaraf, Y.G.; Chen, Z.-S.; Liu, H.-M. Chemical Molecular-Based Approach to Overcome Multidrug Resistance in Cancer by Targeting P-Glycoprotein (P-Gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef] [PubMed]
- Rocco, A.; Compare, D.; Liguori, E.; Cianflone, A.; Pirozzi, G.; Tirino, V.; Bertoni, A.; Santoriello, M.; Garbi, C.; D’Armiento, M.; et al. MDR1-P-Glycoprotein Behaves as an Oncofetal Protein That Promotes Cell Survival in Gastric Cancer Cells. Lab. Investig. J. Tech. Methods Pathol. 2012, 92, 1407–1418. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Cretney, E.; Smyth, M.J. P-Glycoprotein Protects Leukemia Cells against Caspase-Dependent, but Not Caspase-Independent, Cell Death. Blood 1999, 93, 1075–1085. [Google Scholar] [CrossRef]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-Glycoprotein Inhibitors of Natural Origin as Potential Tumor Chemo-Sensitizers: A Review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, X.; Chen, L.; Yin, Z.; Zuo, Z. Discovery and Evaluation of Cytisine N-Isoflavones as Novel EGFR/HER2 Dual Inhibitors. Bioorg. Chem. 2022, 127, 105868. [Google Scholar] [CrossRef]
- Zhang, W.; Fan, Y.; Zhang, J.; Shi, D.; Yuan, J.; Ashrafizadeh, M.; Li, W.; Hu, M.; Abd El-Aty, A.M.; Hacimuftuoglu, A.; et al. Cell Membrane-Camouflaged Bufalin Targets NOD2 and Overcomes Multidrug Resistance in Pancreatic Cancer. Drug Resist. Updat. 2023, 71, 101005. [Google Scholar] [CrossRef]
- Petrikaite, V.; D’Avanzo, N.; Celia, C.; Fresta, M. Nanocarriers Overcoming Biological Barriers Induced by Multidrug Resistance of Chemotherapeutics in 2D and 3D Cancer Models. Drug Resist. Updat. 2023, 68, 100956. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, H.; Wang, X.; Aoki, Y.; Wang, X.; Yang, Y.; Cheng, X.; Wang, Z.; Wang, X. Aurora-A/SOX8/FOXK1 Signaling Axis Promotes Chemoresistance via Suppression of Cell Senescence and Induction of Glucose Metabolism in Ovarian Cancer Organoids and Cells. Theranostics 2020, 10, 6928–6945. [Google Scholar] [CrossRef] [PubMed]
- Carolus, H.; Pierson, S.; Muñoz, J.F.; Subotić, A.; Cruz, R.B.; Cuomo, C.A.; Van Dijck, P. Genome-Wide Analysis of Experimentally Evolved Candida Auris Reveals Multiple Novel Mechanisms of Multidrug Resistance. mBio 2021, 12, 10–1128. [Google Scholar] [CrossRef]
- Cao, Y.; Li, Z.; Mao, L.; Cao, H.; Kong, J.; Yu, B.; Yu, C.; Liao, W. The Use of Proteomic Technologies to Study Molecular Mechanisms of Multidrug Resistance in Cancer. Eur. J. Med. Chem. 2019, 162, 423–434. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Sun, Y. Unraveling the Role of Scutellaria Baicalensis for the Treatment of Breast Cancer Using Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2023, 24, 3594. [Google Scholar] [CrossRef]
- Chen, R.; Liu, H.; Meng, W.; Sun, J. Analysis of Action of 1,4-Naphthoquinone Scaffold-Derived Compounds against Acute Myeloid Leukemia Based on Network Pharmacology, Molecular Docking and Molecular Dynamics Simulation. Sci. Rep. 2024, 14, 21043. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Huang, W.; Wang, X.; Zhang, J.; Tao, T.; Zheng, Y.; Liu, S.; Yang, J.; Chen, Z.-S.; Cai, C.-Y.; et al. Hsa-miR-3178/RhoB/PI3K/Akt, a Novel Signaling Pathway Regulates ABC Transporters to Reverse Gemcitabine Resistance in Pancreatic Cancer. Mol. Cancer 2022, 21, 112. [Google Scholar] [CrossRef]
- Duan, W.; Liu, X. PSAT1 Upregulation Contributes to Cell Growth and Cisplatin Resistance in Cervical Cancer Cells via Regulating PI3K/AKT Signaling Pathway. Ann. Clin. Lab. Sci. 2020, 50, 512–518. [Google Scholar]
- Guo, Y.; Ashrafizadeh, M.; Tambuwala, M.M.; Ren, J.; Orive, G.; Yu, G. P-Glycoprotein (P-Gp)-Driven Cancer Drug Resistance: Biological Profile, Non-Coding RNAs, Drugs and Nanomodulators. Drug Discov. Today 2024, 29, 104161. [Google Scholar] [CrossRef] [PubMed]
- Brüschweiler, R. Efficient RMSD Measures for the Comparison of Two Molecular Ensembles. Proteins Struct. Funct. Bioinform. 2003, 50, 26–34. [Google Scholar] [CrossRef]
- Lobanov, M.I.; Bogatyreva, N.S.; Galzitskaia, O.V. Radius of gyration is indicator of compactness of protein structure. Mol. Biol. 2008, 42, 701–706. [Google Scholar] [CrossRef]
