
5-Substituted 4-Thiouridines, 4-Thio-2′-deoxyuridines and Their Oligoglycol Carbonate Prodrugs as Promising Antimicrobial Agents

 , , , , , , , ,
, , , , , , , ,  , , and
, , and
Abstract
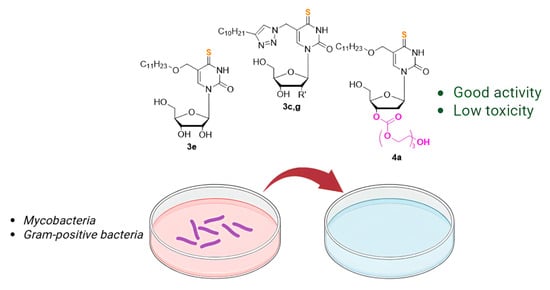
1. Introduction
2. Results
2.1. Chemistry
2.2. Water Solubility
2.3. Cytotoxicity
2.4. In Vitro Study of Antibacterial Activity of the Obtained Compounds
2.5. In Vitro Study of Antimycobacterial Activity of the Obtained Compounds
3. Discussion
4. Materials and Methods
4.1. Chemistry
Study of Water Solubility of Synthesized Compounds
4.2. General Method for the Synthesis of 3′-O-Acetyl-5′-O-Monomethoxytrityl-5-Alkyloxymethyl- and 5-[4-Alkyl-(1,2,3-Triazol-1-yl)Methyl]-2′-Deoxyuridines 5a–d
4.3. General Method for the Synthesis of 5′-O-Monomethoxytrityl-5-Alkyloxymethyl- and 5-[4-Alkyl-(1,2,3-Triazol-1-yl)Methyl]-4-Thio-2′-Deoxyuridines 6a–d
4.4. General Method for the Synthesis of 5-Alkyloxymethyl-and 5-[4-Alkyl-(1,2,3-Triazol-1-yl)Methyl]-4-Thio-2′-Deoxyuridines 3a–d
4.5. General Method for the Synthesis of 3′-O-(8-Hydroxy-3,6-Dioxaoct-1-Yloxy)Carbonyl-5-Alkyloxymethyl- and 5-[4-Alkyl-(1,2,3-Triazol-1-yl)Methyl]-4-Thio-2′-Deoxyuridines 4a–d
4.6. General Method for the Synthesis of 5-Alkyloxymethyl-and 5-[4-Alkyl-(1,2,3-Triazol-1-yl)Methyl]-4-Thiouridines 3e–h
4.7. Bacterial Strains and In Vitro Study of the Antibacterial Effect
4.8. Mycobacterial Strains and Susceptibility Testing of Mycobacteria
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aminov, R.I. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Henderson, I.R.; Capon, R.J.; Blaskovich, M.A.T. Antibiotics in the Clinical Pipeline as of December 2022. J. Antibiot. 2023, 76, 431–473, Erratum in J. Antibiot. 2024, 77, 71. [Google Scholar] [CrossRef]
- Cook, M.A.; Wright, G.D. The Past, Present, and Future of Antibiotics. Sci. Transl. Med. 2022, 14, eabo7793. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Sengupta, S.; Chattopadhyay, M.K.; Grossart, H.-P. The Multifaceted Roles of Antibiotics and Antibiotic Resistance in Nature. Front. Microbiol. 2013, 4, 47. [Google Scholar] [CrossRef]
- World Health Organization. Global Antibiotic Resistance Surveillance Report 2025; World Health Organization: Geneva, Switzerland, 2025; ISBN 978-92-4-011633-7. [Google Scholar]
- Ralhan, K.; Iyer, K.A.; Diaz, L.L.; Bird, R.; Maind, A.; Zhou, Q.A. Navigating Antibacterial Frontiers: A Panoramic Exploration of Antibacterial Landscapes, Resistance Mechanisms, and Emerging Therapeutic Strategies. ACS Infect. Dis. 2024, 10, 1483–1519. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef]
- Weiner-Lastinger, L.M.; Abner, S.; Edwards, J.R.; Kallen, A.J.; Karlsson, M.; Magill, S.S.; Pollock, D.; See, I.; Soe, M.M.; Walters, M.S.; et al. Antimicrobial-Resistant Pathogens Associated with Adult Healthcare-Associated Infections: Summary of Data Reported to the National Healthcare Safety Network, 2015–2017. Infect. Control Hosp. Epidemiol. 2020, 41, 1–18. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report. 2022. Available online: www.who.int/health-topics/tuberculosis (accessed on 27 October 2025).
