
Network Theory Analysis of Allosteric Drug-Rescue Mechanisms in the Tumor Suppressor Protein p53 Y220C Mutant


Abstract
1. Introduction
2. Results
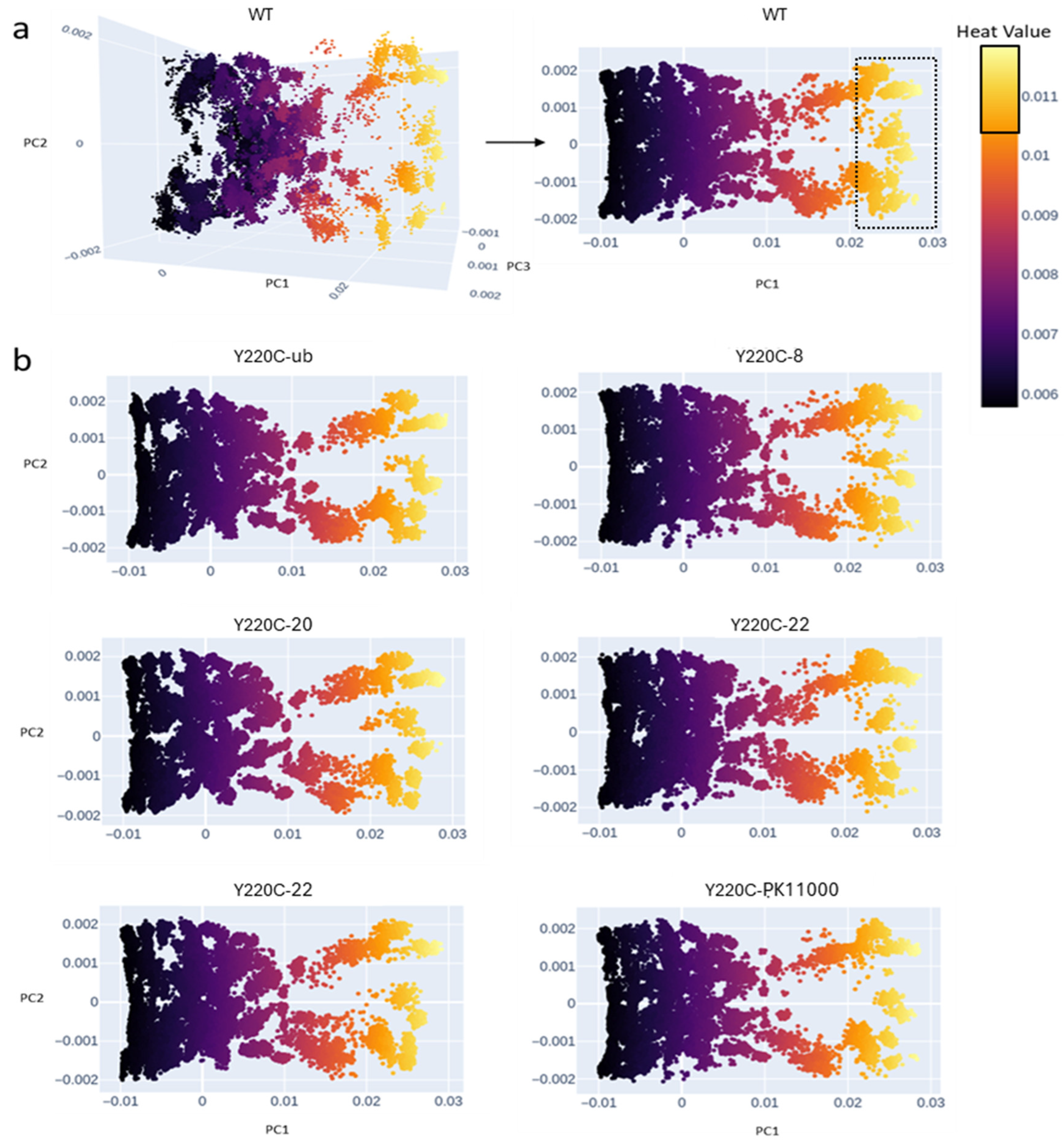
2.1. Energetic Network Generation and Electrostatic Heat Kernel PCA Projection
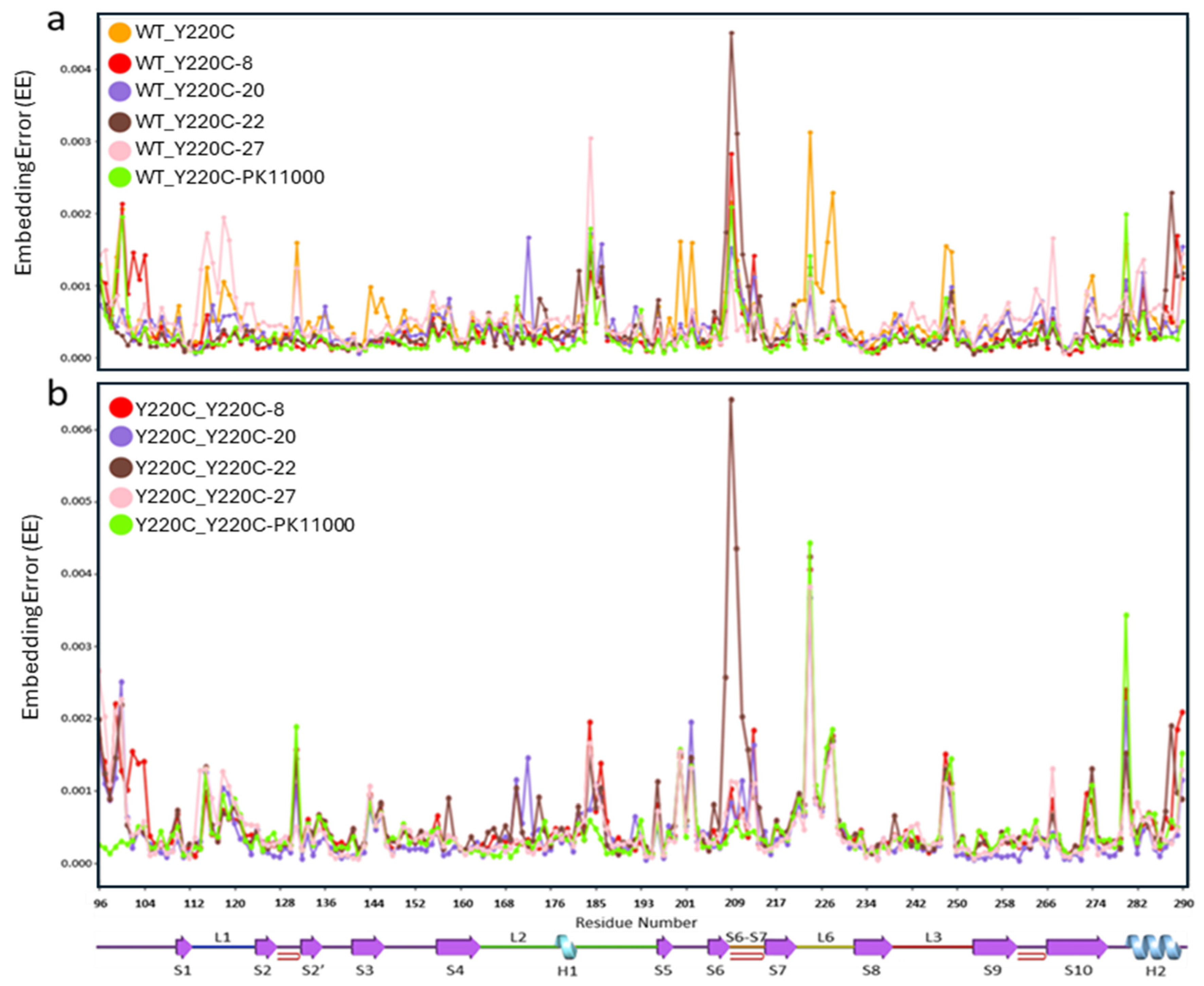
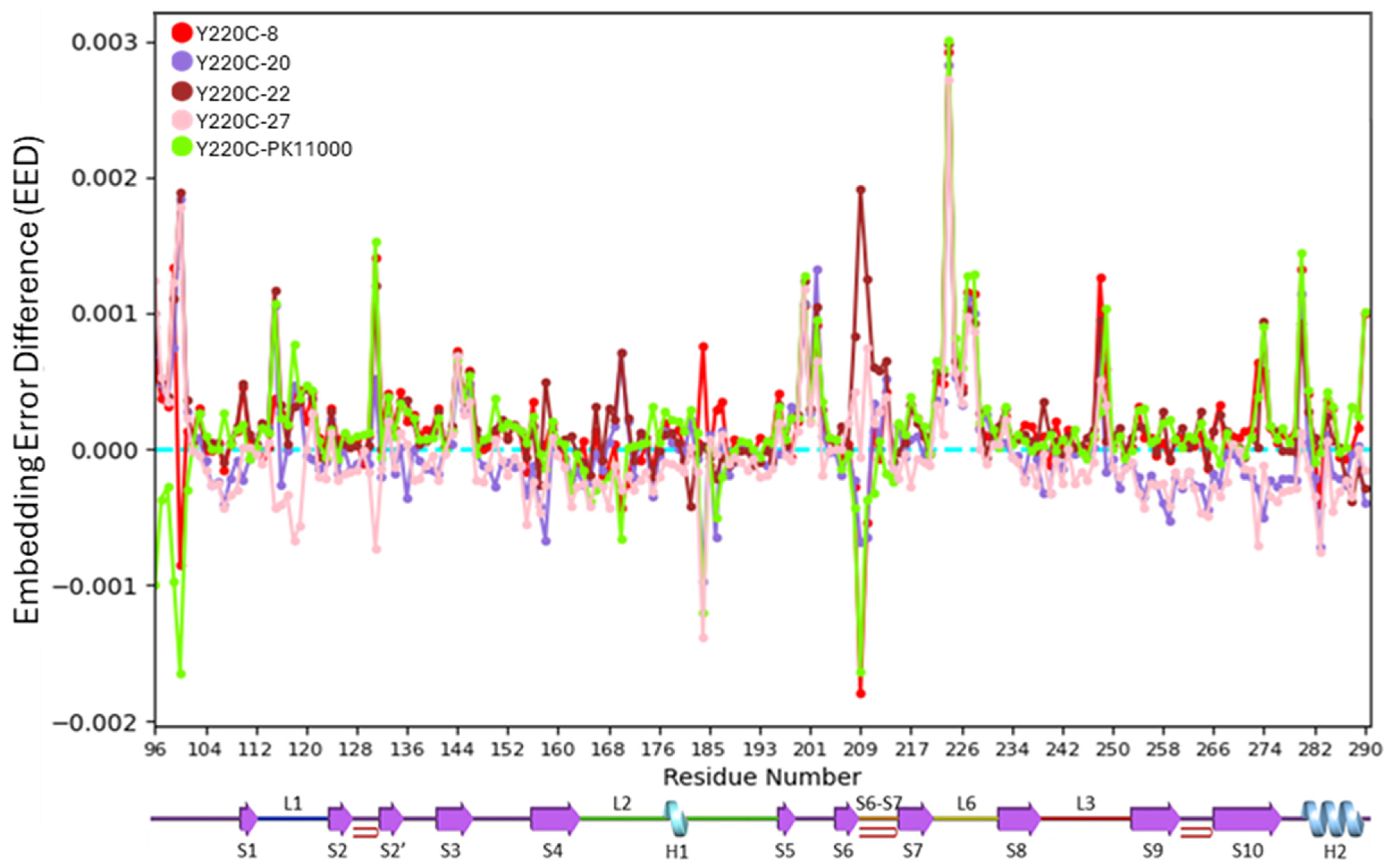
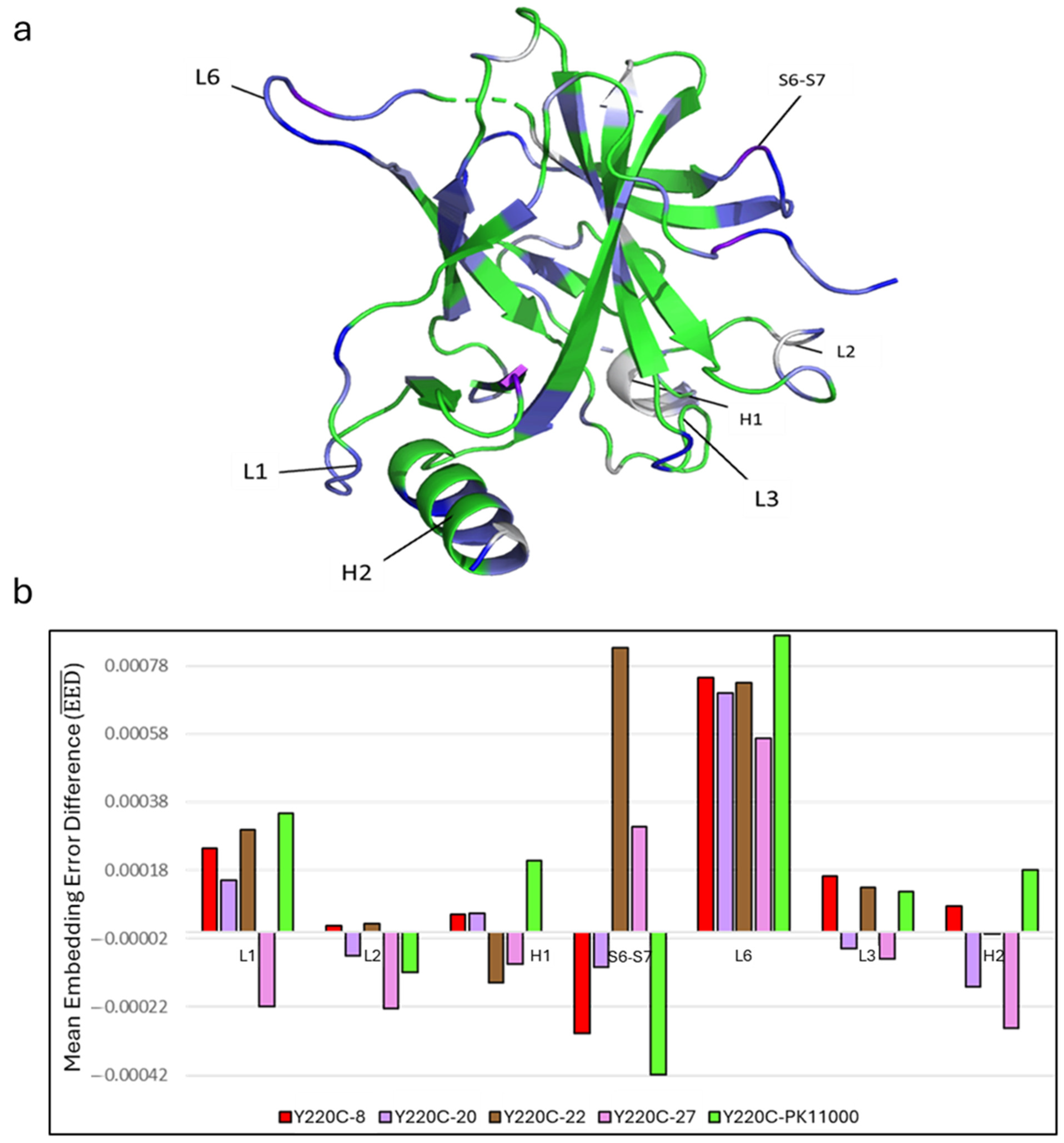
2.2. Embedding Error Analyses
3. Discussion
3.1. Heat Kernel Analyses Capture Dominant Energetic Connections Across Constructs
3.2. Wasserstein Embedding Error Analyses Reveal Long-Range Reorganizations
3.3. Allosteric Disruption and Rescue in p53-DBD Dynamics
3.4. Regional Analysis of Allosteric Rescue in p53-DBD
3.5. Mechanistic Insights into Allosteric Rescue
3.6. Implications for Drug Design and Therapeutic Development
4. Materials and Methods
4.1. MD Simulation Trajectory Specifications for p53-DBD Constructs
4.2. Locally Thresholded Interaction Network Generation
4.3. Heat Kernel Generation from Locally Thresholded Networks
4.4. Determination of the Diffusion Parameter for Heat Kernel Computation
4.5. Principal Component Analysis (PCA) of Heat Kernels
4.6. Calculation of Embedding Error Metrics
4.7. Code Development
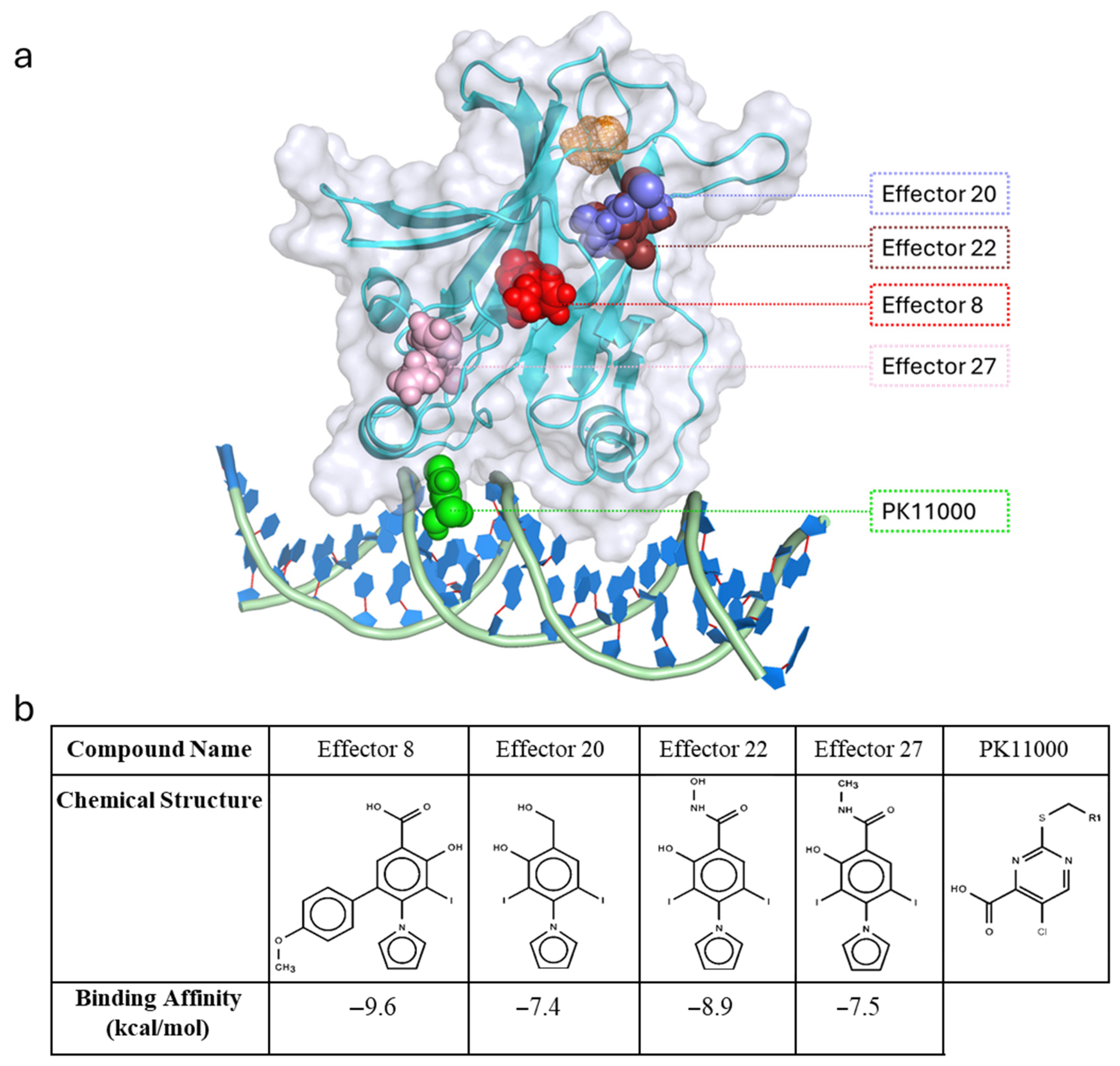
4.8. Molecular and Chemical Structure Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- George, P. p53 how crucial is its role in cancer. Int. J. Curr. Pharm. Res. 2011, 3, 19–25. [Google Scholar]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Luo, Q.; Beaver, J.M.; Liu, Y.; Zhang, Z. Dynamics