
A Framework Integrating GWAS and Genomic Selection to Enhance Prediction Accuracy of Economical Traits in Common Carp
 , ,
, ,
Abstract
1. Introduction
2. Results
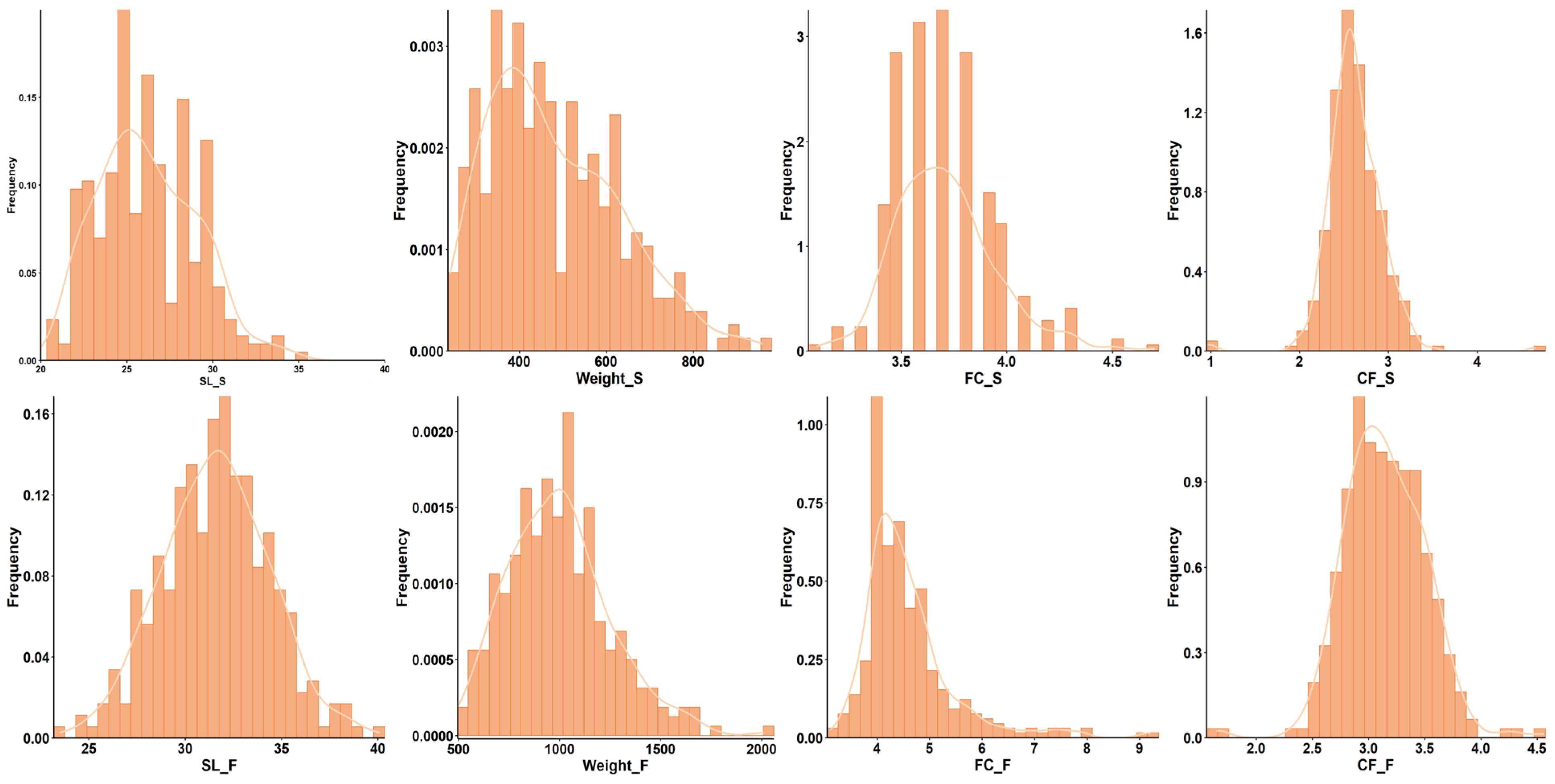
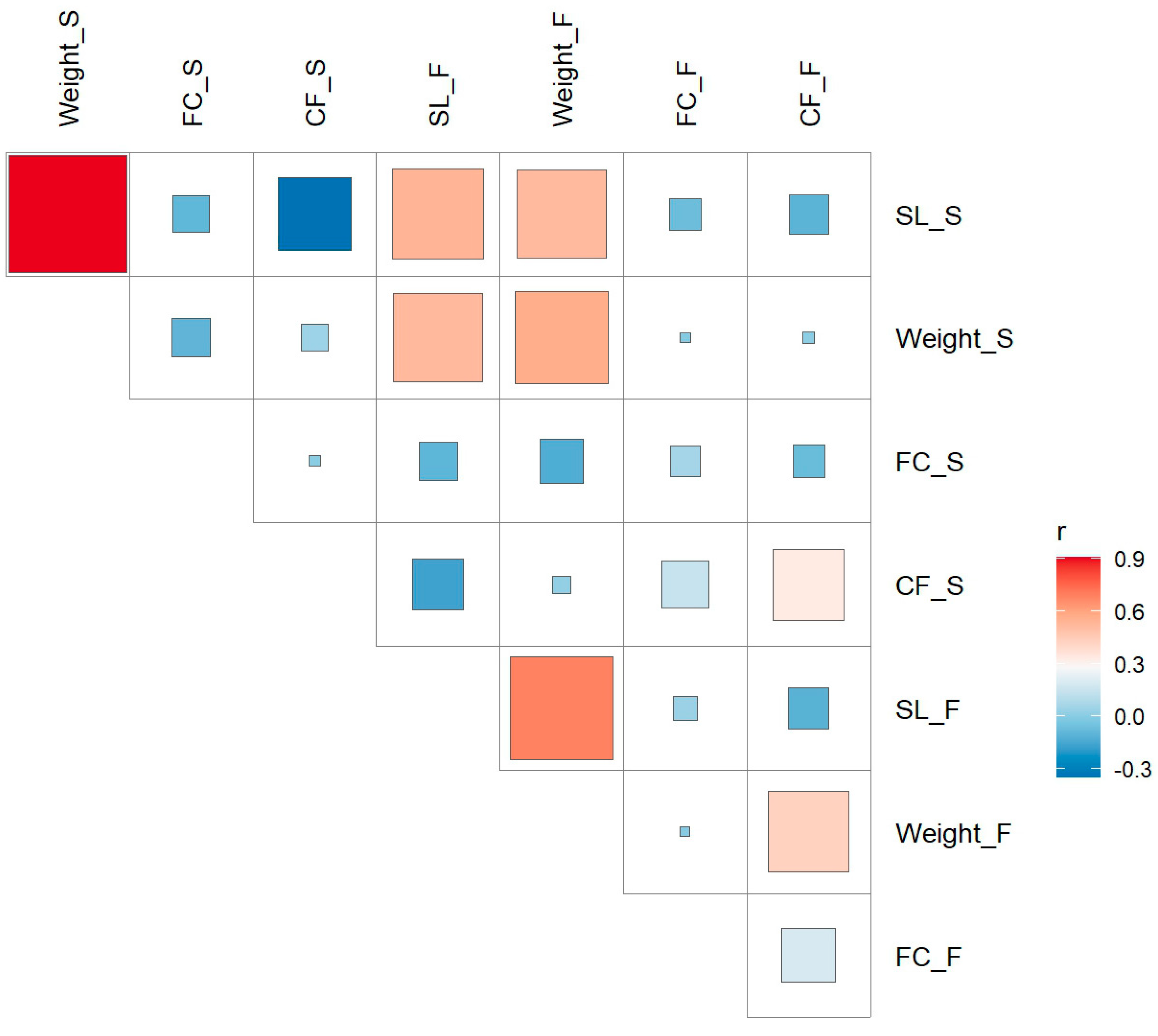
2.1. Phenotypic Analysis
2.2. Seasonal Heritability of Growth Traits via fastGWA-REML
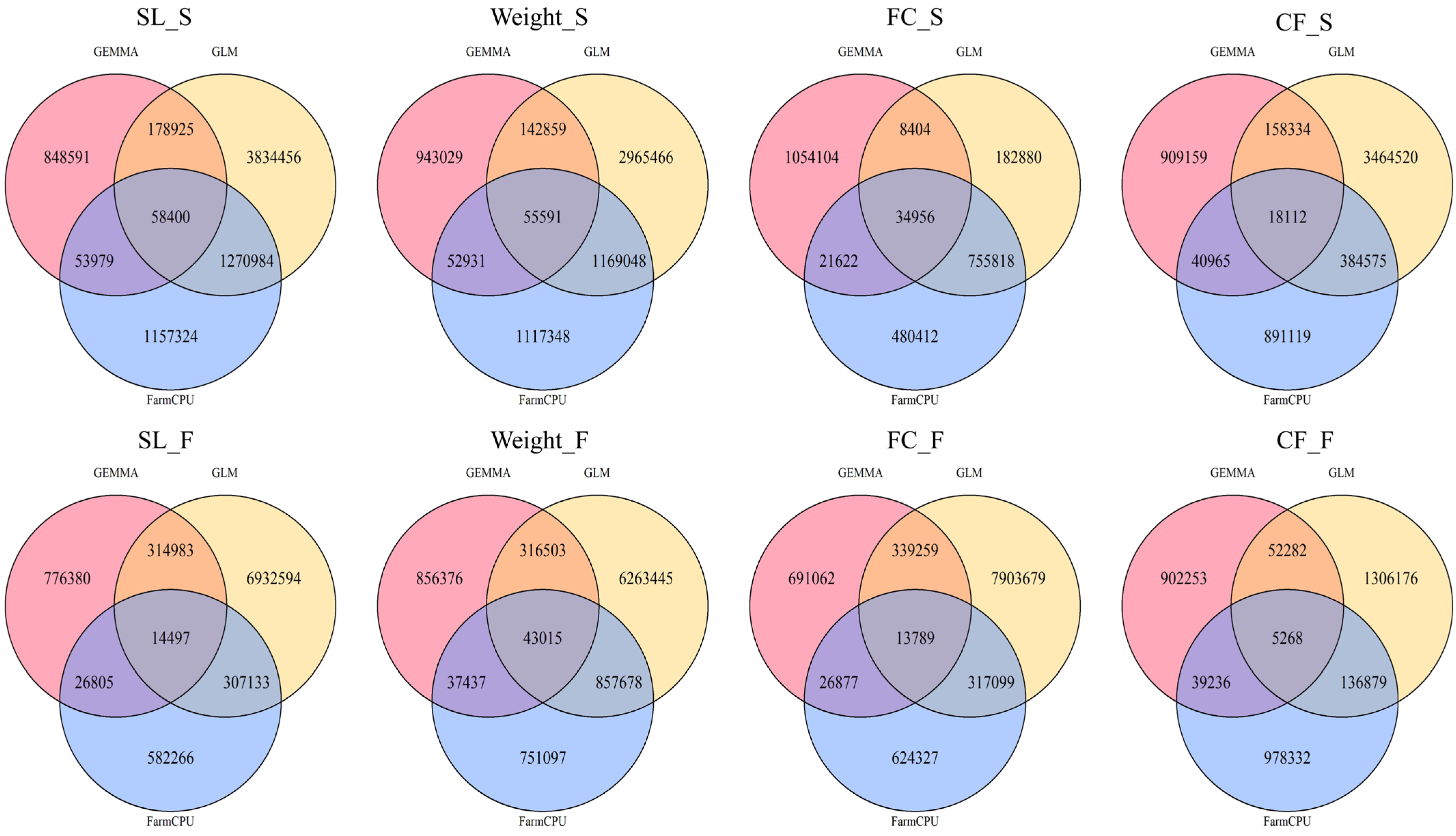
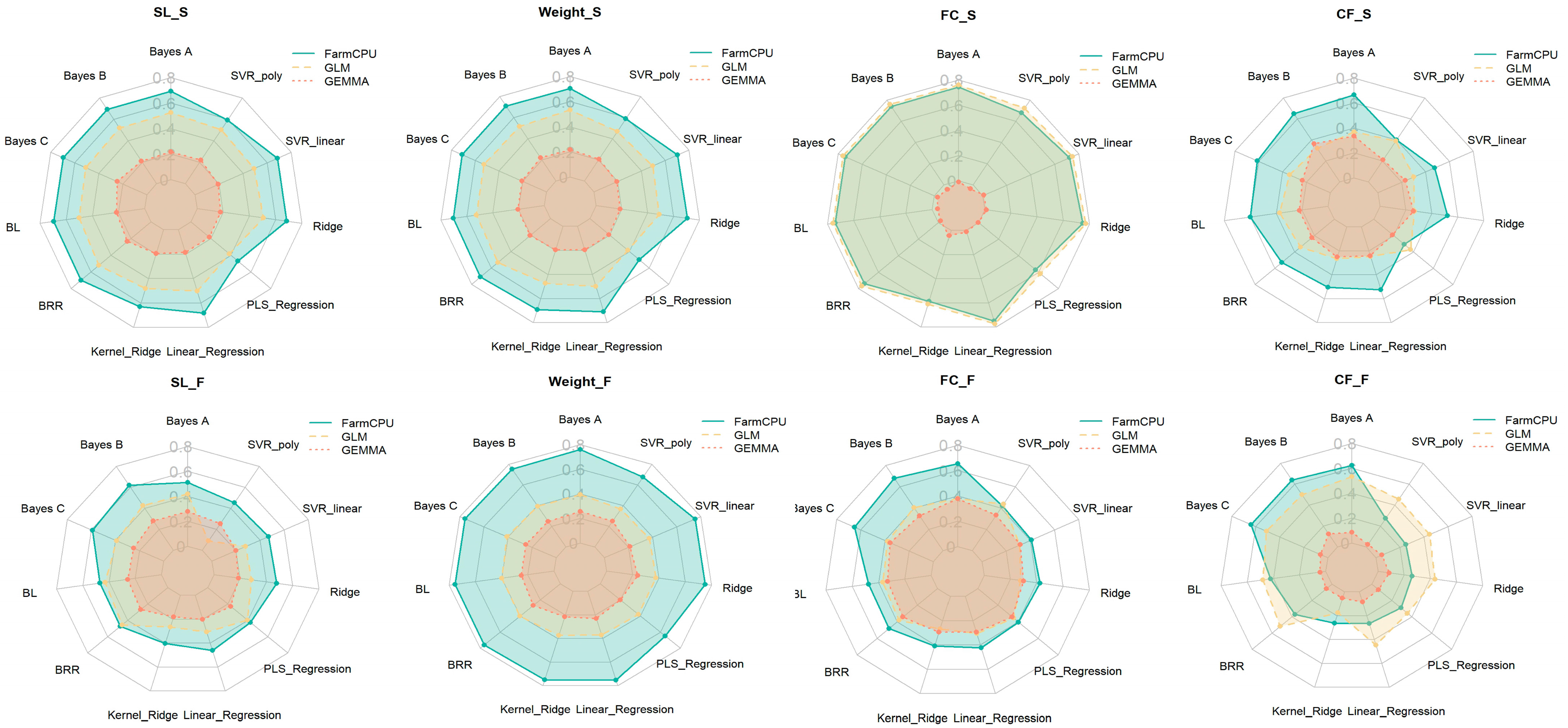
2.3. Comparison of Significant SNP Detection Results for Growth Traits Across Spring and Autumn Seasons Using Three GWAS Methods
2.4. Comparison of the Distribution of Significant SNP β Effect Sizes Across Different GWAS Methods
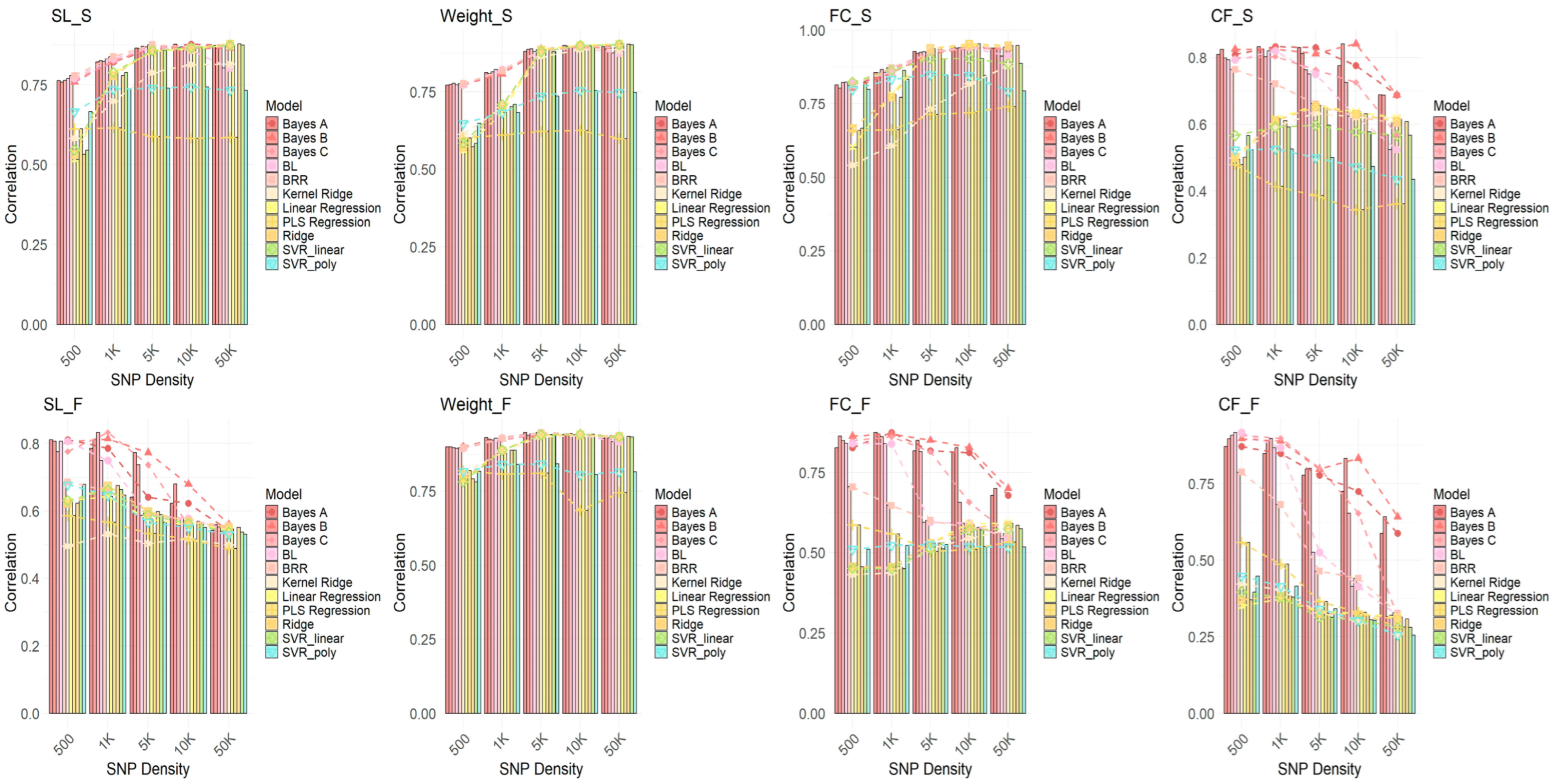
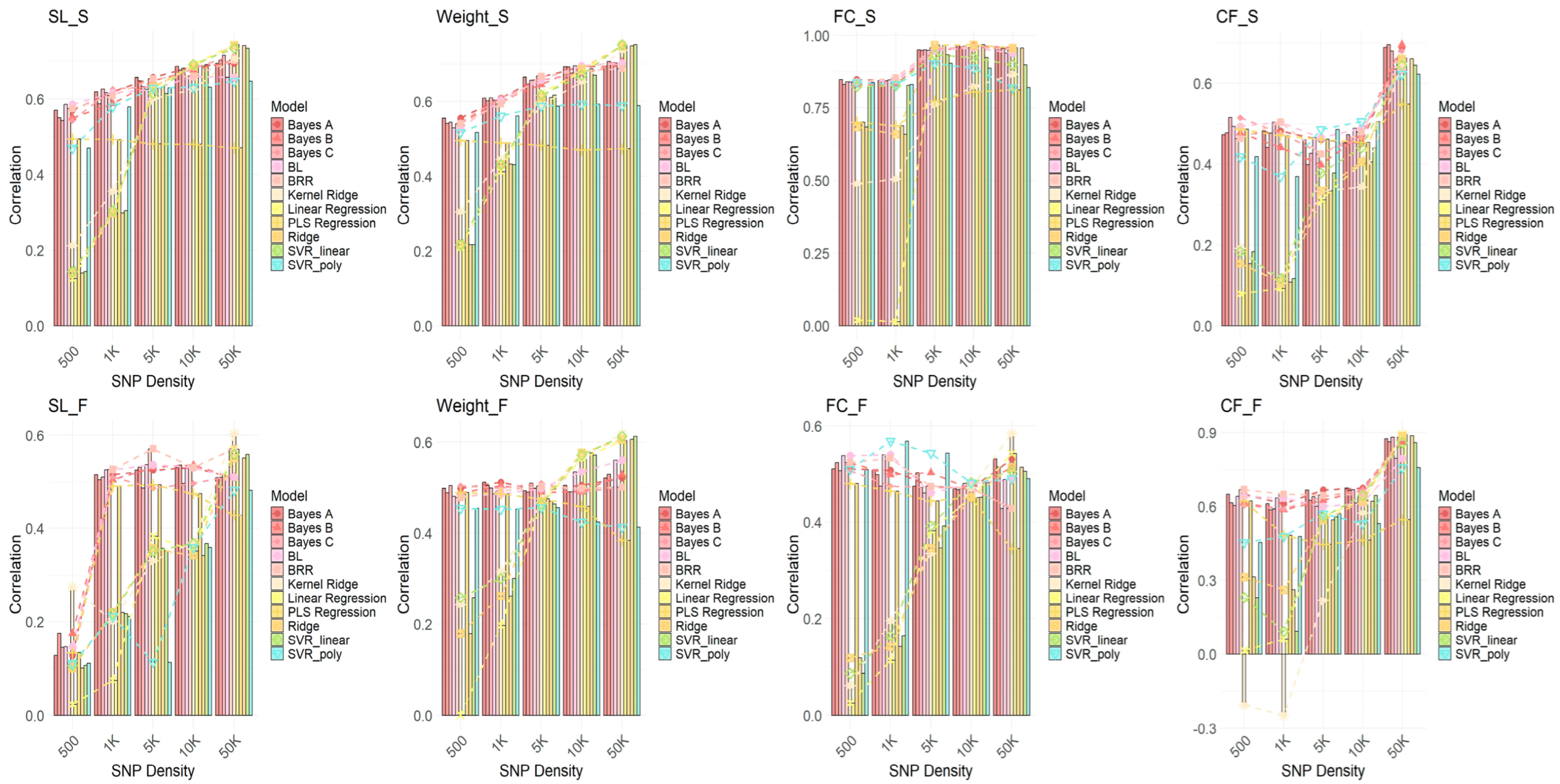
2.5. Comparison of Genomic Selection Performance of Different GWAS Methods and GS Models at Different SNP Densities
2.6. Prediction Accuracy of GWAS-GS Models for Seasonal Traits Using 5K SNPs
3. Discussion
3.1. Performance Differences Across GWAS Methods and the Impact of Algorithms
3.2. The Impact of SNP Density on Genetic Prediction Accuracy
3.3. Environmental and Seasonal Effects on Model Performance
3.4. Implications of Results for Genomic Selection in Carp
4. Materials and Methods
4.1. Source of Fish and Phenotypic Collection
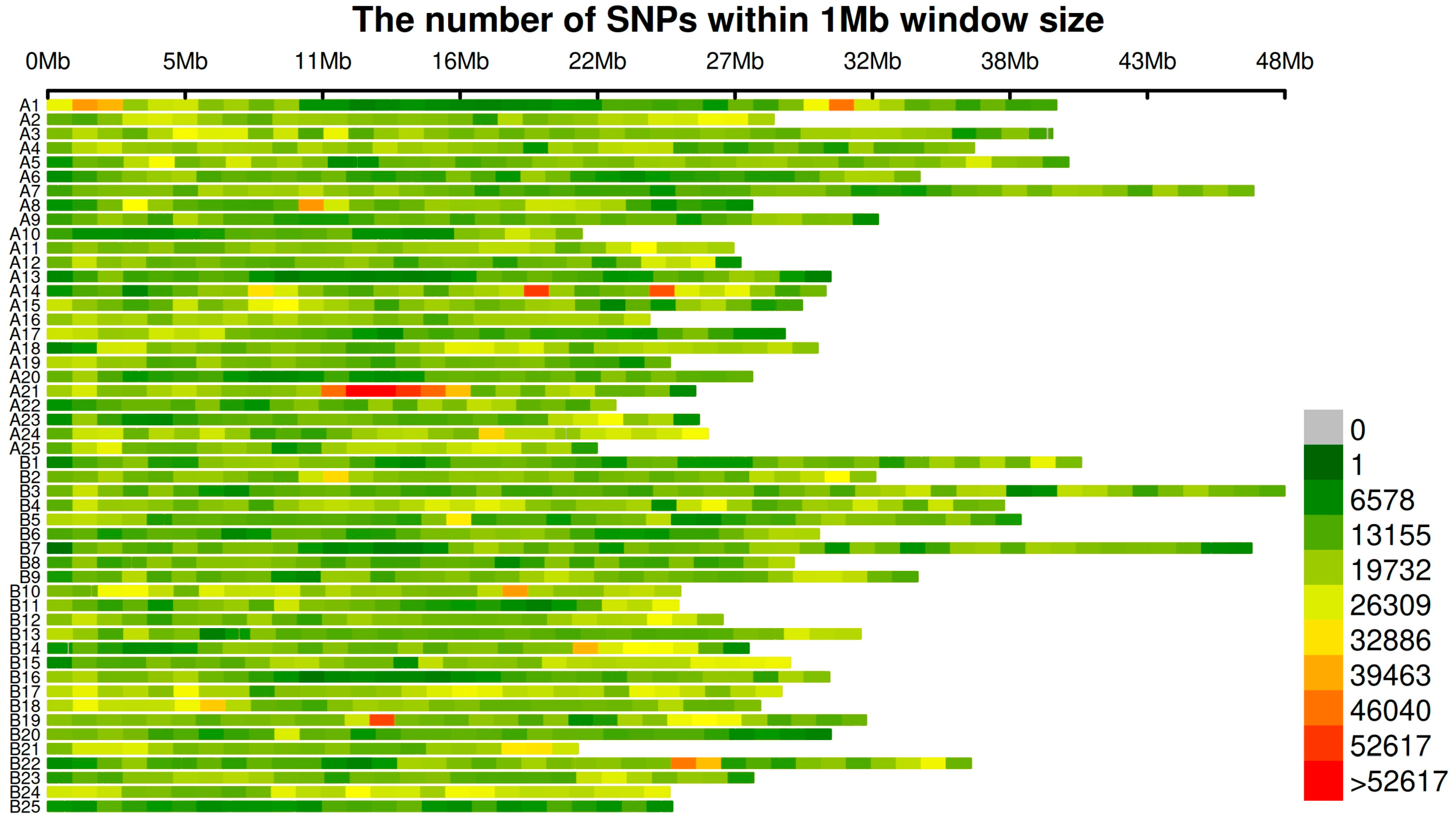
4.2. Genotyping and SNP Calling
4.3. Heritability Analysis
4.4. GWAS Analysis
4.5. Genomic Selection (GS) Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, M.H.; Wang, Q.; Zhao, R.; Li, Q.S.; Cui, M.S.; Zhang, Y.; Li, J.T. Cyprinus carpio (common carp). Trends Genet. 2022, 38, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Xu, J.; Liu, G.; Chen, L.; Zhou, Z.; Peng, W.; Jiang, Y.; Zhao, Z.; Jia, Z.; Sun, Y.; et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 2019, 10, 4625. [Google Scholar] [CrossRef] [PubMed]
