

Low-Dose Docetaxel Is Effective in Reducing Atherogenic Lipids and Atherosclerosis
 , , , , and
, , , , and
Abstract

1. Introduction
2. Results
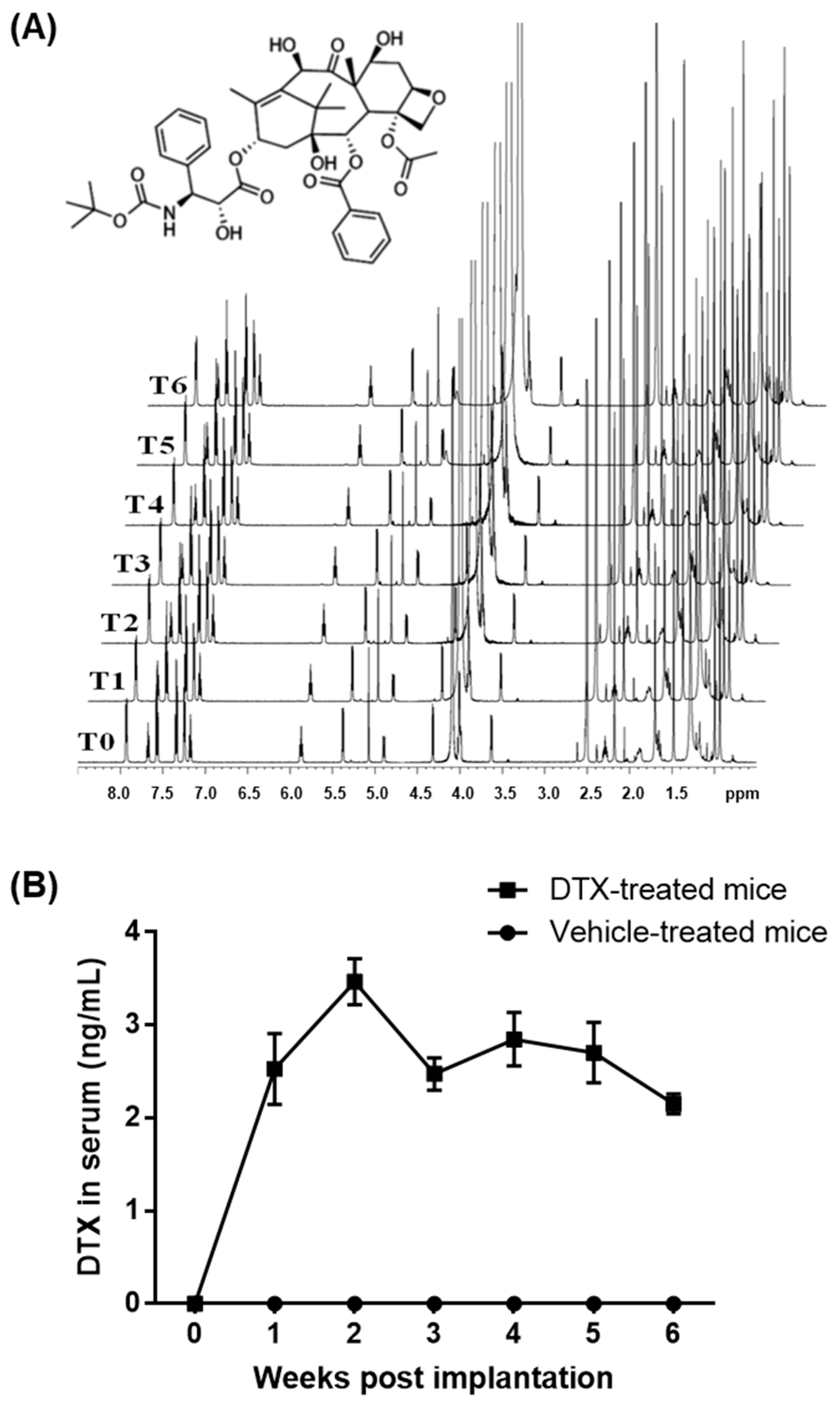
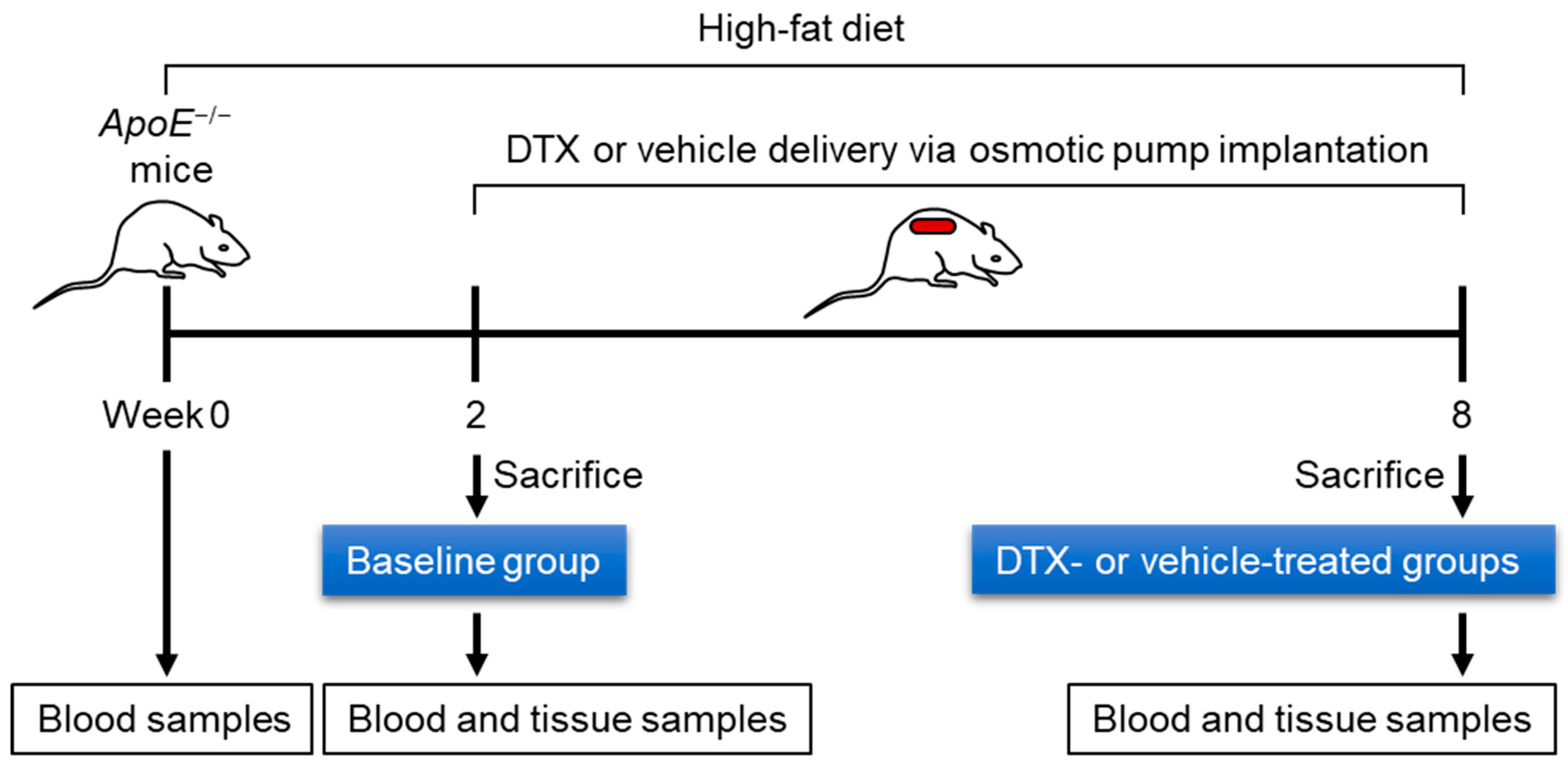
2.1. Establishment of a DTX Administration Protocol for Atherosclerosis Studies in Mice
2.2. DTX Exhibits No Hematologic Toxicity at Low-Nanomolar Concentrations
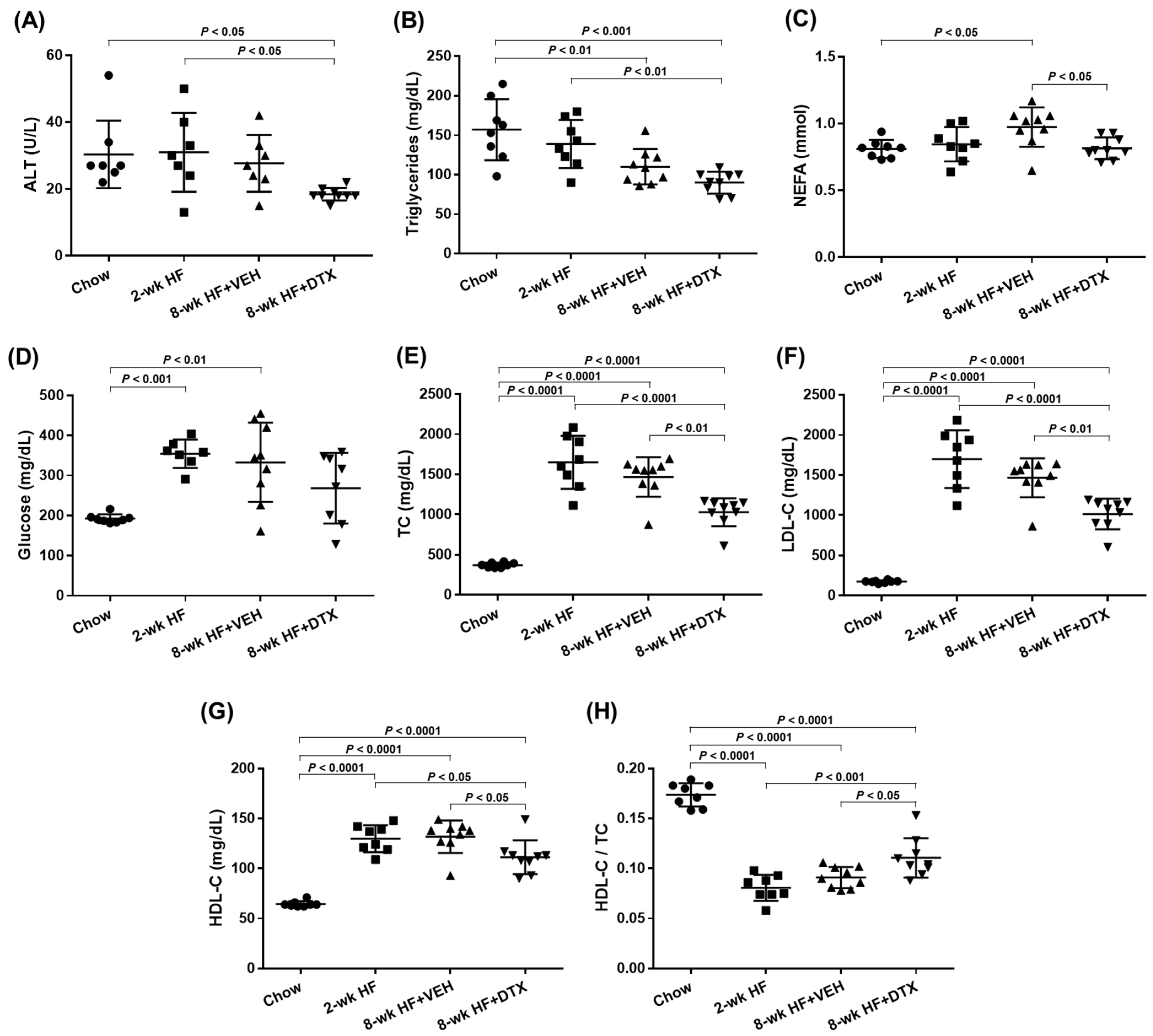
2.3. DTX Improves the Circulating Lipid Profile
2.4. DTX Does Not Directly Affect the Modulation of Inflammatory Cytokines
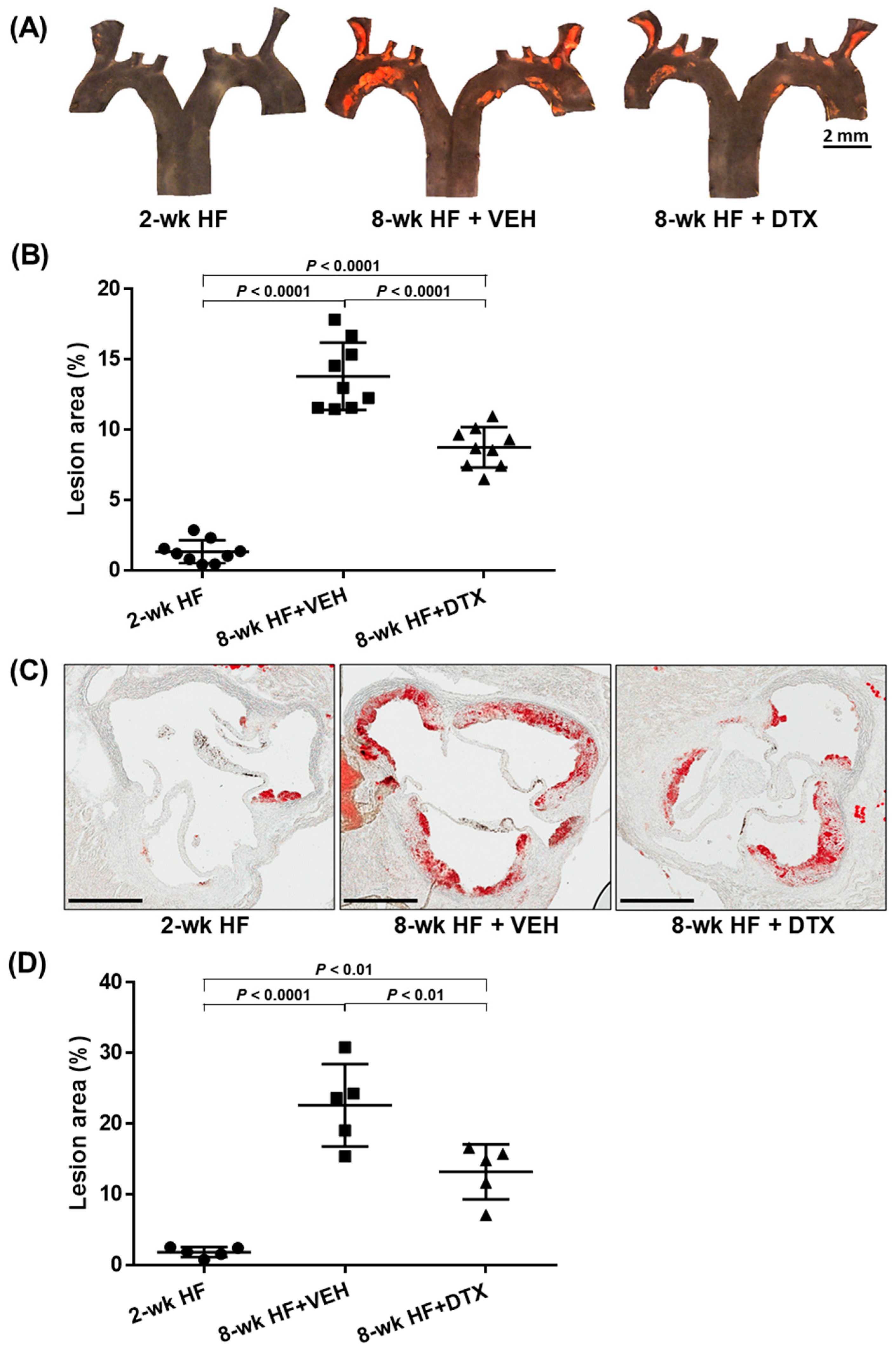
2.5. DTX Effectively Reduces Atherosclerosis Development
2.6. Limitations
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Husbandry
4.3. Analysis of the Chemical Stability of DTX
4.4. Measurement of DTX Concentration in Mouse Serum
4.5. Subcutaneous Implantation of Osmotic Pumps
4.6. Complete Blood Count
4.7. Measurement of Serum Biochemical Parameters Associated with Lipid Metabolism
4.8. Measurement of Cytokine Levels in Plasma
4.9. Quantification of Atherosclerotic Lesions
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations and Acronyms
References
- Choi, H.Y.; Hafiane, A.; Schwertani, A.; Genest, J. High-Density Lipoproteins: Biology, Epidemiology, and Clinical Management. Can. J. Cardiol. 2017, 33, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Ruel, I.; Choi, S.; Genest, J. New Strategies to Promote Macrophage Cholesterol Efflux. Front. Cardiovasc. Med. 2021, 8, 795868. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef]
- Pownall, H.J.; Rosales, C.; Gillard, B.K.; Gotto, A.M., Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat. Rev. Cardiol. 2021, 18, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Ronda, N.; Bernini, F.; Zimetti, F. High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives. Cells 2021, 10, 574. [Google Scholar] [CrossRef]
