Routine CT Diagnostics Cause Dose-Dependent Gene Expression Changes in Peripheral Blood Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
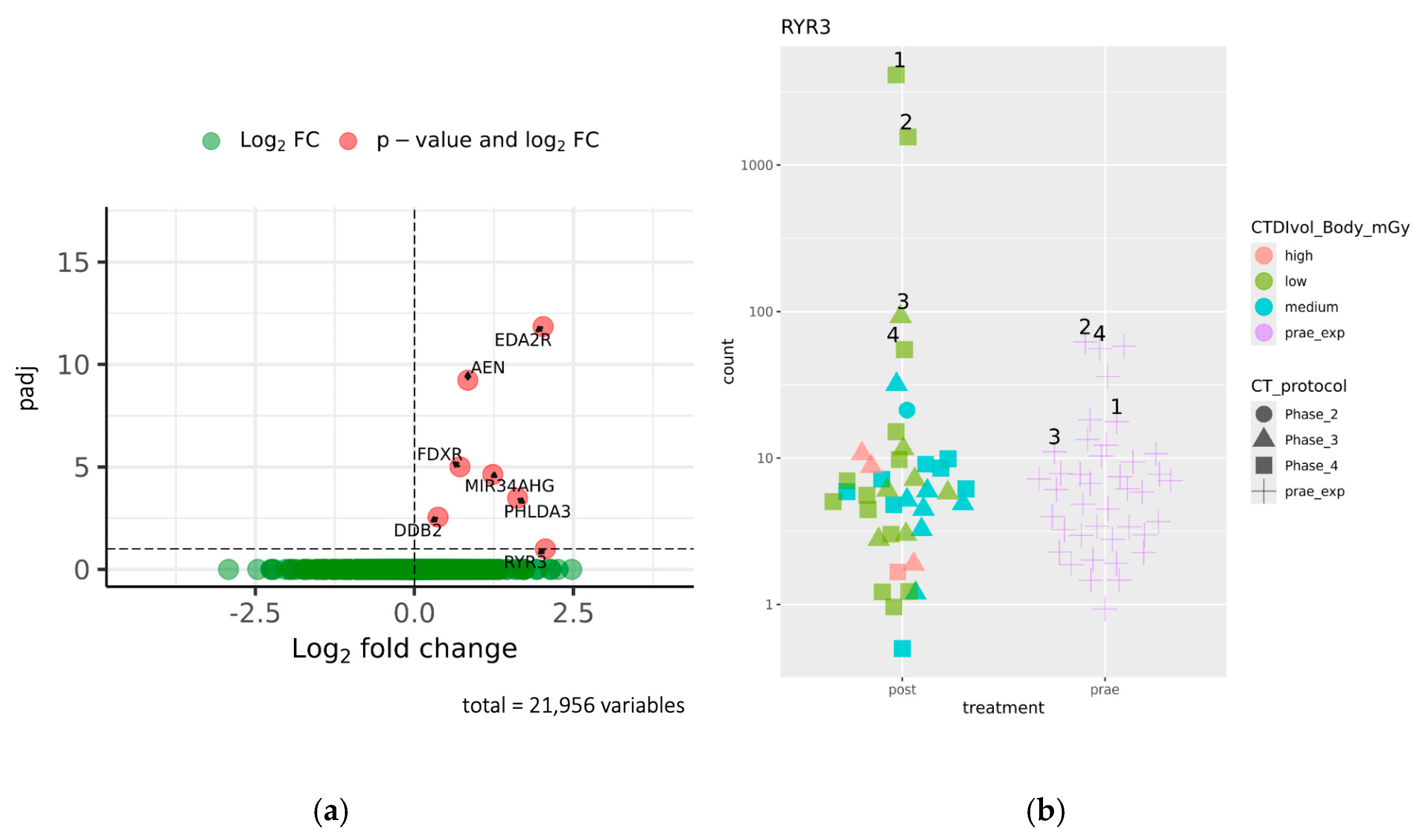
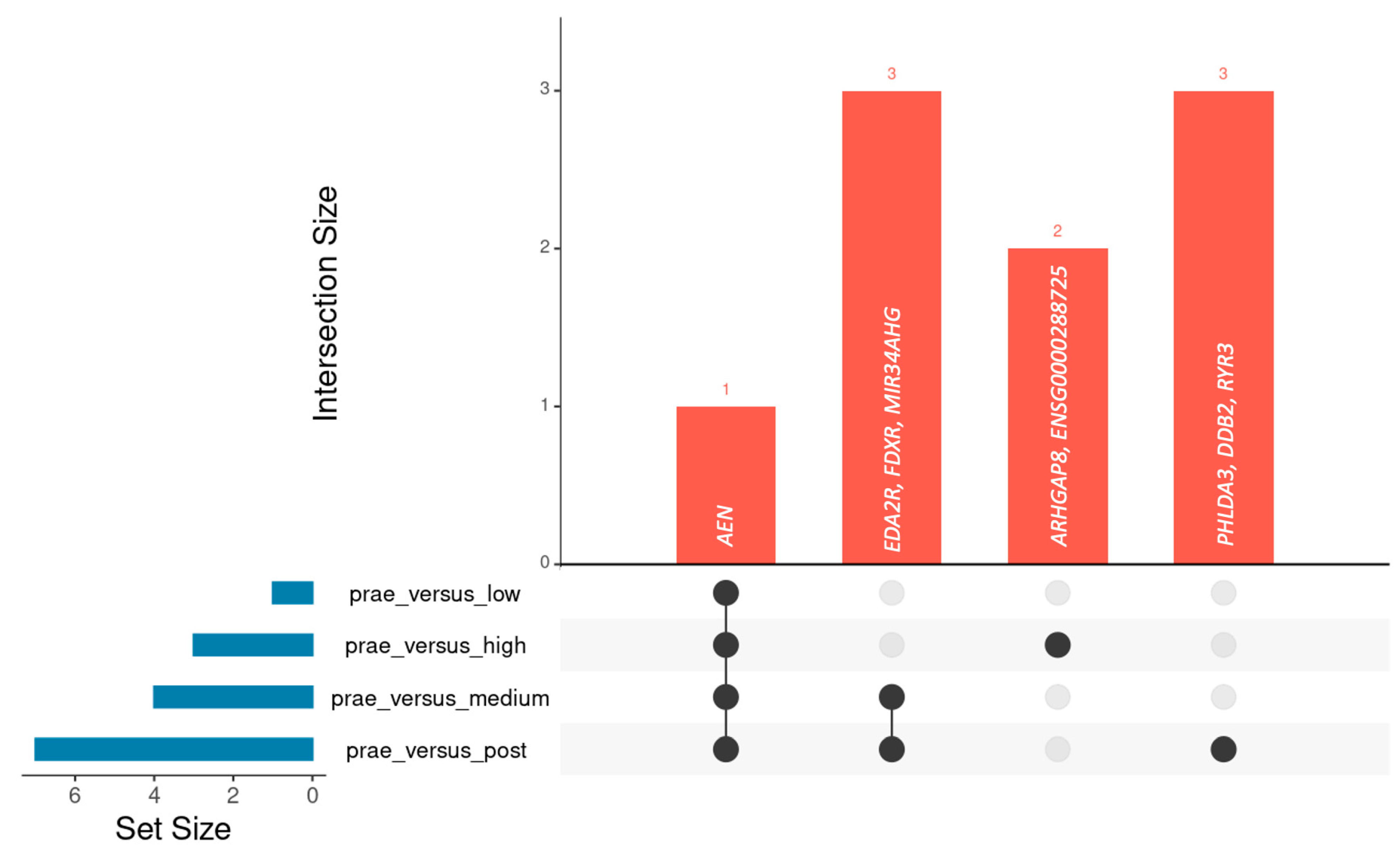
2.1. Identification of Genes Differentially Expressed Before and After CT Exposure
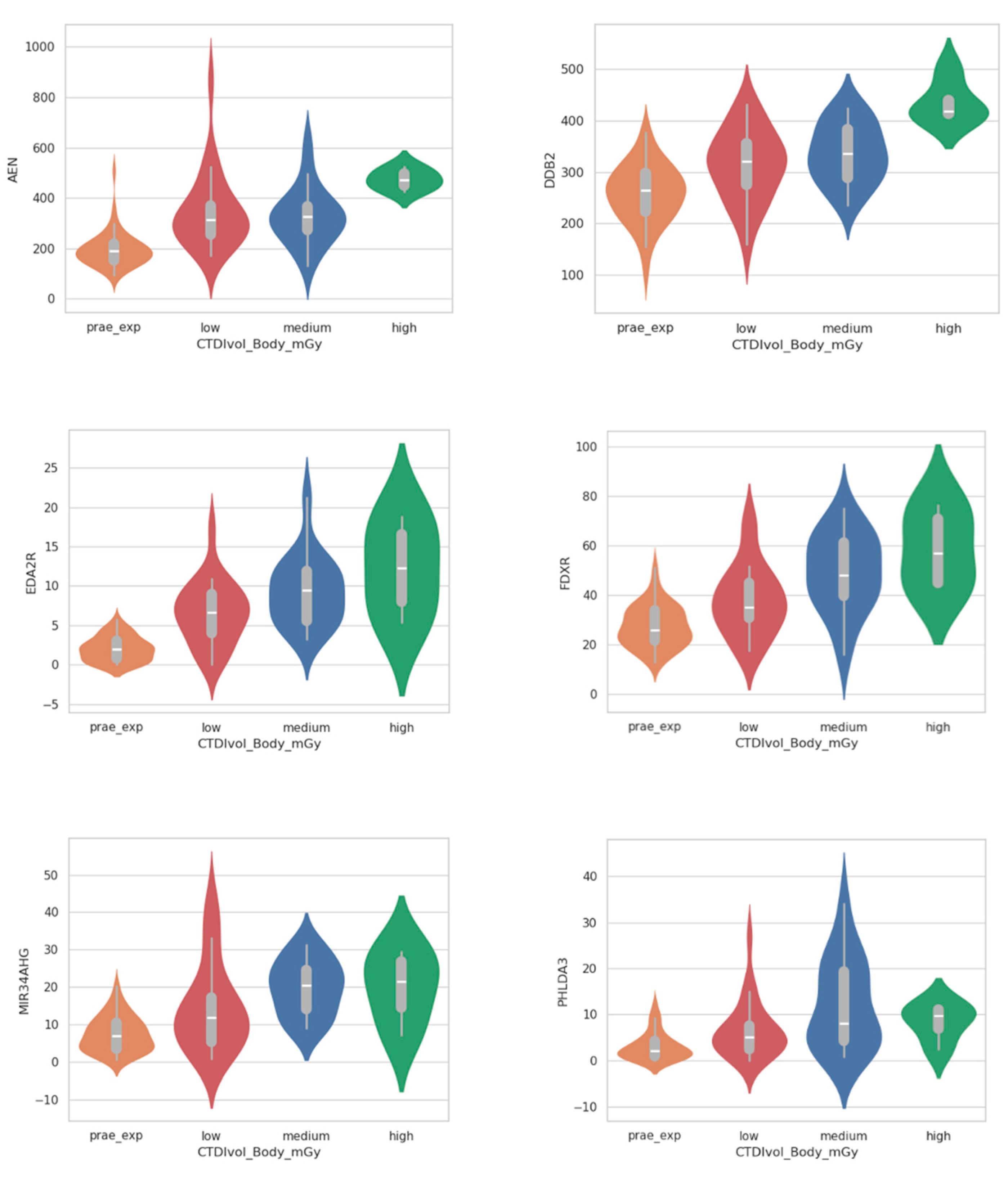
2.2. Analysis of Dose-Dependent Effects
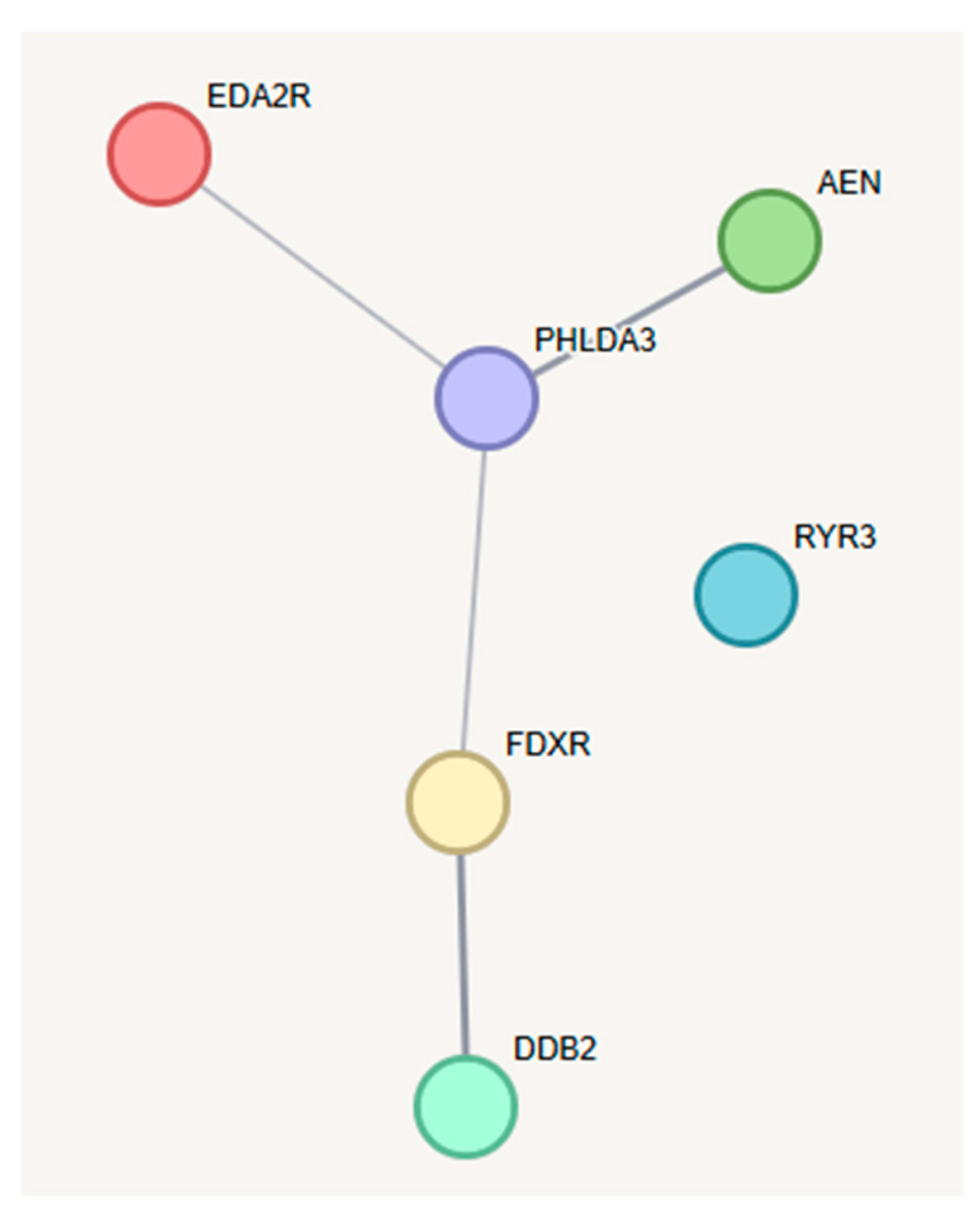