- Bahuguna, A.; Rawat, D.S. An Overview of New Antitubercular Drugs, Drug Candidates, and Their Targets. Med. Res. Rev. 2020, 40, 263–292. [Google Scholar] [CrossRef]
- Brode, S.K.; Daley, C.L.; Marras, T.K. The Epidemiologic Relationship between Tuberculosis and Non-Tuberculous Mycobacterial Disease: A Systematic Review. Int. J. Tuberc. Lung Dis. 2014, 18, 1370–1377. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023; ISBN 9789240083851. [Google Scholar]
- Dahl, V.N.; Mølhave, M.; Fløe, A.; van Ingen, J.; Schön, T.; Lillebaek, T.; Andersen, A.B.; Wejse, C. Global Trends of Pulmonary Infections with Nontuberculous Mycobacteria: A Systematic Review. Int. J. Infect. Dis. 2022, 125, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.M.; Furuya-Kanamori, L.; Coffey, C.; Bell, S.C.; Knibbs, L.D.; Lau, C.L. Influence of Climate Variables on the Rising Incidence of Nontuberculous Mycobacterial (NTM) Infections in Queensland, Australia 2001–2016. Sci. Total Environ. 2020, 740, 139796. [Google Scholar] [CrossRef]
- Behra, P.R.K.; Pettersson, B.M.F.; Ramesh, M.; Das, S.; Dasgupta, S.; Kirsebom, L.A. Comparative Genome Analysis of Mycobacteria Focusing on TRNA and Non-Coding RNA. BMC Genom. 2022, 23, 704. [Google Scholar] [CrossRef]
- Kumar, K.; Ponnuswamy, A.; Capstick, T.G.; Chen, C.; McCabe, D.; Hurst, R.; Morrison, L.; Moore, F.; Gallardo, M.; Keane, J.; et al. Non-Tuberculous Mycobacterial Pulmonary Disease (NTM-PD): Epidemiology, Diagnosis and Multidisciplinary Management. Clin. Med. 2024, 24, 100017. [Google Scholar] [CrossRef]
- Brode, S.K.; Jamieson, F.B.; Ng, R.; Campitelli, M.A.; Kwong, J.C.; Paterson, J.M.; Li, P.; Marchand-Austin, A.; Bombardier, C.; Marras, T.K. Increased Risk of Mycobacterial Infections Associated with Anti-Rheumatic Medications. Thorax 2015, 70, 677–682. [Google Scholar] [CrossRef]
- Wu, U.-I.; Holland, S.M. Host Susceptibility to Non-Tuberculous Mycobacterial Infections. Lancet Infect. Dis. 2015, 15, 968–980. [Google Scholar] [CrossRef]
- Wagner, D.; van Ingen, J.; van der Laan, R.; Obradovic, M.; NTM-NET. Non-Tuberculous Mycobacterial Lung Disease in Patients with Bronchiectasis: Perceived Risk, Severity and Guideline Adherence in a European Physician Survey. BMJ open Respir. Res. 2020, 7, e000498, Correction in BMJ Open Respir. Res. 2020, 7, e000498corr1. [Google Scholar] [CrossRef]
- Prieto, M.D.; Alam, M.E.; Franciosi, A.N.; Quon, B.S. Global Burden of Nontuberculous Mycobacteria in the Cystic Fibrosis Population: A Systematic Review and Meta-Analysis. ERJ open Res. 2023, 9, 00336-2022. [Google Scholar] [CrossRef]
- Nie, C.X.; Zhang, J.B.; Ji, H.L.; Li, X.R.; Luo, J.F.; Fu, A.S.; Ge, Y.L.; Liu, T.J.; Chen, T. Multiple Postoperative Lung Infections after Thymoma Surgery Diagnosed as Nontuberculous Mycobacterial Infection. Clin. Lab. 2024, 70, 1579. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, X.-Y.; Liu, J.-W. Demographic and Clinical Features of Nontuberculous Mycobacteria Infection Resulting from Cosmetic Procedures: A Systematic Review. Int. J. Infect. Dis. 2024, 149, 107259. [Google Scholar] [CrossRef]