- Ndraha, N.; Wong, H.C.; Hsiao, H.I. Managing the risk of Vibrio parahaemolyticus infections associated with oyster consumption: A review. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1187–1217. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Q.; Zang, S.; Liu, X.; Liu, H.; Li, Z.; Fan, Q.; Tan, S.; Shi, K.; Xia, Y.; et al. Inflammatory reaction and immune response of half-smooth tongue sole (Cynoglossus semilaevis) after infection with Vibrio anguillarum. Fish Shellfish Immunol. 2023, 141, 109043. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Peng, W.; Kong, S.; Pu, F.; Chen, B.; Zhou, Z.; Feng, J.; Li, X.; Xu, P. Genetic Mapping of Head Size Related Traits in Common Carp (Cyprinus carpio). Front Genet. 2018, 9, 448. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Qu, A.; Liu, Y.; Chen, X.; Wang, J.; Zhao, J.; Ke, Q.; Chen, L.; Chi, H.; Gong, H.; et al. Integrative analysis of GWAS and transcriptome reveals p53 signaling pathway mediates resistance to visceral white-nodules disease in large yellow croaker. Fish Shellfish Immunol. 2022, 130, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.O.; Ali, A.; Al-Tobasei, R.; Leeds, T.; Kenney, B.; Salem, M. Weighted Single-Step GWAS Identifies Genes Influencing Fillet Color in Rainbow Trout. Genes 2022, 13, 1331. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.P.; Yáñez, J.M.; Fukui, S.; Swift, B.; Davidson, W.S. Genome-wide association study (GWAS) for growth rate and age at sexual maturation in Atlantic salmon (Salmo salar). PLoS ONE 2015, 10, e0119730. [Google Scholar] [CrossRef] [PubMed]