2.3. Analysis of Gene Interactions and Biological Pathways
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. CT Examination
4.3. Dose Calculations
4.4. Sample Acquisition, RNA-Isolation, and Whole-Genome RNA Sequencing
4.5. Differential Gene Expression Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2MDPI | Multidisciplinary Digital Publishing Institute |
| CT | Computed Tomography |
| CTDIvol | Computed Tomography Dose Index per Volume |
| SD | Standard deviation |
| GSEA | Gene Set Enrichment Analysis |
| ALARA | As Low As Reasonably Achievable |
| DNA | Deoxyribonucleic Acid |
| RNA | Ribonucleic Acid |
References
- Schulz, R.A.; Stein, J.A.; Pelc, N.J. How CT happened: The early development of medical computed tomography. J. Med. Imaging 2021, 8, 052110. [Google Scholar]
- Hsieh, J.; Flohr, T. Computed tomography recent history and future perspectives. J. Med. Imaging 2021, 8, 052109. [Google Scholar] [CrossRef]
- European Council Directive 2013/59/Euratom on Basic Safety Standards for Protection Against the Dangers Srising from Exposure to Ionising Radiation and Repealing Directives 89/618/Euratom, 90/641/Euratom, 96/29/Euratom, 97/43/Euratom and 2003/122/Euratom. OJ of the EU. L13; 57: 1–73. 2014. Available online: https://eur-lex.europa.eu/eli/dir/2013/59/oj/eng (accessed on 25 March 2025).