of p53: A Master Decider of Cell Fate. Genes 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Barton, M.C. p53: Emerging roles in stem cells, development and beyond. Development 2018, 145, dev158360. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Kenzelmann Broz, D.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef]
- Candau, R.; Scolnick, D.M.; Darpino, P.; Ying, C.Y.; Halazonetis, T.D.; Berger, S.L. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 1997, 15, 807–816. [Google Scholar] [CrossRef]
- Walker, K.K.; Levine, A.J. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc. Natl. Acad. Sci. USA 1996, 93, 15335–15340. [Google Scholar] [CrossRef]
- Berger, M.; Vogt Sionov, R.; Levine, A.J.; Haupt, Y. A role for the polyproline domain of p53 in its regulation by Mdm2. J. Biol. Chem. 2001, 276, 3785–3790. [Google Scholar] [CrossRef]
- Brandt, T.; Petrovich, M.; Joerger, A.C.; Veprintsev, D.B. Conservation of DNA-binding specificity and oligomerisation properties within the p53 family. BMC Genom. 2009, 10, 628. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Harris, C.C. Structure and function of the p53 tumor suppressor gene: Clues for rational cancer therapeutic strategies. J. Natl. Cancer Inst. 1996, 88, 1442–1455. [Google Scholar] [CrossRef]
- Han, I.S.M.; Thayer, K.M. Reconnaissance of Allostery via the Restoration of Native p53 DNA-Binding Domain Dynamics in Y220C Mutant p53 Tumor Suppressor Protein. ACS Omega 2024, 9, 19837–19847. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef]
