

Genetic Analysis of the Awn Length Gene in the Rice Chromosome Segment Substitution Line CSSL29


 ,
,
Abstract
1. Introduction
2. Result
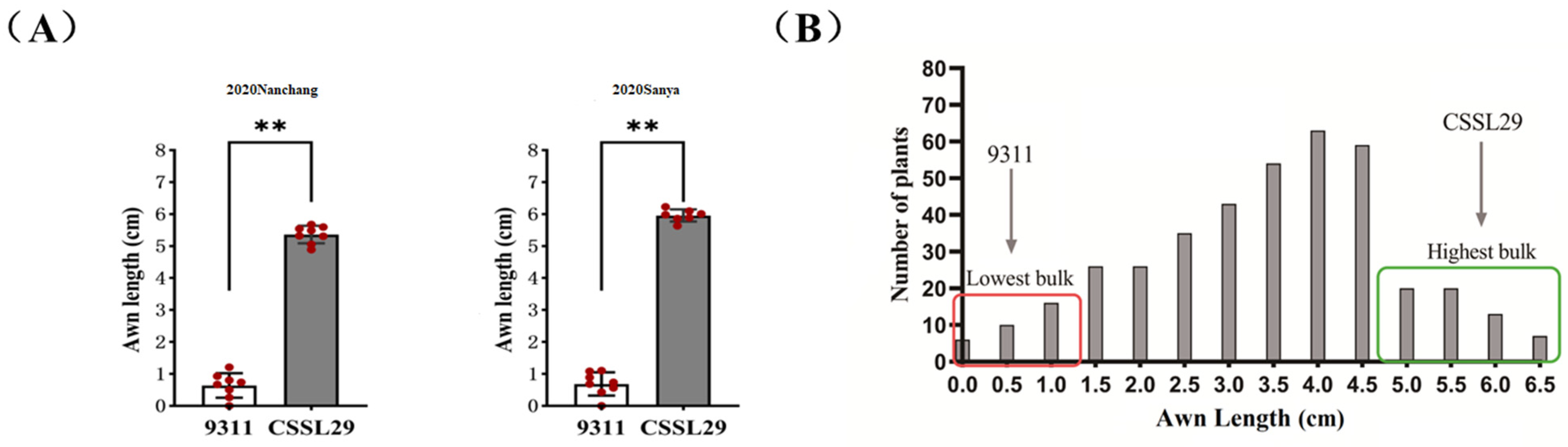
2.1. Phenotypic Analysis and Evaluation of Awn
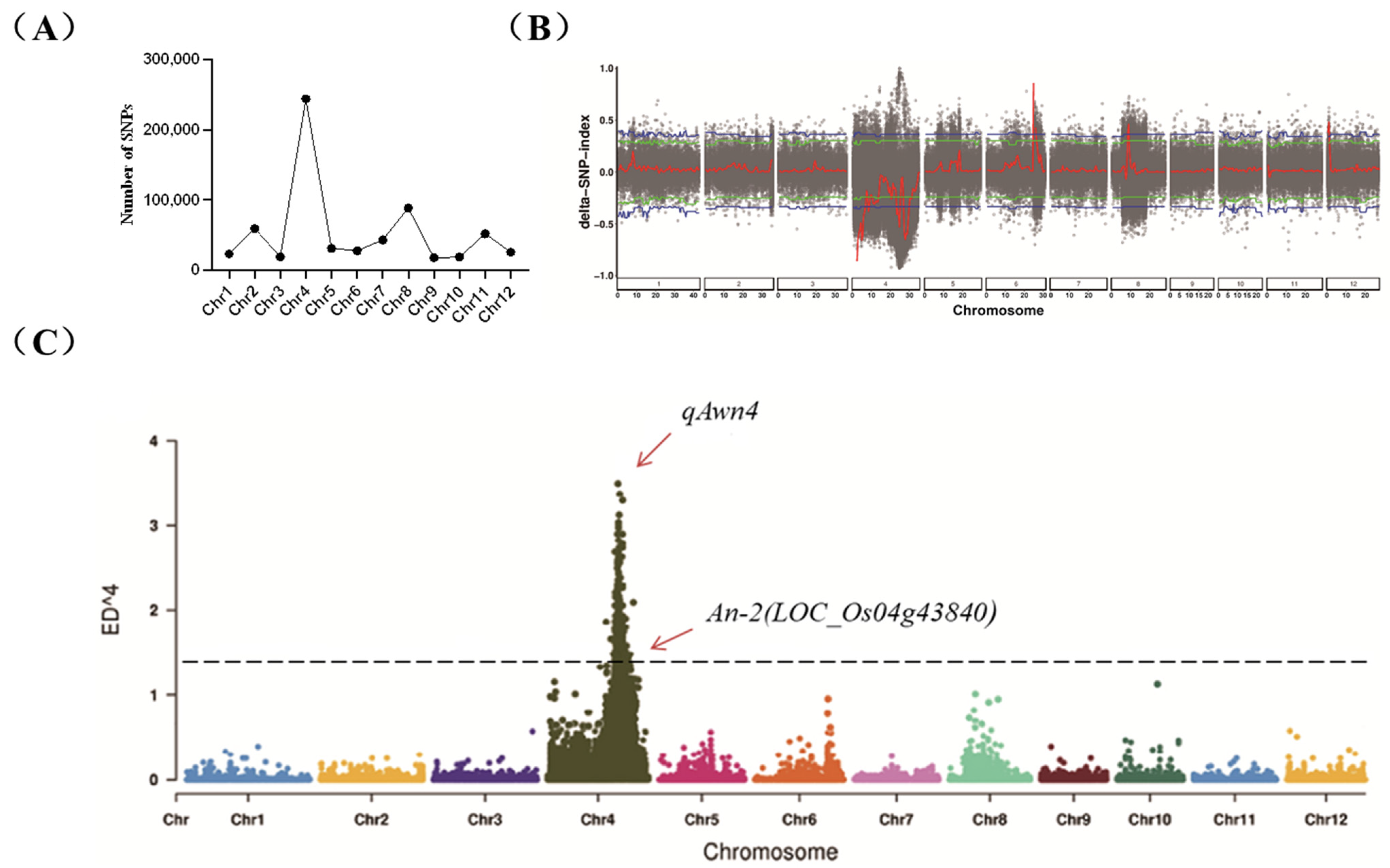
2.2. QTL-Seq Data Analysis
2.3. Genetic Analysis of CSSL Population
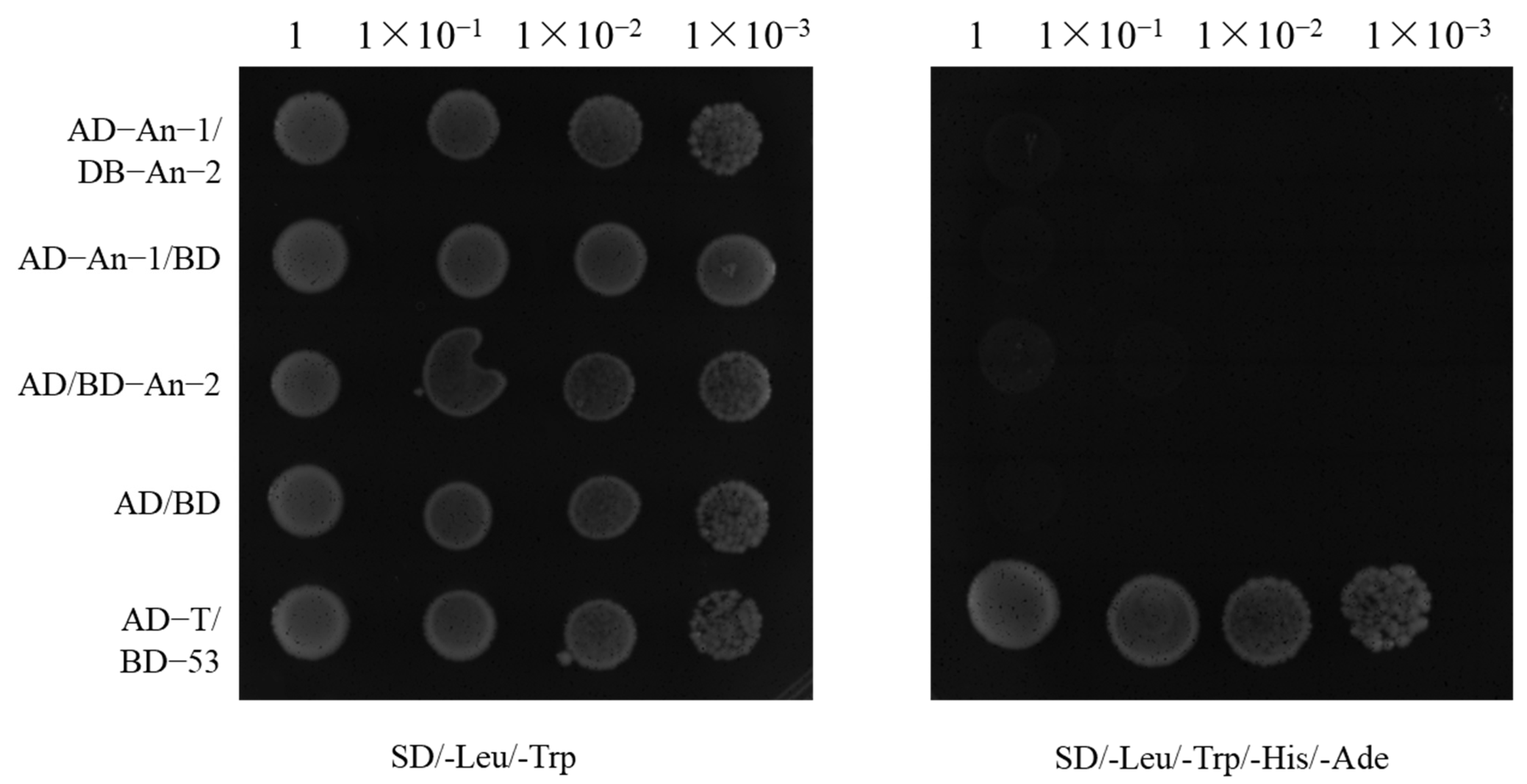
2.4. Relationship Between An-1 and An-2
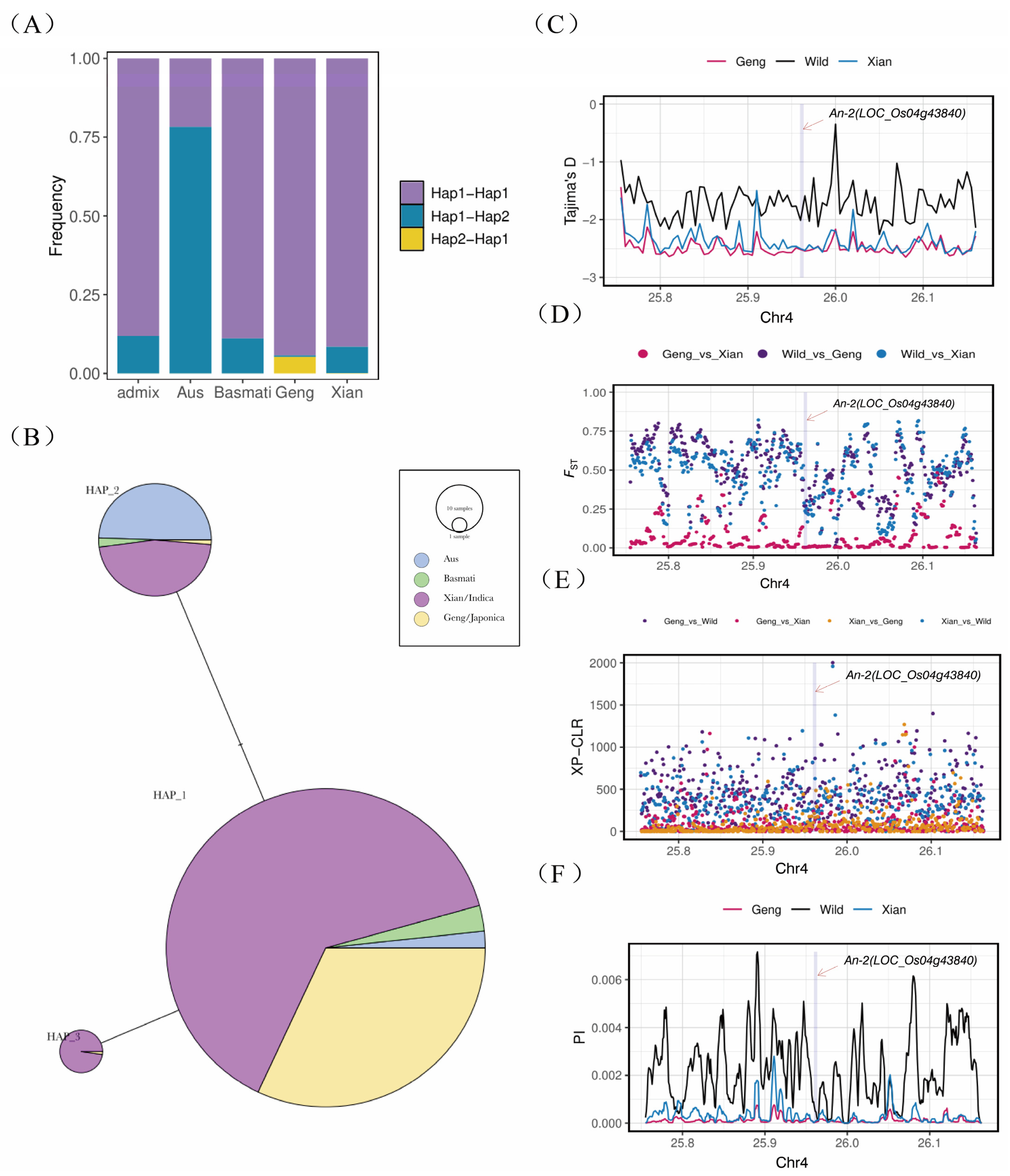
2.5. Haplotype and Population Genetic Analysis
3. Discussion
4. Materials and Methods
4.1. Test Materials
4.1.1. Plant Materials and Field Treatments
4.1.2. Strains and Vectors
4.1.3. Experimental Design
4.2. Grain Awn Length Measurement
4.3. Whole-Genome Resequencing
4.4. QTL-Seq Analysis
4.5. Yeast Two-Hybrid Assay
4.6. Data Analysis of Rice Population Resources
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SNP | Single Nucleotide Polymorphism |
| QTL | Quantitative Trait Locus |
| BSA | Bulk Segregant Analysis |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ED | Euclidean Distance |
| RIL | Recombinant Inbred Lines |
| CSSL | Chromosome Segment Substitution Lines |
| NIL | Near Isogenic Lines |
| LD | Linkage Disequilibrium |
References
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The Molecular Genetics of Crop Domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef]
- Fornasiero, A.; Wing, R.A.; Ronald, P. Rice Domestication. Curr. Biol. 2022, 32, R20–R24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guo, Z.; Zou, T.; Zhang, Z.; Zhang, J.; He, P.; Song, R.; Liu, Z.; Zhu, H.; Zhang, G.; et al. Substitution Mapping and Allelic Variations of the Domestication Genes from O. Rufipogon and O. Nivara. Rice 2023, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Wambugu, P.W.; Ndjiondjop, M.-N.; Henry, R. Genetics and Genomics of African Rice (Oryza Glaberrima Steud) Domestication. Rice 2021, 14, 6. [Google Scholar] [CrossRef]