- Tsapaki, V. Radiation dose optimization in diagnostic and interventional radiology: Current issues and future perspectives. Phys. Medica 2020, 79, 16–21. [Google Scholar]
- Mettler, F.A., Jr.; Mahesh, M.; Bhargavan-Chatfield, M.; Chambers, C.E.; Elee, J.G.; Frush, D.P.; Miller, D.L.; Royal, H.D.; Milano, M.T.; Milano, M.T.; et al. Patient exposure from radiologic and nuclear medicine procedures in the United States: Procedure volume and effective dose for the period 2006–2016. Radiology 2020, 295, 418–427. [Google Scholar] [PubMed]
- United Nations Scientific Committee on the Effects of Atomic Radiation, UNSCEAR 2020/2021 Report Volume I, Scientific Annex A: “Medical Exposure to Lonizing Radiation”; United Nations: New York, NY, USA, 2022.
- NCRP. Medical Radiation Exposure of Patients in the United States: Recommendations of the National Council on Radiation Protection and Measurements; NCRP: Bethesda, MA, USA, 2019. [Google Scholar]
- Schegerer, A.A.; Nagel, H.D.; Stamm, G.; Adam, G.; Brix, G. Current CT practice in Germany: Results and implications of a nationwide survey. Eur. J. Radiol. 2017, 90, 114–128. [Google Scholar] [CrossRef]
- de Basea Gomez, B.; Thierry-Chef, I.; Harbron, R.; Hauptmann, M.; Byrnes, G.; Bernier, M.O.; Le Cornet, L.; Dabin, J.; Ferro, G.; Istad, T.S.; et al. Risk of hematological malignancies from CT radiation exposure in children, adolescents and young adults. Nat. Med. 2023, 29, 3111–3119. [Google Scholar] [CrossRef]
- Pearce, M.S.; Salotti, J.A.; Little, M.P.; McHugh, K.; Lee, C.; Kim, K.P.; Howe, N.L.; Ronckers, C.M.; Rajaraman, P.; Sir Craft, A.W.; et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. Lancet 2012, 380, 499–505. [Google Scholar]
- Mathews, J.D.; Forsythe, A.V.; Brady, Z.; Butler, M.W.; Goergen, S.K.; Byrnes, G.B.; Giles, G.G.; Wallace, A.B.; Anderson, P.R.; Guiver, T.A.; et al. Cancer risk in 680,000 people exposed to computed tomography scans in childhood or adolescence: Data linkage study of 11 million Australians. BMJ 2013, 346, f2360. [Google Scholar]
- Foucault, A.; Ancelet, S.; Dreuil, S.; Caër-Lorho, S.; Ducou Le Pointe, H.; Brisse, H.; Chateil, J.F.; Lee, C.; Leuraud, K.; Bernier, M.O. Childhood cancer risks estimates following CT scans: An update of the French CT cohort study. Eur. Radiol. 2022, 32, 5491–5498. [Google Scholar]
- Nikkilä, A.; Raitanen, J.; Lohi, O.; Auvinen, A. Radiation exposure from computerized tomography and risk of childhood leukemia: Finnish register-based case-control study of childhood leukemia (FRECCLE). Haematologica 2018, 103, 1873–1880. [Google Scholar]
- Berrington de Gonzalez, A.; Pasqual, E.; Veiga, L. Epidemiological studies of CT scans and cancer risk: The state of the science. Br. J. Radiol. 2021, 94, 20210471. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Sung, C.Y.; Wang, S.C.; Shao, Y.J. Risks of leukemia, intracranial tumours and lymphomas in childhood and early adulthood after pediatric radiation exposure from computed tomography. CMAJ 2023, 195, E575–E583. [Google Scholar]