- Gibson, C.M.; Duffy, D.; Korjian, S.; Bahit, M.C.; Chi, G.; Alexander, J.H.; Lincoff, A.M.; Heise, M.; Tricoci, P.; Deckelbaum, L.I.; et al. Apolipoprotein A1 Infusions and Cardiovascular Outcomes after Acute Myocardial Infarction. N. Engl. J. Med. 2024, 390, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Choi, S.; Iatan, I.; Ruel, I.; Genest, J. Biomedical Advances in ABCA1 Transporter: From Bench to Bedside. Biomedicines 2023, 11, 561. [Google Scholar] [CrossRef] [PubMed]
- Genest, J.; Choi, H.Y. Novel Approaches for HDL-Directed Therapies. Curr. Atheroscler. Rep. 2017, 19, 55. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Ruel, I.; Malina, A.; Garrod, D.R.; Oda, M.N.; Pelletier, J.; Schwertani, A.; Genest, J. Desmocollin 1 is abundantly expressed in atherosclerosis and impairs high-density lipoprotein biogenesis. Eur. Heart J. 2018, 39, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Ruel, I.; Genest, J. Identification of Docetaxel as a Potential Drug to Promote HDL Biogenesis. Front. Pharmacol. 2021, 12, 679456. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Yoo, J.M.; Lim, Y.; Tudev, M.; Yoo, H.S.; Hong, J.T.; Yun, Y.P. Inhibitory effects of docetaxel on platelet-derived growth factor (PDGF)-BB-induced proliferation of vascular smooth muscle cells through blocking PDGF-receptor beta phosphorylation. J. Pharmacol. Sci. 2011, 116, 204–213. [Google Scholar] [CrossRef]
- Boucher, P.; Gotthardt, M.; Li, W.P.; Anderson, R.G.; Herz, J. LRP: Role in vascular wall integrity and protection from atherosclerosis. Science 2003, 300, 329–332. [Google Scholar] [CrossRef]