- Zimenkov, D.; Ushtanit, A. Comparative Analysis of Evolutionary Distances Using the Genus Mycobacterium. Int. J. Mol. Sci. 2025, 26, 10471. [Google Scholar] [CrossRef]
- Hoffman, P.S. Antibacterial Discovery: 21st Century Challenges. Antibiotics 2020, 9, 213. [Google Scholar] [CrossRef]
- Belay, W.Y.; Getachew, M.; Tegegne, B.A.; Teffera, Z.H.; Dagne, A.; Zeleke, T.K.; Abebe, R.B.; Gedif, A.A.; Fenta, A.; Yirdaw, G.; et al. Mechanism of Antibacterial Resistance, Strategies and next-Generation Antimicrobials to Contain Antimicrobial Resistance: A Review. Front. Pharmacol. 2024, 15, 1444781. [Google Scholar] [CrossRef]
- De Clercq, E. Human Viral Diseases: What Is next for Antiviral Drug Discovery? Curr. Opin. Virol. 2012, 2, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Khandazhinskaya, A.L.; Matyugina, E.S.; Makarov, D.A.; Kochetkov, S.N. Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors. Microorganisms 2022, 10, 1299. [Google Scholar] [CrossRef]
- Yssel, A.E.J.; Vanderleyden, J.; Steenackers, H.P. Repurposing of Nucleoside- and Nucleobase-Derivative Drugs as Antibiotics and Biofilm Inhibitors. J. Antimicrob. Chemother. 2017, 72, 2156–2170. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the Development of Nucleoside and Nucleotide Analogues for Cancer and Viral Diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Osada, H. Discovery and Applications of Nucleoside Antibiotics beyond Polyoxin. J. Antibiot. 2019, 72, 855–864. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Kerr, R.V. Mechanism of Action of Nucleoside Antibacterial Natural Product Antibiotics. J. Antibiot. 2019, 72, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Serpi, M.; Ferrari, V.; Pertusati, F. Nucleoside Derived Antibiotics to Fight Microbial Drug Resistance: New Utilities for an Established Class of Drugs? J. Med. Chem. 2016, 59, 10343–10382. [Google Scholar] [CrossRef]
- Ferrari, V.; Serpi, M. Nucleoside Analogs and Tuberculosis: New Weapons against an Old Enemy. Future Med. Chem. 2015, 7, 291–314. [Google Scholar] [CrossRef]
- Platonova, Y.B.; Volov, A.N.; Tomilova, L.G. The Synthesis and Antituberculosis Activity of 5-Alkynyl Uracil Derivatives. Bioorg. Med. Chem. Lett. 2020, 30, 127351. [Google Scholar] [CrossRef]
- Platonova, Y.B.; Kirillova, V.A.; Volov, A.N.; Savilov, S.V. Synthesis and Antitubercular Activity of New 5-Alkynyl Derivatives of 2-Thiouridine. Russ. J. Org. Chem. 2023, 59, 2083–2091. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Bagiński, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A Database of RNA Modification Pathways. 2017 Update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Wnuk, S.F. Sulfur- and Seleno-Sugar Modified Nucleosides. Synthesis, Chemical Transformations and Biological Properties. Tetrahedron 1993, 49, 9877–9936. [Google Scholar] [CrossRef]