- Monod, J.; Wyman, J.; Changeux, J.P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Cooper, A.; Dryden, D.T. Allostery without conformational change. A plausible model. Eur. Biophys. J. EBJ 1984, 11, 103–109. [Google Scholar] [CrossRef]
- Tsai, C.-J.; del Sol, A.; Nussinov, R. Allostery: Absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 2008, 378, 1–11. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Nussinov, R. A Unified View of “How Allostery Works”. PLoS Comput. Biol. 2014, 10, e1003394. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Ma, B.; Nussinov, R. Is allostery an intrinsic property of all dynamic proteins? Proteins 2004, 57, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nussinov, R. Energetic redistribution in allostery to execute protein function. Proc. Natl. Acad. Sci. USA 2017, 114, 7480–7482. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X.; Pang, X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A. Electrostatic basis of structure-function correlation in proteins. Acc. Chem. Res. 1981, 14, 284–290. [Google Scholar] [CrossRef]
- Perutz, M.F. Electrostatic effects in proteins. Science 1978, 201, 1187–1191. [Google Scholar] [CrossRef]
- Kumawat, A.; Chakrabarty, S. Hidden electrostatic basis of dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. USA 2017, 114, E5825–E5834. [Google Scholar] [CrossRef]
- Cowan, B.S.; Beveridge, D.L.; Thayer, K.M. Allosteric Signaling in PDZ Energetic Networks: Embedding Error Analysis. J. Phys. Chem. B 2023, 127, 623–633. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Sengupta, J. Hidden electrostatic energy contributions define dynamic allosteric communications within p53 during molecular recognition. Biophys. J. 2021, 120, 4512–4524. [Google Scholar] [CrossRef]
- Bauer, M.R.; Joerger, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef]
- Han, I.S.M.; Abramson, D.; Thayer, K.M. Insights into Rational Design of a New Class of Allosteric Effectors with Molecular Dynamics Markov State Models and Network Theory. ACS Omega 2022, 7, 2831–2841. [Google Scholar] [CrossRef]
- Amitai, G.; Shemesh, A.; Sitbon, E.; Shklar, M.; Netanely, D.; Venger, I.; Pietrokovski, S. Network Analysis of Protein Structures Identifies Functional Residues. J. Mol. Biol. 2004, 344, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- del Sol, A.; Fujihashi, H.; Amoros, D.; Nussinov, R. Residue centrality, functionally important residues, and active site shape: Analysis of enzyme and non-enzyme families. Protein Sci. Publ. Protein Soc. 2006, 15, 2120–2128. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.J.; Rader, A.J.; Kuhn, L.A.; Thorpe, M.F. Protein flexibility predictions using graph theory. Proteins Struct. Funct. Bioinform. 2001, 44, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Brinda, K.V.; Vishveshwara, S. A network representation of protein structures: Implications for protein stability. Biophys. J. 2005, 89, 4159–4170. [Google Scholar] [CrossRef]