- Basford, J.E.; Moore, Z.W.; Zhou, L.; Herz, J.; Hui, D.Y. Smooth muscle LDL receptor-related protein-1 inactivation reduces vascular reactivity and promotes injury-induced neointima formation. Arter. Thromb. Vasc. Biol. 2009, 29, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Braun-Dullaeus, R.C.; Sedding, D.G. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat. Med. 2002, 8, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arter. Thromb. Vasc. Biol. 2008, 28, 812–819. [Google Scholar] [CrossRef]
- Kenmotsu, H.; Tanigawara, Y. Pharmacokinetics, dynamics and toxicity of docetaxel: Why the Japanese dose differs from the Western dose. Cancer Sci. 2015, 106, 497–504. [Google Scholar] [CrossRef]
- Baker, S.D.; Zhao, M.; Lee, C.K.; Verweij, J.; Zabelina, Y.; Brahmer, J.R.; Wolff, A.C.; Sparreboom, A.; Carducci, M.A. Comparative pharmacokinetics of weekly and every-three-weeks docetaxel. Clin. Cancer Res. 2004, 10, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, C.; Han, X.; Jia, X.; Li, Y.; Liu, C.; Li, N.; Liu, L.; Liu, P.; Jiang, X.; et al. E17241 as a Novel ABCA1 (ATP-Binding Cassette Transporter A1) Upregulator Ameliorates Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2021, 41, e284–e298. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Puisset, F.; Alexandre, J.; Treluyer, J.M.; Raoul, V.; Roche, H.; Goldwasser, F.; Chatelut, E. Clinical pharmacodynamic factors in docetaxel toxicity. Br. J. Cancer 2007, 97, 290–296. [Google Scholar] [CrossRef]