- Sinclair-Waters, M.; Ødegård, J.; Korsvoll, S.A.; Moen, T.; Lien, S.; Primmer, C.R.; Barson, N.J. Beyond large-effect loci: Large-scale GWAS reveals a mixed large-effect and polygenic architecture for age at maturity of Atlantic salmon. Genet. Sel. Evol. 2020, 52, 9. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Sha, J.; Liu, S.; Bao, L.; Zhang, J.; Wang, R.; Yao, J.; Li, C.; Feng, J.; Sun, F.; et al. A genome-wide association study in catfish reveals the presence of functional hubs of related genes within QTLs for columnaris disease resistance. BMC Genom. 2015, 16, 196. [Google Scholar] [CrossRef] [PubMed]
- Mhalhel, K.; Levanti, M.; Abbate, F.; Laurà, R.; Guerrera, M.C.; Aragona, M.; Porcino, C.; Pansera, L.; Sicari, M.; Cometa, M.; et al. Skeletal Morphogenesis and Anomalies in Gilthead Seabream: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 16030. [Google Scholar] [CrossRef] [PubMed]
- Palaiokostas, C.; Kocour, M.; Prchal, M.; Houston, R.D. Accuracy of Genomic Evaluations of Juvenile Growth Rate in Common Carp (Cyprinus carpio) Using Genotyping by Sequencing. Front Genet. 2018, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Kuang, Y.; Lv, W.; Cao, D.; Sun, Z.; Sun, X. Genome-Wide Association Study for Muscle Fat Content and Abdominal Fat Traits in Common Carp (Cyprinus carpio). PLoS ONE 2016, 11, e0169127. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Roncancio, C.; García, B.; Gallardo-Hidalgo, J.; Yáñez, J.M. GWAS on Imputed Whole-Genome Sequence Variants Reveal Genes Associated with Resistance to Piscirickettsia salmonis in Rainbow Trout (Oncorhynchus mykiss). Genes 2022, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Kusmec, A.; Schnable, P.S. FarmCPUpp: Efficient large-scale genomewide association studies. Plant Direct. 2018, 2, e00053. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zeng, Y.; Wang, J.; Wang, T.; Li, X.; Shen, Z.; Meng, J.; Yao, X. A genome-wide association study of the racing performance traits in Yili horses based on Blink and FarmCPU models. Sci. Rep. 2024, 14, 27648. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Li, J.; Su, P.; Duan, H.; Sun, L.; Xu, S.; Sun, Y.; Zhao, H.; Chen, X.; Ding, D.; et al. Genetic dissection of maize (Zea mays L.) chlorophyll content using multi-locus genome-wide association studies. BMC Genom. 2023, 24, 384. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Zheng, X.; Kuang, Y.; Cao, D.; Yan, Y.; Sun, X. QTL variations for growth-related traits in eight distinct families of common carp (Cyprinus carpio). BMC Genet. 2016, 17, 65. [Google Scholar] [CrossRef] [PubMed]
- Lillehammer, M.; Meuwissen, T.H.; Sonesson, A.K. A low-marker density implementation of genomic selection in aquaculture using within-family genomic breeding values. Genet. Sel. Evol. 2013, 45, 39. [Google Scholar] [CrossRef] [PubMed]
- Pe’er, I.; Yelensky, R.; Altshuler, D.; Daly, M.J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. Off. Publ. Int. Genet. Epidemiol. Soc. 2008, 32, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Palaiokostas, C.; Ferraresso, S.; Franch, R.; Houston, R.D.; Bargelloni, L. Genomic prediction of resistance to pasteurellosis in gilthead sea bream (Sparus aurata) using 2b-RAD sequencing. G3 Genes Genomes Genet. 