- Halder, A.; Anto, A.; Subramanyan, V.; Bhattacharyya, M.; Vishveshwara, S.; Vishveshwara, S. Surveying the Side-Chain Network Approach to Protein Structure and Dynamics: The SARS-CoV-2 Spike Protein as an Illustrative Case. Front. Mol. Biosci. 2020, 7, 596945. [Google Scholar] [CrossRef]
- Abramson, D.I. The Colors of a Protein: Protein Dynamics Through the Lens of Spectral Graph Theory. Bachelor’s Thesis, Wesleyan University, Middletown, CT, USA, 2021. [Google Scholar]
- Vijayabaskar, M.S.; Vishveshwara, S. Interaction energy based protein structure networks. Biophys. J. 2010, 99, 3704–3715. [Google Scholar] [CrossRef]
- Charles, M.; Roth, B.L.N.; Abraham, M. Lenhoff. Van der Waals Interactions Involving Proteins. Biophys. J. 1996, 70, 977–987. [Google Scholar]
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019, 47, 1637–1652. [Google Scholar] [CrossRef]
- Kar, G.; Keskin, O.; Gursoy, A.; Nussinov, R. Allostery and population shift in drug discovery. Curr. Opin. Pharmacol. 2010, 10, 715–722. [Google Scholar] [CrossRef]
- Sokalingam, S.; Raghunathan, G.; Soundrarajan, N.; Lee, S.-G. A Study on the Effect of Surface Lysine to Arginine Mutagenesis on Protein Stability and Structure Using Green Fluorescent Protein. PLoS ONE 2012, 7, e40410. [Google Scholar] [CrossRef]
- Martínez, L.; Figueira, A.C.M.; Webb, P.; Polikarpov, I.; Skaf, M.S. Mapping the Intramolecular Vibrational Energy Flow in Proteins Reveals Functionally Important Residues. J. Phys. Chem. Lett. 2011, 2, 2073–2078. [Google Scholar] [CrossRef]