- Scott, K.A.; Njardarson, J.T. Analysis of US FDA-Approved Drugs Containing Sulfur Atoms; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–34. [Google Scholar]
- Mustafa, M.; Winum, J.-Y. The Importance of Sulfur-Containing Motifs in Drug Design and Discovery. Expert Opin. Drug Discov. 2022, 17, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of Sulphur-Heterocycles in Medicinal Chemistry: An Update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef]
- Huang, G.; Cierpicki, T.; Grembecka, J. Thioamides in Medicinal Chemistry and as Small Molecule Therapeutic Agents. Eur. J. Med. Chem. 2024, 277, 116732. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Debnath, B.; Panda, S.; Manna, T.; Maity, A.; Dayaramani, R.; Nath, R.; Khan, S.A.; Akhtar, M.J. Anticancer Potential of the S -Heterocyclic Ring Containing Drugs and Its Bioactivation to Reactive Metabolites. Chem. Biodivers. 2024, 21, e202400473. [Google Scholar] [CrossRef] [PubMed]
- Brancale, A.; McGuigan, C.; Algain, B.; Savy, P.; Benhida, R.; Fourrey, J.-L.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. Bicyclic Anti-VZV Nucleosides: Thieno Analogues Retain Full Antiviral Activity. Bioorg. Med. Chem. Lett. 2001, 11, 2507–2510. [Google Scholar] [CrossRef]
- Shakya, N.; Srivastav, N.C.; Bhavanam, S.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Discovery of Novel 5-(Ethyl or Hydroxymethyl) Analogs of 2′-‘up’ Fluoro (or Hydroxyl) Pyrimidine Nucleosides as a New Class of Mycobacterium Tuberculosis, Mycobacterium Bovis and Mycobacterium Avium Inhibitors. Bioorg. Med. Chem. 2012, 20, 4088–4097. [Google Scholar] [CrossRef]
- Shmalenyuk, E.R.; Chernousova, L.N.; Karpenko, I.L.; Kochetkov, S.N.; Smirnova, T.G.; Andreevskaya, S.N.; Chizhov, A.O.; Efremenkova, O.V.; Alexandrova, L.A. Inhibition of Mycobacterium Tuberculosis Strains H37Rv and MDR MS-115 by a New Set of C5 Modified Pyrimidine Nucleosides. Bioorg. Med. Chem. 2013, 21, 4874–4884. [Google Scholar] [CrossRef]
- Makarov, D.A.; Negrya, S.D.; Jasko, M.V.; Karpenko, I.L.; Solyev, P.N.; Chekhov, V.O.; Kaluzhny, D.N.; Efremenkova, O.V.; Vasilyeva, B.F.; Chizhov, A.O.; et al. 5-Substituted Uridines with Activity against Gram-Positive Bacteria. ChemMedChem 2023, 18, e202300366. [Google Scholar] [CrossRef] [PubMed]
- Ostroumova, O.S.; Efimova, S.S.; Zlodeeva, P.D.; Alexandrova, L.A.; Makarov, D.A.; Matyugina, E.S.; Sokhraneva, V.A.; Khandazhinskaya, A.L.; Kochetkov, S.N. Derivatives of Pyrimidine Nucleosides Affect Artificial Membranes Enriched with Mycobacterial Lipids. Pharmaceutics 2024, 16, 1110. [Google Scholar] [CrossRef] [PubMed]