- Little, M.P.; Bazyka, D.; Berrington de Gonzalez, A.; Brenner, A.V.; Chumak, V.V.; Cullings, H.M.; Daniels, R.D.; French, B.; Grant, E.; Hamada, N.; et al. A Historical Survey of Key Epidemiological Studies of Ionizing Radiation Exposure. Radiat. Res. 2024, 202, 432–487. [Google Scholar] [PubMed]
- Kaatsch, H.L.; Becker, B.V.; Schüle, S.; Ostheim, P.; Nestler, K.; Jakobi, J.; Schäfer, B.; Hantke, T.; Brockmann, M.A.; Abend, M.; et al. Gene expression changes and DNA damage after ex vivo exposure of peripheral blood cells to various CT photon spectra. Sci. Rep. 2021, 11, 12060. [Google Scholar]
- Becker, B.V.; Kaatsch, H.L.; Nestler, K.; Overhoff, D.; Schneider, J.; Dillinger, D.; Piechotka, J.; Brockmann, M.A.; Ullmann, R.; Port, M.; et al. Initial experience on abdominal photon-counting computed tomography in clinical routine: General image quality and dose exposure. Eur. Radiol. 2023, 33, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.V.; Kaatsch, H.L.; Nestler, K.; Jakobi, J.; Schäfer, B.; Hantke, T.; Brockmann, M.A.; Waldeck, S.; Port, M.; Ullmann, R. Impact of medical imaging on the epigenome—Low-dose exposure in the course of computed tomography does not induce detectable changes of DNA-methylation profiles in peripheral blood cells. Int. J. Radiat. Biol. 2022, 98, 980–985. [Google Scholar]
- Kaatsch, H.L.; Majewski, M.; Schrock, G.; Obermair, R.; Seidel, J.; Nestler, K.; Abend, M.; Waldeck, S.; Port, M.; Ullmann, R.; et al. CT Irradiation-induced Changes of Gene Expression within Peripheral Blood Cells. Health Phys. 2020, 119, 44–51. [Google Scholar]
- O’Brien, G.; Cruz-Garcia, L.; Majewski, M.; Grepl, J.; Abend, M.; Port, M.; Tichý, A.; Sirak, I.; Malkova, A.; Donovan, E.; et al. FDXR is a biomarker of radiation exposure in vivo. Sci. Rep. 2018, 8, 684. [Google Scholar]
- Nguyen, P.K.; Lee, W.H.; Li, Y.F.; Hong, W.X.; Hu, S.; Chan, C.; Liang, G.; Nguyen, I.; Ong, S.G.; Churko, J.; et al. Assessment of the Radiation Effects of Cardiac CT Angiography Using Protein and Genetic Biomarkers. JACC Cardiovasc. Imaging 2015, 8, 873–884. [Google Scholar] [CrossRef]
- Löbrich, M.; Rief, N.; Kühne, M.; Heckmann, M.; Fleckenstein, J.; Rübe, C.; Uder, M. In vivo formation and repair of DNA double-strand breaks after computed tomography examinations. Proc. Natl. Acad. Sci. USA 2005, 102, 8984–8989. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Amundson, S.A.; Lee, R.A.; Koch-Paiz, C.A.; Bittner, M.L.; Meltzer, P.; Trent, J.M.; Fornace, A.J., Jr. Differential responses of stress genes to low dose-rate gamma irradiation. Mol. Cancer Res. 2003, 1, 445–452. [Google Scholar]
- Amundson, S.A.; Bittner, M.; Meltzer, P.; Trent, J.; Fornace, A.J., Jr. Induction of gene expression as a monitor of exposure to ionizing radiation. Radiat. Res. 2001, 156 Pt 2, 657–661. [Google Scholar] [CrossRef]
- Long, X.H.; Zhao, Z.Q.; He, X.P.; Wang, H.P.; Xu, Q.Z.; An, J.; Bai, B.; Sui, J.L.; Zhou, P.K. Dose-dependent expression changes of early response genes to ionizing radiation in human lymphoblastoid cells. Int. J. Mol. Med. 2007, 19, 607–615. [Google Scholar] [CrossRef]