2016, 6, 3693–3700. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, C.; Shang, M.; Ge, Y.; Jia, Z.; Wang, S.; Zhang, Q.; Shi, L. Inheritance of growth traits in Songpu mirror carp (Cyprinus carpio L.) cultured in Northeast China. Aquaculture 2017, 477, 1–5. [Google Scholar] [CrossRef]
- Gustavsson, E.K.; Zhang, D.; Reynolds, R.H.; Garcia-Ruiz, S.; Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 2022, 38, 3844–3846. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; McCarthy, S.A. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.; Tu, Y.; Lv, X. Accelerating BWA-MEM Read Mapping on GPUs. Ics 2023, 2023, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Slifer, S.H. PLINK: Key Functions for Data Analysis. Curr. Protoc. Hum. Genet. 2018, 97, e59. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Wray, N.R.; Goddard, M.E.; Visscher, P.M. GCTA-GREML accounts for linkage disequilibrium when estimating genetic variance from genome-wide SNPs. Proc. Natl. Acad. Sci. USA 2016, 113, E4579–E4580. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yin, L.; Zhang, H.; Li, X.; Zhao, S. Performing Genome-Wide Association Studies Using rMVP. Methods Mol. Biol. 2022, 2481, 219–245. [Google Scholar] [CrossRef] [PubMed]
- Vogt, F.; Shirsekar, G.; Weigel, D. vcf2gwas: Python API for comprehensive GWAS analysis using GEMMA. Bioinformatics 2022, 38, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Guo, W.; Wang, Y.; Xiong, Y.; Ge, S.; Gong, G.; Lin, Q.; Xu, Z.; Gui, J.F.; Mei, J. Genome-wide association study reveals the genetic basis of growth trait in yellow catfish with sexual size dimorphism. Genomics 2022, 114, 110380. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association mapping in plants in the post-GWAS genomics era. Adv. Genet. 2019, 104, 75–154. [Google Scholar] [CrossRef] [PubMed]