- Naganathan, A.N. Modulation of allosteric coupling by mutations: From protein dynamics and packing to altered native ensembles and function. Curr. Opin. Struct. Biol. 2019, 54, 1–9. [Google Scholar] [CrossRef]
- Barros, E.P.; Demir, Ö.; Soto, J.; Cocco, M.J.; Amaro, R.E. Markov state models and NMR uncover an overlooked allosteric loop in p53. Chem. Sci. 2021, 12, 1891–1900. [Google Scholar] [CrossRef]
- Lukman, S.; Lane, D.P.; Verma, C.S. Mapping the structural and dynamical features of multiple p53 DNA binding domains: Insights into loop 1 intrinsic dynamics. PLoS ONE 2013, 8, e80221. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a p53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Lambrughi, M.; De Gioia, L.; Gervasio, F.L.; Lindorff-Larsen, K.; Nussinov, R.; Urani, C.; Bruschi, M.; Papaleo, E. DNA-binding protects p53 from interactions with cofactors involved in transcription-independent functions. Nucleic Acids Res. 2016, 44, 9096–9109. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, M.-C.; Xu, X.-K.; Zhao, Y.; Mahanand, C.; Zhu, T.; Deng, H.; Nevo, E.; Du, J.-Z.; Chen, X.-Q. Functional Diversity of p53 in Human and Wild Animals. Front. Endocrinol. 2019, 10, 152. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.A.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Chem. 2019, 19, 2491–2504. [Google Scholar] [CrossRef]
- Zupnick, A.; Prives, C. Mutational Analysis of the p53 Core Domain L1 Loop. J Biol Chem. 2006, 281, 20464–20473. [Google Scholar] [CrossRef]