- Laurier, D.; Billarand, Y.; Klokov, D.; Leuraud, K. The scientific basis for the use of the linear no-threshold (LNT) model at low doses and dose rates in radiological protection. J. Radiol. Prot. 2023, 43, 024003. [Google Scholar]
- Broustas, C.G.; Xu, Y.; Harken, A.D.; Chowdhury, M.; Garty, G.; Amundson, S.A. Impact of Neutron Exposure on Global Gene Expression in a Human Peripheral Blood Model. Radiat. Res. 2017, 187, 433–440. [Google Scholar]
- Sokolov, M.; Neumann, R. Global Gene Expression Alterations as a Crucial Constituent of Human Cell Response to Low Doses of Ionizing Radiation Exposure. Int. J. Mol. Sci. 2016, 17, 55. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, J.; Pouliot, M.; Authier, S.; Zhou, D.; Loose, D.S.; Hauer-Jensen, M. Gene expression profiling in non-human primate jejunum, ileum and colon after total-body irradiation: A comparative study of segment-specific molecular and cellular responses. BMC Genom. 2015, 16, 984. [Google Scholar] [CrossRef]
- Lacombe, J.; Sima, C.; Amundson, S.A.; Zenhausern, F. Candidate gene biodosimetry markers of exposure to external ionizing radiation in human blood: A systematic review. PLoS ONE 2018, 13, e0198851. [Google Scholar] [CrossRef] [PubMed]
- Kultova, G.; Tichy, A.; Rehulkova, H.; Myslivcova-Fucikova, A. The hunt for radiation biomarkers: Current situation. Int. J. Radiat. Biol. 2020, 96, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell. 2007, 26, 745–752. [Google Scholar] [CrossRef]
- Allen, M.A.; Andrysik, Z.; Dengler, V.L.; Mellert, H.S.; Guarnieri, A.; Freeman, J.A.; Sullivan, K.D.; Galbraith, M.D.; Luo, X.; Kraus, W.L.; et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 2014, 3, e02200. [Google Scholar] [CrossRef]
- Jeay, S.; Gaulis, S.; Ferretti, S.; Bitter, H.; Ito, M.; Valat, T.; Murakami, M.; Ruetz, S.; Guthy, D.A.; Rynn, C.; et al. A distinct p53 target gene set predicts for response to the selective p53–HDM2 inhibitor NVP-CGM097. eLife 2015, 4, e06498. [Google Scholar]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [PubMed]
- Pashaei-Asl, R.; Pashaiasl, M.; Ebrahimie, E.; Lale Ataei, M.; Paknejad, M. Apoptotic effects of human amniotic fluid mesenchymal stem cells conditioned medium on human MCF-7 breast cancer cell line. Bioimpacts 2023, 13, 191–206. [Google Scholar]
- Dressman, H.K.; Muramoto, G.G.; Chao, N.J.; Meadows, S.; Marshall, D.; Ginsburg, G.S.; Nevins, J.R.; Chute, J.P. Gene expression signatures that predict radiation exposure in mice and humans. PLoS Med. 2007, 4, e106. [Google Scholar] [CrossRef]
- Jing, S.; Michael, G.W.; Michael, G.W. Gene Set Enrichment Analysis (GSEA) for Interpreting Gene Expression Profiles. Curr. Bioinform. 2007, 2, 133–137. [Google Scholar]