- Azman, S.; Pathmanathan, D. The GLM framework of the Lee-Carter model: A multi-country study. J. Appl. Stat. 2022, 49, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Xiao, S.; Wang, Q.; Wang, Z. Comparative analysis of the GBLUP, emBayesB, and GWAS algorithms to predict genetic values in large yellow croaker (Larimichthys crocea). BMC Genom. 2016, 17, 460. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, P.; de Los Campos, G. Multitrait Bayesian shrinkage and variable selection models with the BGLR-R package. Genetics 2022, 222, iyac112. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, X.; Ma, H.; Li, J. Transformers-sklearn: A toolkit for medical language understanding with transformer-based models. BMC Med. Inform. Decis. Mak. 2021, 21, 90. [Google Scholar] [CrossRef] [PubMed]
- Bastien, P.; Bertrand, F.; Meyer, N.; Maumy-Bertrand, M. Deviance residuals-based sparse PLS and sparse kernel PLS regression for censored data. Bioinformatics 2015, 31, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Nengsih, T.A.; Bertrand, F.; Maumy-Bertrand, M.; Meyer, N. Determining the number of components in PLS regression on incomplete data set. Stat. Appl. Genet. Mol. Biol. 2019, 18, 20180059. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.; Meei, T.I.; Ling, G.C. Managing Fibrous Maxillary Ridge: A Case Series of Impression Techniques. Prim. Dent. J. 2023, 12, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Zhu, C.C.; Chen, X. Ensemble of kernel ridge regression-based small molecule-miRNA association prediction in human disease. Brief Bioinform. 