- Khandazhinskaya, A.L.; Matyugina, E.S.; Alexandrova, L.A.; Kezin, V.A.; Chernousova, L.N.; Smirnova, T.G.; Andreevskaya, S.N.; Popenko, V.I.; Leonova, O.G.; Kochetkov, S.N. Interaction of 5-Substituted Pyrimidine Nucleoside Analogues and M.tuberculosis: A View through an Electron Microscope. Biochimie 2020, 171–172, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, L.A.; Chekhov, V.O.; Shmalenyuk, E.R.; Kochetkov, S.N.; El-Asrar, R.A.; Herdewijn, P. Synthesis and Evaluation of C-5 Modified 2′-Deoxyuridine Monophosphates as Inhibitors of M. tuberculosis Thymidylate Synthase. Bioorg. Med. Chem. 2015, 23, 7131–7137. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Solyev, P.N.; Karpenko, I.L.; Efremenkova, O.V.; Vasilyeva, B.F.; Sumarukova, I.G.; Kochetkov, S.N.; Alexandrova, L.A. Synthesis of Water-Soluble Prodrugs of 5-Modified 2′-Deoxyuridines and Their Antibacterial Activity. J. Antibiot. 2020, 73, 236–246. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Makarov, D.A.; Solyev, P.N.; Karpenko, I.L.; Shevchenko, O.V.; Chekhov, O.V.; Glukhova, A.A.; Vasilyeva, B.F.; Efimenko, T.A.; et al. Glycol and Phosphate Depot Forms of 4- and/or 5-Modified Nucleosides Exhibiting Antibacterial Activity. Mol. Biol. 2021, 55, 143–153. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Makarov, D.A.; Karpenko, I.L.; Solyev, P.N.; Chekhov, V.O.; Efremenkova, O.V.; Vasilieva, B.F.; Efimenko, T.A.; Kochetkov, S.N.; et al. Oligoglycol Carbonate Prodrugs of 5-Modified 2′-Deoxyuridines: Synthesis and Antibacterial Activity. Mendeleev Communictions 2022, 32, 433–435. [Google Scholar] [CrossRef]
- Khatoon, H.; Abdulmalek, E. A Focused Review of Synthetic Applications of Lawesson’s Reagent in Organic Synthesis. Molecules 2021, 26, 6937. [Google Scholar] [CrossRef] [PubMed]
- Divakar, K.J.; Reese, C.B. 4-(1,2,4-Triazol-1-yl)- and 4-(3-Nitro-1,2,4-Triazol-1-yl)-1-(β-D-2,3,5-Tri-O-Acetylarabinofuranosyl)Pyrimidin-2(1H)-Ones. Valuable Intermediates in the Synthesis of Derivatives of 1-(β-D-Arabinofuranosyl)Cytosine (Ara-C). J. Chem. Soc. Perkin Trans. 1 1982, 1, 1171–1176. [Google Scholar] [CrossRef]
- Lin, T.S.; Gao, Y.S.; Mancini, W.R. Synthesis and Biological Activity of Various 3′-Azido and 3′-Amino Analogs of 5-Substituted Pyrimidine Deoxyribonucleosides. J. Med. Chem. 1983, 26, 1691–1696. [Google Scholar] [CrossRef]
- Žemlička, J.; Šorm, F. Nucleic Acids Components and Their Analogues. LXI. The Reaction of Dimethylchloromethyleneammonium Chloride with the 2′,3′,5′-Tri-O-Acyl Derivatives of Uridine and 6-Azauridine; A New Synthesis of 6-Azacytidine. Collect. Czechoslov. Chem. Commun. 1965, 30, 2052–2067. [Google Scholar] [CrossRef]
- Matsuda, A.; Itoh, H.; Takenuki, K.; Sasaki, T.; Ueda, T. Alkyl Addition Reaction of Pyrimidine 2′-Ketonucleosides: Synthesis of 2′-Branched-Chain Sugar Pyrimidine Nucleosides. Nucleodides and Nucleotides. LXXXI. Chem. Pharm. Bull. 1988, 36, 945–953. [Google Scholar] [CrossRef]