- Brace, L.E.; Vose, S.C.; Stanya, K.; Gathungu, R.M.; Marur, V.R.; Longchamp, A.; Treviño-Villarreal, H.; Mejia, P.; Vargas, D.; Inouye, K.; et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech. Dis. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Lall, R.; Ganapathy, S.; Yang, M.; Xiao, S.; Xu, T.; Su, H.; Shadfan, M.; Asara, J.M.; Ha, C.S.; Ben-Sahra, I.; et al. Low-dose radiation exposure induces a HIF-1-mediated adaptive and protective metabolic response. Cell Death Differ. 2014, 21, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Schüle, S.; Bunert, F.; Hackenbroch, C.; Beer, M.; Ostheim, P.; Stewart, S.; Port, M.; Scherthan, H.; Abend, M. The Influence of Computed Tomography Contrast Agent on Radiation-Induced Gene Expression and Double-Strand Breaks. Radiat. Res. 2024, 201, 396–405. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 March 2025).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar]
- Love, M.I.; Anders, S.; Kim, V.; Huber, W. RNA-Seq workflow: Gene-level exploratory analysis and differential expression. F1000Research 2015, 4, 1070. [Google Scholar]
- Fang, Z.; Liu, X.; Peltz, G. GSEApy: A comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 2023, 39, btac757. [Google Scholar] [CrossRef]
- Schegerer, A.; Loose, R.; Heuser, L.J.; Brix, G. Diagnostic Reference Levels for Diagnostic and Interventional X-Ray Procedures in Germany: Update and Handling. In RöFo-Fortschritte auf dem Gebiet der Röntgenstrahlen und der bildgebenden Verfahren; © Georg Thieme Verlag KG: Stuttgart, Germany, 2019; Volume 191, pp. 739–751. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. R Package Version 1.24.0. 2024. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 25 March 2025).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaatsch, H.L.; Kubitscheck, L.; Wagner, S.; Hantke, T.; Preiss, M.; Ostheim, P.; Nestler, T.; Piechotka, J.; Overhoff, D.; Brockmann, M.A.; et al. Routine CT Diagnostics Cause Dose-Dependent Gene Expression Changes in Peripheral Blood Cells. Int. J. Mol. Sci. 2025, 26, 3185. https://doi.org/10.3390/ijms26073185
Kaatsch HL, Kubitscheck L, Wagner S, Hantke T, Preiss M, Ostheim P, Nestler T, Piechotka J, Overhoff D, Brockmann MA, et al. Routine CT Diagnostics Cause Dose-Dependent Gene Expression Changes in Peripheral Blood Cells. International Journal of Molecular Sciences. 2025; 26(7):3185. https://doi.org/10.3390/ijms26073185
Chicago/Turabian StyleKaatsch, Hanns Leonhard, Laura Kubitscheck, Simon Wagner, Thomas Hantke, Maximilian Preiss, Patrick Ostheim, Tim Nestler, Joel Piechotka, Daniel Overhoff, Marc A. Brockmann, and et al. 2025. "Routine CT Diagnostics Cause Dose-Dependent Gene Expression Changes in Peripheral Blood Cells" International Journal of Molecular Sciences 26, no. 7: 3185. https://doi.org/10.3390/ijms26073185
APA StyleKaatsch, H. L., Kubitscheck, L., Wagner, S., Hantke, T., Preiss, M., Ostheim, P., Nestler, T., Piechotka, J., Overhoff, D., Brockmann, M. A., Waldeck, S., Port, M., Ullmann, R., & Becker, B. V. (2025). Routine CT Diagnostics Cause Dose-Dependent Gene Expression Changes in Peripheral Blood Cells. International Journal of Molecular Sciences, 26(7), 3185. https://doi.org/10.3390/ijms26073185