- Ma, Y.; Lin, Q.; Yang, Y.; Liang, W.; Salamone, S.J.; Li, Y.; Lin, Y.; Zhao, H.; Zhao, Y.; Fang, W.; et al. Clinical pharmacokinetics and drug exposure-toxicity correlation study of docetaxel based chemotherapy in Chinese head and neck cancer patients. Ann. Transl. Med. 2020, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.E.; Mikkola, A.M.; Stepanek, A.M.; Vernet, A.; Hall, C.D.; Sun, C.C.; Yildirim, E.; Staropoli, J.F.; Lee, J.T.; Brown, D.E. Practical murine hematopathology: A comparative review and implications for research. Comp. Med. 2015, 65, 96–113. [Google Scholar] [PubMed]
- Andrade, R.J.; Chalasani, N.; Bjornsson, E.S.; Suzuki, A.; Kullak-Ublick, G.A.; Watkins, P.B.; Devarbhavi, H.; Merz, M.; Lucena, M.I.; Kaplowitz, N.; et al. Drug-induced liver injury. Nat. Rev. Dis. Primers 2019, 5, 58. [Google Scholar] [CrossRef]
- Hadizadeh, F.; Faghihimani, E.; Adibi, P. Nonalcoholic fatty liver disease: Diagnostic biomarkers. World J. Gastrointest. Pathophysiol. 2017, 8, 11–26. [Google Scholar] [CrossRef]
- Johnston, L.W.; Harris, S.B.; Retnakaran, R.; Giacca, A.; Liu, Z.; Bazinet, R.P.; Hanley, A.J. Association of NEFA composition with insulin sensitivity and beta cell function in the Prospective Metabolism and Islet Cell Evaluation (PROMISE) cohort. Diabetologia 2018, 61, 821–830. [Google Scholar] [CrossRef]
- Choi, H.Y.; Iatan, I.; Ruel, I.; Brown, L.; Hales, L.; Choi, S.; Genest, J. Docetaxel as a Model Compound to Promote HDL (High-Density Lipoprotein) Biogenesis and Reduce Atherosclerosis. Arter. Thromb. Vasc. Biol. 2023, 43, 609–617. [Google Scholar] [CrossRef]
- van Diepen, J.A.; Berbee, J.F.; Havekes, L.M.; Rensen, P.C. Interactions between inflammation and lipid metabolism: Relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis 2013, 228, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Malekmohammad, K.; Bezsonov, E.E.; Rafieian-Kopaei, M. Role of Lipid Accumulation and Inflammation in Atherosclerosis: Focus on Molecular and Cellular Mechanisms. Front. Cardiovasc. Med. 2021, 8, 707529. [Google Scholar] [CrossRef]