- Bessho-Uehara, K.; Yamagata, Y.; Takashi, T.; Makino, T.; Yasui, H.; Yoshimura, A.; Ashikari, M. Exploring the Loci Responsible for Awn Development in Rice through Comparative Analysis of All AA Genome Species. Plants 2021, 10, 725. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zou, T.; He, Z.; Yuan, G.; Luo, T.; Zhu, J.; Liang, Y.; Deng, Q.; Wang, S.; Zheng, A.; et al. GRAIN LENGTH AND AWN 1 Negatively Regulates Grain Size in Rice. J. Integr. Plant Biol. 2019, 61, 1036–1042. [Google Scholar] [CrossRef]
- Ntakirutimana, F.; Xie, W. Unveiling the Actual Functions of Awns in Grasses: From Yield Potential to Quality Traits. Int. J. Mol. Sci. 2020, 21, 7593. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Lu, T.; Xu, Y.; Shen, W.; Liu, F.; Xie, X.; Li, Y.; Wang, K.; Li, R. Identification and Characterization of An-4, a Potential Quantitative Trait Locus for Awn Development in Rice. BMC Plant Biol. 2021, 21, 298. [Google Scholar] [CrossRef]
- Luong, N.H.; Balkunde, S.G.; Shim, K.-C.; Adeva, C.; Lee, H.-S.; Kim, H.-J.; Ahn, S.-N. Characterization of Domestication Loci Associated with Awn Development in Rice. Rice 2022, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Iehisa, J.C.M.; Ohno, R.; Kimura, T.; Enoki, H.; Nishimura, S.; Nasuda, S.; Takumi, S. Three Dominant Awnless Genes in Common Wheat: Fine Mapping, Interaction and Contribution to Diversity in Awn Shape and Length. PLoS ONE 2017, 12, e0176148. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Xing, P.; Bao, Y.; Wang, H.; Li, X. Advances in genetic studies of the awn in cereal crops. J. Bot. 2020, 55, 613–622. [Google Scholar]