2022, 23, bbab431. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; He, F.; He, M.; Yang, J.; Huang, X. Decentralized Kernel Ridge Regression Based on Data-Dependent Random Feature. IEEE Trans Neural. Netw. Learn Syst. 2025, 36, 7945–7954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qu, H.; Zhou, Z.; Chen, S.; Ngando, F.J.; Yang, F.; Xiao, J.; Guo, Y.; Cai, J.; Zhang, C. Age Determination of Chrysomya megacephala Pupae through Reflectance and Machine Learning Analysis. Insects 2024, 15, 184. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.A.; Lee, Y.M.; Ha, J.J.; Jin, S.; Park, B.; Kim, N.Y.; Won, J.I.; Kim, J.J. Genomic Predictions in Korean Hanwoo Cows: A Comparative Analysis of Genomic BLUP and Bayesian Methods for Reproductive Traits. Animals 2023, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wang, Z. Genomic prediction using an iterative conditional expectation algorithm for a fast BayesC-like model. Genetica 2018, 146, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, P.; de Los Campos, G.; Wu, H.; Vazquez, A.I.; Jones, K. Fast analysis of biobank-size data and meta-analysis using the BGLR R-package. G3 2025, 15, jkae288. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Murphy, D. Application of ggplot2 to Pharmacometric Graphics. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e79. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Heritability | Pval | Vg | Ve |
|---|---|---|---|---|
| SL_S | 0.3367 | 0.0111 | 2.7868 ± 1.0972 | 5.4904 ± 0.8285 |