- Ray, M.; Autieri, M.V. Regulation of pro- and anti-atherogenic cytokines. Cytokine 2019, 122, 154175. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef]
- Nakashima, Y.; Plump, A.S.; Raines, E.W.; Breslow, J.L.; Ross, R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arter. Thromb. 1994, 14, 133–140. [Google Scholar] [CrossRef]
- Reddick, R.L.; Zhang, S.H.; Maeda, N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arter. Thromb. 1994, 14, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, F.; Zeigerer, A.; Kory, N.; Krahmer, N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin. Cell Dev. Biol. 2020, 108, 72–81. [Google Scholar] [CrossRef]
- Carotti, S.; Aquilano, K.; Valentini, F.; Ruggiero, S.; Alletto, F.; Morini, S.; Picardi, A.; Antonelli-Incalzi, R.; Lettieri-Barbato, D.; Vespasiani-Gentilucci, U. An overview of deregulated lipid metabolism in nonalcoholic fatty liver disease with special focus on lysosomal acid lipase. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G469–G480. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Pratico, D.; Lin, L.; Mantzoros, C.S.; Bahijri, S.; Tuomilehto, J.; Ren, J. Inflammation in atherosclerosis: Pathophysiology and mechanisms. Cell Death Dis. 2024, 15, 817. [Google Scholar] [CrossRef] [PubMed]
- Bjorkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.F.; Rehman, M.; Sarwar, H.S.; Naveed, S.; Salman, O.; Bukhari, N.I.; Hussain, I.; Webster, T.J.; Shahnaz, G. Advancements in the oral delivery of Docetaxel: Challenges, current state-of-the-art and future trends. Int. J. Nanomed. 2018, 13, 3145–3161. [Google Scholar] [CrossRef]