- Shen, W.; Tan, W.; Han, Y.; He, J.; Xu, Y.; Huang, H.; Zhou, S.; Qin, B.; Luo, J.; Li, R. Genetic analysis and gene mapping of AWN-3 gene in rice. Genom. Appl. Biol. 2023, 42, 1338–1343. [Google Scholar] [CrossRef]
- Bessho-Uehara, K.; Masuda, K.; Wang, D.R.; Angeles-Shim, R.B.; Obara, K.; Nagai, K.; Murase, R.; Aoki, S.-I.; Furuta, T.; Miura, K.; et al. Regulator of Awn Elongation 3, an E3 Ubiquitin Ligase, Is Responsible for Loss of Awns during African Rice Domestication. Proc. Natl. Acad. Sci. USA 2023, 120, e2207105120. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Liu, H.; Zhou, T.; Gu, B.; Huang, X.; Shangguan, Y.; Zhu, J.; Li, Y.; Zhao, Y.; Wang, Y.; et al. An-1 Encodes a Basic Helix-Loop-Helix Protein That Regulates Awn Development, Grain Size, and Grain Number in Rice. Plant Cell 2013, 25, 3360–3376. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Wang, D.R.; Tan, L.; Fu, Y.; Liu, F.; Xiao, L.; Zhu, Z.; Fu, Q.; Sun, X.; Gu, P.; et al. LABA1, a Domestication Gene Associated with Long, Barbed Awns in Wild Rice. Plant Cell 2015, 27, 1875–1888. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Zhou, T.; Luo, J.; Liu, H.; Wang, Y.; Shangguan, Y.; Zhu, J.; Li, Y.; Sang, T.; Wang, Z.; et al. An-2 Encodes a Cytokinin Synthesis Enzyme That Regulates Awn Length and Grain Production in Rice. Mol. Plant 2015, 8, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Hua, L.; Zhu, Z.; Tan, L.; Zhao, X.; Zhang, W.; Liu, F.; Fu, Y.; Cai, H.; Sun, X.; et al. GAD1 Encodes a Secreted Peptide That Regulates Grain Number, Grain Length, and Awn Development in Rice Domestication. Plant Cell 2016, 28, 2453–2463. [Google Scholar] [CrossRef] [PubMed]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin Oxidase Regulates Rice Grain Production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Sharma, A.; Prakash, S.; Chattopadhyay, D. Killing Two Birds with a Single Stone—Genetic Manipulation of Cytokinin Oxidase/Dehydrogenase (CKX) Genes for Enhancing Crop Productivity and Amelioration of Drought Stress Response. Front. Genet. 