- Nikš, M.; Otto, M. Towards an Optimized MTT Assay. J. Immunol. Methods 1990, 130, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Zimenkov, D. Variability of Mycobacterium Avium Complex Isolates Drug Susceptibility Testing by Broth Microdilution. Antibiotics 2022, 11, 1756. [Google Scholar] [CrossRef]
- Sampathkumar, P.; Turley, S.; Ulmer, J.E.; Rhie, H.G.; Sibley, C.H.; Hol, W.G.J. Structure of the Mycobacterium Tuberculosis Flavin Dependent Thymidylate Synthase (MtbThyX) at 2.0Å Resolution. J. Mol. Biol. 2005, 352, 1091–1104. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2012. [Google Scholar]
- Ozturk, T.; Ertas, E.; Mert, O. Use of Lawesson’s Reagent in Organic Syntheses. Chem. Rev. 2007, 107, 5210–5278. [Google Scholar] [CrossRef]
- Kaneko, K.; Katayama, H.; Wakabayashi, T.; Kumonaka, T. Pyrimidine Derivatives; 1. The Highly Regioselective 4-Thionation of Pyrimidine-2,4(1H,3H)-Dione Derivatives with Lawesson Reagent. Synthesis 1988, 1988, 152–154. [Google Scholar] [CrossRef]
- Jacobi, P.A.; Guo, J.; Zheng, W. An Unequivocal Synthesis of the Ring-A,B Dihydropyrromethenone of Phytochrome. Tetrahedron Lett. 1995, 36, 1197–1200. [Google Scholar] [CrossRef]
- El-Kateb, A.A.; Abd El-Rahman, N.M. Synthesis of New Heterocyclic Compounds Using Lawesson Reagent. Phosphorus. Sulfur. Silicon Relat. Elem. 2006, 181, 249–254. [Google Scholar] [CrossRef]
- Amblard, F.; Cho, J.H.; Schinazi, R.F. Cu (I)-catalyzed Huisgen azide− alkyne 1, 3-dipolar cycloaddition reaction in nucleoside, nucleotide, and oligonucleotide chemistry. Chem. Rev. 2009, 109, 4207. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Zimenkov, D.; Zhuravlev, V.; Ushtanit, A.; Filippova, M.; Semenova, U.; Solovieva, N.; Sviridenko, M.; Khakhalina, A.; Safonova, S.; Makarova, M.; et al. Biochip-Based Identification of Mycobacterial Species in Russia. Int. J. Mol. Sci. 2024, 25, 13200. [Google Scholar] [CrossRef]
- CLSI. Susceptibility Testing of Mycobacteria, Nocardia Spp., and Other Aerobic Actinomycetes, 3rd ed.; CLSI: Wayne, PA, USA, 2018; ISBN 9781684400263. [Google Scholar]
- Parish, T.; Kumar, A. (Eds.) Methods in Molecular Biology. In Mycobacteria Protocols; Springer: New York, NY, USA, 2021; Volume 2314, ISBN 978-1-0716-1459-4. [Google Scholar]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Lipophilic Substituent | Sugar Substituent | Water Solubility (mg/mL) |
|---|---|---|---|
| 3a | -CH2OC11H23 | - | <0.013 |
| 3b | -CH2OC12H25 | - | 0.013 |
| 3c | -CH2TriC10H21 | - | 0.014 |
| 3d | -CH2TriC12H25 | - | 0.005 |
| 3e | -CH2OC11H23 | - | 0.2 |
| 3f | -CH2OC12H25 | - | 0.013 |
| 3g | -CH2TriC10H21 | - | 0.01 |
| 3h | -CH2TriC12H25 | - | <0.01 |
| 4a | -CH2OC11H23 | 3′-O-C(O)-OTEG | 0.1 |
| 4b | -CH2OC12H25 | 3′-O-C(O)-OTEG | 0.028 |
| 4c | -CH2TriC10H21 | 3′-O-C(O)-OTEG | 0.6 |
| 4d | -CH2TriC12H25 | 3′-O-C(O)-OTEG | <0.1 |
| Compound | MIC [mg/L] | ||||
|---|---|---|---|---|---|