- Vermunt, M.A.C.; Robbrecht, D.G.J.; Devriese, L.A.; Janssen, J.M.; Thijssen, B.; Keessen, M.; van Eijk, M.; Kessels, R.; Eskens, F.; Beijnen, J.H.; et al. ModraDoc006, an oral docetaxel formulation in combination with ritonavir (ModraDoc006/r), in metastatic castration-resistant prostate cancer patients: A phase Ib study. Cancer Rep. 2021, 4, e1367. [Google Scholar] [CrossRef] [PubMed]
- Jibodh, R.A.; Lagas, J.S.; Nuijen, B.; Beijnen, J.H.; Schellens, J.H. Taxanes: Old drugs, new oral formulations. Eur. J. Pharmacol. 2013, 717, 40–46. [Google Scholar] [CrossRef]
- Bardelmeijer, H.A.; Ouwehand, M.; Buckle, T.; Huisman, M.T.; Schellens, J.H.; Beijnen, J.H.; van Tellingen, O. Low systemic exposure of oral docetaxel in mice resulting from extensive first-pass metabolism is boosted by ritonavir. Cancer Res. 2002, 62, 6158–6164. [Google Scholar] [PubMed]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.B.; Cassagnol, M. HMG-CoA Reductase Inhibitors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- National Institute of Diabetes and Digestive and Kidney Diseases. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney diseases: Bethesda, MD, USA, 2021. [Google Scholar]
- Robbins, N.; Thompson, A.; Mann, A.; Blomkalns, A.L. Isolation and excision of murine aorta; a versatile technique in the study of cardiovascular disease. J. Vis. Exp. 2014, 93, e52172. [Google Scholar] [CrossRef]
- Centa, M.; Ketelhuth, D.F.J.; Malin, S.; Gistera, A. Quantification of Atherosclerosis in Mice. J. Vis. Exp. 2019, 148, e59828. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Reference Range | Non-Treated Group Mean ± SEM (n = 5) | Vehicle-Treated Group Mean ± SEM (n = 5) | DTX-Treated Group Mean ± SEM (n = 5) |