2022, 13, 941595. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, S.; Liang, Y.; Zhang, R.; Liu, L.; Qin, P.; Zhang, Z.; Wang, Y.; Zhou, J.; Tang, X.; et al. Loss-Function Mutants of OsCKX Gene Family Based on CRISPR-Cas Systems Revealed Their Diversified Roles in Rice. Plant Genome 2023, 16, e20283. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, Y.P.J.; Kuwata, R.; Nishimura, A.; Phan, P.D.T.; Ishikawa, R.; Ishii, T. Evaluation of Domestication Loci Associated with Awnlessness in Cultivated Rice, Oryza Sativa. Rice 2020, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Paiano, A.; Margiotta, A.; De Luca, M.; Bucci, C. Yeast Two-Hybrid Assay to Identify Interacting Proteins. Curr. Protoc. Protein Sci. 2019, 95, e70. [Google Scholar] [CrossRef]
- Brachmann, R.K.; Boeke, J.D. Tag Games in Yeast: The Two-Hybrid System and Beyond. Curr. Opin. Biotechnol. 1997, 8, 561–568. [Google Scholar] [CrossRef]
- Smirnova, E.V.; Rakitina, T.V.; Saratov, G.A.; Kudriaeva, A.A.; Belogurov, A.A. Deconvolution of the MBP-Bri2 Interaction by a Yeast Two Hybrid System and Synergy of the AlphaFold2 and High Ambiguity Driven Protein-Protein Docking. Crystals 2022, 12, 197. [Google Scholar] [CrossRef]
- Amarasinghe, Y.P.J.; Otsuka, M.; Lim, S.; Ishikawa, R.; Ishii, T. The Role of Wild Rice (Oryza Rufipogon) Awns in Seed Dispersal. Seed Sci. Res. 2020, 30, 319–324. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, C.; Li, G.; Shi, J.; Yuan, Y.; Ye, L.; Song, Q.; Shi, J.; Zhang, D. MADS1-Regulated Lemma and Awn Development Benefits Barley Yield. Nat. Commun. 2024, 15, 301. [Google Scholar] [CrossRef]