| S. aureus FDA 209P (MSSA) | S. aureus INA 00761 (MRSA) | Mic. luteus NCTC 8340 | M. smegmatis VKPM Ac-1339 | M. smegmatis mc2 155 | |
| 4a | 5 | 5 | 5 | 5 | 5 |
| 4b | 21 | 21 | 21 | 21 | 21 |
| 2a | n/a | n/a | n/a | 24 | n/a |
| 2b | 50 | 50 | 50 | 25 | 25 |
| 2c | 78 | n/a | n/a | n/a | n/a |
| 2d, 3e–3h, 4c, 4d | n/a | n/a | n/a | n/a | n/a |
| Amikacin | 2 | 30 | 0.5 | 30 | - |
| Ciprofloxacin | 4 | 4 | - | 4 | - |
| Isoniazid | - | - | - | 0.25 | 4 |
| Rifampicin | - | - | - | 8 | 4 |
| Oxacillin | 1 | 32 | - | - | - |
| Compound | MIC [mg/L] | |||
|---|---|---|---|---|
| M. abscessus | M. fortuitum | M. intracellulare | M. avium | |
| 3a | 80 | >80 | >80 | >80 |
| 3b | 40 | >80 | >80 | >80 |
| 3c | 40 | 20 | 10 | 20 |
| 3d | >80 | >80 | >80 | >80 |
| 3e | 20 | 20 | 10 | 20 |
| 3f | >80 | >80 | 10 | 40 |
| 3g | >80 | 80 | 40 | 40 |
| 3h | >80 | >80 | >80 | >80 |
| 4a | >80 | >80 | 10 | 40 |
| 4b | >80 | >80 | 20 | 40 |
| 4c | >80 | >80 | 20 | 20 |
| 4d | >80 | >80 | 40 | 80 |
| 1b | >80 | >80 | 80 | >80 |
| 1e | >80 | >80 | 80 | 80 |
| 1a,c,d,f–h | >80 | >80 | >80 | >80 |
| Clarithromycin | 1.25 | 20 | >80 | >80 |
| Ethambutol | 80 | 10 | 80 | 40 |
| Rifampicin | >80 | 20 | 1.25 | 1.25 |
| Amikacin | 40 | 1.25 | 2.5 | 2.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarov, D.A.; Jasko, M.V.; Negrya, S.D.; Karpenko, I.L.; Urbina, E.V.; Chekhov, V.O.; Efremenkova, O.V.; Vasilyeva, B.F.; Zimenkov, D.V.; Ushtanit, A.I.; et al. 5-Substituted 4-Thiouridines, 4-Thio-2′-deoxyuridines and Their Oligoglycol Carbonate Prodrugs as Promising Antimicrobial Agents. Int. J. Mol. Sci. 2025, 26, 11712. https://doi.org/10.3390/ijms262311712
Makarov DA, Jasko MV, Negrya SD, Karpenko IL, Urbina EV, Chekhov VO, Efremenkova OV, Vasilyeva BF, Zimenkov DV, Ushtanit AI, et al. 5-Substituted 4-Thiouridines, 4-Thio-2′-deoxyuridines and Their Oligoglycol Carbonate Prodrugs as Promising Antimicrobial Agents. International Journal of Molecular Sciences. 2025; 26(23):11712. https://doi.org/10.3390/ijms262311712
Chicago/Turabian StyleMakarov, Dmitry A., Maxim V. Jasko, Sergey D. Negrya, Inna L. Karpenko, Elizabeth V. Urbina, Vladimir O. Chekhov, Olga V. Efremenkova, Byazilya F. Vasilyeva, Danila V. Zimenkov, Anastasia I. Ushtanit, and et al. 2025. "5-Substituted 4-Thiouridines, 4-Thio-2′-deoxyuridines and Their Oligoglycol Carbonate Prodrugs as Promising Antimicrobial Agents" International Journal of Molecular Sciences 26, no. 23: 11712. https://doi.org/10.3390/ijms262311712
APA StyleMakarov, D. A., Jasko, M. V., Negrya, S. D., Karpenko, I. L., Urbina, E. V., Chekhov, V. O., Efremenkova, O. V., Vasilyeva, B. F., Zimenkov, D. V., Ushtanit, A. I., Kochetkov, S. N., & Alexandrova, L. A. (2025). 5-Substituted 4-Thiouridines, 4-Thio-2′-deoxyuridines and Their Oligoglycol Carbonate Prodrugs as Promising Antimicrobial Agents. International Journal of Molecular Sciences, 26(23), 11712. https://doi.org/10.3390/ijms262311712