|---|---|---|---|---|
| WBC count (×109/L) | 2–10 | 7.8 ± 1.48 | 8.3 ± 1.32 | 7.7 ± 1.73 |
| RBC count (×1012/L) | 7.8–10.6 | 9.8 ± 0.31 | 9.4 ± 0.27 | 9.3 ± 0.25 |
| Hemoglobin (g/L) | 136–164 | 145.4 ± 2.50 | 142.2 ± 3.15 | 140.2 ± 2.75 |
| Hematocrit (L/L) | 0.35–0.52 | 0.5 ± 0.01 | 0.5 ± 0.01 | 0.5 ± 0.01 |
| MCV (fL) | 45–55 | 48.6 ± 1.12 | 50.4 ± 0.40 | 50.2 ± 0.58 |
| MCH (pg) | 14–17 | 14.8 ± 0.44 | 15.2 ± 0.23 | 15.2 ± 0.18 |
| MCHC (g/L) | 300–380 | 304.6 ± 4.87 | 301.2 ± 2.20 | 303.0 ± 2.49 |
| Platelet count (×109/L) | 900–1600 | 1107.8 ± 143.41 | 1075.8 ± 94.35 | 1169.2 ± 129.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.Y.; Ruel, I.; Choi, S.; Iatan, I.; Choi, S.; Lee, J.-Y.; Genest, J. Low-Dose Docetaxel Is Effective in Reducing Atherogenic Lipids and Atherosclerosis. Int. J. Mol. Sci. 2025, 26, 1484. https://doi.org/10.3390/ijms26041484
Choi HY, Ruel I, Choi S, Iatan I, Choi S, Lee J-Y, Genest J. Low-Dose Docetaxel Is Effective in Reducing Atherogenic Lipids and Atherosclerosis. International Journal of Molecular Sciences. 2025; 26(4):1484. https://doi.org/10.3390/ijms26041484
Chicago/Turabian StyleChoi, Hong Y., Isabelle Ruel, Shiwon Choi, Iulia Iatan, Senna Choi, Jyh-Yeuan Lee, and Jacques Genest. 2025. "Low-Dose Docetaxel Is Effective in Reducing Atherogenic Lipids and Atherosclerosis" International Journal of Molecular Sciences 26, no. 4: 1484. https://doi.org/10.3390/ijms26041484
APA StyleChoi, H. Y., Ruel, I., Choi, S., Iatan, I., Choi, S., Lee, J.-Y., & Genest, J. (2025). Low-Dose Docetaxel Is Effective in Reducing Atherogenic Lipids and Atherosclerosis. International Journal of Molecular Sciences, 26(4), 1484. https://doi.org/10.3390/ijms26041484