| Weight_S | 0.3145 | 0.0133 | 7020.45 ± 2836.48 | 15,299.9 ± 2204.44 |
| FC_S | 0.0000 | 1.0000 | 3.4787 × 10−18 ± 0.0054 | 0.0564 ± 0.0061 |
| CF_S | 0.3196 | 0.0257 | 0.0311 ± 0.0139 | 0.0662 ± 0.0105 |
| SL_F | 0.5298 | 0.0005 | 5.8179 ± 1.6799 | 5.164 ± 1.0661 |
| Weight_F | 0.3554 | 0.0089 | 27,012 ± 10,329 | 48,996.1 ± 7624.17 |
| FC_F | 0.5426 | 0.0004 | 0.4877 ± 0.1368 | 0.4111 ± 0.0848 |
| CF_F | 0.0234 | 0.7751 | 0.0066 ± 0.0233 | 0.2788 ± 0.0291 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Fu, Y.; Zhu, X.; Zhang, R.; Shu, Y.; Zheng, X.; Hu, G. A Framework Integrating GWAS and Genomic Selection to Enhance Prediction Accuracy of Economical Traits in Common Carp. Int. J. Mol. Sci. 2025, 26, 7009. https://doi.org/10.3390/ijms26147009
Sun Z, Fu Y, Zhu X, Zhang R, Shu Y, Zheng X, Hu G. A Framework Integrating GWAS and Genomic Selection to Enhance Prediction Accuracy of Economical Traits in Common Carp. International Journal of Molecular Sciences. 2025; 26(14):7009. https://doi.org/10.3390/ijms26147009
Chicago/Turabian StyleSun, Zhipeng, Yuhan Fu, Xiaoyue Zhu, Ruixin Zhang, Yongjun Shu, Xianhu Zheng, and Guo Hu. 2025. "A Framework Integrating GWAS and Genomic Selection to Enhance Prediction Accuracy of Economical Traits in Common Carp" International Journal of Molecular Sciences 26, no. 14: 7009. https://doi.org/10.3390/ijms26147009
APA StyleSun, Z., Fu, Y., Zhu, X., Zhang, R., Shu, Y., Zheng, X., & Hu, G. (2025). A Framework Integrating GWAS and Genomic Selection to Enhance Prediction Accuracy of Economical Traits in Common Carp. International Journal of Molecular Sciences, 26(14), 7009. https://doi.org/10.3390/ijms26147009