- Ariharasutharsan, G.; Karthikeyan, A.; Geetha, S.; Saraswathi, R.; Raveendran, M.; Krishna-Surendar, K.; Ananda-Lekshmi, L.-D.; Kailappan, A.; Suresh, R.; Devasena, N. Refining the Major-Effect QTL and Candidate Genes Associated with Grain Number per Panicle by QTL-Seq in Rice (Oryza sativa L.). Euphytica 2024, 220, 154. [Google Scholar] [CrossRef]
- Yang, L.; Wang, J.; Han, Z.; Lei, L.; Liu, H.L.; Zheng, H.; Xin, W.; Zou, D. Combining QTL-Seq and Linkage Mapping to Fine Map a Candidate Gene in qCTS6 for Cold Tolerance at the Seedling Stage in Rice. BMC Plant Biol. 2021, 21, 278. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Thomas, J.; Gill, N.; Dwiningsih, Y.; Ruiz, C.; Famoso, A.; Pereira, A. Molecular Mapping and Characterization of QTLs for Grain Quality Traits in a RIL Population of US Rice under High Nighttime Temperature Stress. Sci. Rep. 2023, 13, 4880. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, J.; Zhang, Y.; Li, J.; Liu, M.; Li, L.; Li, S.; Wang, T.; Shaw, R.K.; Jiang, F.; et al. Hotspot Regions of Quantitative Trait Loci and Candidate Genes for Ear-Related Traits in Maize: A Literature Review. Genes 2024, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Zhong, F.; Liu, L. Enhancing Recognition and Interpretation of Functional Phenotypic Sequences through Fine-Tuning Pre-Trained Genomic Models. J. Transl. Med. 2024, 22, 756. [Google Scholar] [CrossRef]
- Wu, G.; Deng, H.; Yu, M.; Cai, Y.; Zhou, D.; Tan, J.; Yu, J.; Luo, X.; Tong, S.; Wang, P.; et al. Genetic Analysis of Rice Seed Recovery under Low-Temperature Conditions Using a New CSSL Population with a High-Density Genetic Map in Rice. Mol. Breeding 2020, 40, 109. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-Seq: Rapid Mapping of Quantitative Trait Loci in Rice by Whole Genome Resequencing of DNA from Two Bulked Populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.G.; Russ, C.; Costello, M.; Hollinger, A.; Lennon, N.J.; Hegarty, R.; Nusbaum, C.; Jaffe, D.B. Characterizing and Measuring Bias in Sequence Data. Genome Biol. 2013, 14, R51. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. BioMed Res. Int. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Su, Y.-C.; Yost, H.J. MMAPPR: Mutation Mapping Analysis Pipeline for Pooled RNA-Seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap+: Genetic Mapping and Mutant Identification without Crossing in Rice. PLoS ONE 2013, 8, e68529. [Google Scholar] [CrossRef]
- Vollert, C.S.; Uetz, P. Two-Hybrid System. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin, Heidelberg, 2006; pp. 1945–1949. ISBN 978-3-540-29623-2. [Google Scholar]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic Variation in 3,010 Diverse Accessions of Asian Cultivated Rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, W.; Li, X.; Zhang, C.; He, H.; Yuan, Q.; Zhang, B.; Zhang, H.; Leng, Y.; Wei, H.; et al. A Rice Variation Map Derived from 10 548 Rice Accessions Reveals the Importance of Rare Variants. Nucleic Acids Res. 2023, 51, 10924–10933. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTL | Chr. | Start | End | Interval (bp) | Δ(SNP Index) | SNP Index | SNP Index |
|---|---|---|---|---|---|---|---|
| qAwn4 | 4 | 23,547,272 | 26,376,809 | 2,829,537 | 0.85 | 0.95 | 0.10 |
| Gene | Marker | Chr. | Pos | 13 | 28 | 29 | 42 | 48 | 65 | 94 | 122 | NIP | 9311 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| An-1 | M279 | 4 | 16,710,416 | 0 | 2 | 2 | 0 | 0 | 0 | 2 | 2 | 2 | 0 |