- Barazorda-Ccahuana, H.L.; Gómez, B.; Mas, F.; Madurga, S. Effect of pH on the Supramolecular Structure of Helicobacter Pylori Urease by Molecular Dynamics Simulations. Polymers 2020, 12, 2713. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinforma. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype–gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef]
- Liu, M.; Thomas, P.D. GO Functional Similarity Clustering Depends on Similarity Measure, Clustering Method, and Annotation Completeness. BMC Bioinform. 2019, 20, 155. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Nayar, D.; Agarwal, M.; Chakravarty, C. Comparison of Tetrahedral Order, Liquid State Anomalies, and Hydration Behavior of mTIP3P and TIP4P Water Models. J. Chem. Theory Comput. 2011, 7, 3354–3367. [Google Scholar] [CrossRef]
- Zuo, Z.; Weng, J.; Wang, W. Insights into the Inhibitory Mechanism of D13-9001 to the Multidrug Transporter AcrB through Molecular Dynamics Simulations. J. Phys. Chem. B 2016, 120, 2145–2154. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT Pathway as a Key Link Modulates the Multidrug Resistance of Cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Docking Score (Kcal/mol) | Hydrogen Bonding | Hydrophobic Interactions |
|---|---|---|---|
| CNI1 | −10.4 | Tyr952 | MET68,Phe71,Ile305, Tyr309,Phe335,Leu338, Ile339,Phe342,Ser343, Gln346,Phe731,Phe982 |
| CNI2 | −10.1 | Tyr309, Tyr952 | Leu64,Met67,Met68, Ile305,Phe335,Phe727, Met948,Tyr949,Phe982 |
| CNI3 | −10.0 | Gln346, Tyr309, Gln989 | Leu64,Met68,Ile339, Ala986 |
| CNI4 | −10.2 | Tyr952 | Phe71,Trp231,Ala301, Phe302,Ile305,Tyr309, Phe335,Leu338,Ile339, Phe342,Phe731,Phe982, Gln989 |
| VRP | −7.9 | Tyr952, Gln989 | Met68,Ile305,Tye309, Phe335,Leu338,Phe342, Gln724,Phe727,Met948, Phe982,Ala986 |
| CNI1 | CNI2 | CNI3 | CNI4 | |
|---|---|---|---|---|
| VDWAALS | −51.0246 | −70.7361 | −59.6757 | −61.6645 |
| EEL | −4.4372 | −12.4815 | −0.6747 | −4.5164 |
| EPB | 34.2307 | 46.2531 | 33.3708 | 36.9572 |
| DELTA G gas | −55.4618 | −83.2176 | −60.3504 | −66.1808 |
| DELTA G solv | 28.8456 | 41.1148 | 27.9166 | 31.4011 |
| DELTA TOTAL | −26.6162 | −42.1028 | −32.4338 | −34.7798 |
| CNI1 | CNI2 | CNI3 | CNI4 | VRP | |
|---|---|---|---|---|---|
| VDWAALS | −63.2064 | −56.8462 | −58.2130 | −66.5229 | −58.8949 |
| EEL | −7.5823 | −1.8142 | −2.4056 | −3.0904 | −8.3045 |
| EPB | 37.7542 | 24.8517 | 29.9995 | 29.6317 | 31.6844 |
| DELTA G gas | −70.7888 | −58.6568 | −60.6186 | −69.6133 | −67.1994 |
| DELTA G solv | 32.1985 | 19.5962 | 24.6478 | 24.1233 | 25.4008 |
| DELTA TOTAL | −38.5902 | −39.0606 | −35.9709 | −45.4900 | −41.7985 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, C.; Yin, X.; Xi, R.; Yuan, C.; Ou, Y. Molecular Mechanisms of Reversal of Multidrug Resistance in Breast Cancer by Inhibition of P-gp by Cytisine N-Isoflavones Derivatives Explored Through Network Pharmacology, Molecular Docking, and Molecular Dynamics. Int. J. Mol. Sci. 2025, 26, 3813. https://doi.org/10.3390/ijms26083813
Xiao C, Yin X, Xi R, Yuan C, Ou Y. Molecular Mechanisms of Reversal of Multidrug Resistance in Breast Cancer by Inhibition of P-gp by Cytisine N-Isoflavones Derivatives Explored Through Network Pharmacology, Molecular Docking, and Molecular Dynamics. International Journal of Molecular Sciences. 2025; 26(8):3813. https://doi.org/10.3390/ijms26083813
Chicago/Turabian StyleXiao, Chuangchuang, Xiaoying Yin, Rui Xi, Chunping Yuan, and Yangsheng Ou. 2025. "Molecular Mechanisms of Reversal of Multidrug Resistance in Breast Cancer by Inhibition of P-gp by Cytisine N-Isoflavones Derivatives Explored Through Network Pharmacology, Molecular Docking, and Molecular Dynamics" International Journal of Molecular Sciences 26, no. 8: 3813. https://doi.org/10.3390/ijms26083813
APA StyleXiao, C., Yin, X., Xi, R., Yuan, C., & Ou, Y. (2025). Molecular Mechanisms of Reversal of Multidrug Resistance in Breast Cancer by Inhibition of P-gp by Cytisine N-Isoflavones Derivatives Explored Through Network Pharmacology, Molecular Docking, and Molecular Dynamics. International Journal of Molecular Sciences, 26(8), 3813. https://doi.org/10.3390/ijms26083813