| M280 | 4 | 17,414,886 | 0 | 2 | 2 | 0 | 0 | 0 | 2 | 2 | 2 | 0 | |
| An-2 | M297 | 4 | 25,751,684 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| M298 | 4 | 26,268,109 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| Sample | No. of Clean Reads (Million) | Clean Base (Gb) | Depth | Properly Paired Reads (million) | Properly Paired Ratio |
|---|---|---|---|---|---|
| Highest bulk | 97.25 | 14.59 | 38.957× | 89.04 | 91.56% |
| Lowest bulk | 96.08 | 14.52 | 38.782× | 89.51 | 92.45% |
| CSSL29 | 95.80 | 14.37 | 38.375× | 87.82 | 91.67% |
| 9311 | 95.72 | 14.36 | 38.340× | 87.88 | 91.81% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Yang, J.; Huang, T.; Chen, Z.; Nyasulu, M.; Zhong, Q.; He, H.; Bian, J. Genetic Analysis of the Awn Length Gene in the Rice Chromosome Segment Substitution Line CSSL29. Int. J. Mol. Sci. 2025, 26, 1436. https://doi.org/10.3390/ijms26041436
Wang Z, Yang J, Huang T, Chen Z, Nyasulu M, Zhong Q, He H, Bian J. Genetic Analysis of the Awn Length Gene in the Rice Chromosome Segment Substitution Line CSSL29. International Journal of Molecular Sciences. 2025; 26(4):1436. https://doi.org/10.3390/ijms26041436
Chicago/Turabian StyleWang, Zhengjie, Jun Yang, Tao Huang, Zhihao Chen, Mvuyeni Nyasulu, Qi Zhong, Haohua He, and Jianmin Bian. 2025. "Genetic Analysis of the Awn Length Gene in the Rice Chromosome Segment Substitution Line CSSL29" International Journal of Molecular Sciences 26, no. 4: 1436. https://doi.org/10.3390/ijms26041436
APA StyleWang, Z., Yang, J., Huang, T., Chen, Z., Nyasulu, M., Zhong, Q., He, H., & Bian, J. (2025). Genetic Analysis of the Awn Length Gene in the Rice Chromosome Segment Substitution Line CSSL29. International Journal of Molecular Sciences, 26(4), 1436. https://doi.org/10.3390/ijms26041436