- Thayer, K.M.; Stetson, S.; Caballero, F.; Chiu, C.; Han, I.S.M. Navigating the complexity of p53-DNA binding: Implications for cancer therapy. Biophys. Rev. 2024, 16, 479–496. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, B.; Thayer, K.M.; Black, E.; Beveridge, D.L. Spectral analysis of molecular dynamics simulations on PDZ: MD sectors. J. Biomol. Struct. Dyn. 2020, 38, 781–790. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 1981, 103, 335–340. [Google Scholar] [CrossRef]
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015.
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M., Jr. Structural Survey of Zinc Containing Proteins and the Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Arvai, K. Kneed: Knee-Point Detection in Python; GitHub Repository: Washington, DC, USA, 2020. [Google Scholar]
- Satopaa, V.; Albrecht, J.; Irwin, D.; Raghavan, B. Finding a “Kneedle” in a Haystack: Detecting Knee Points in System Behavior. In Proceedings of the 2011 31st International Conference on Distributed Computing Systems Workshops, Minneapolis, MN, USA, 20–24 June 2011; pp. 166–171. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: North Charleston, SC, USA, 1995. [Google Scholar]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks—A publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas; IOS Press: Amsterdam, The Netherlands, 2016; pp. 87–90. [Google Scholar]
- Karulin, B.; Kozhevnikov, M. Ketcher: Web-based chemical structure editor. J. Cheminform. 2011, 3, P3. [Google Scholar] [CrossRef]
- Cowan, B. Interconnected Protein Networks: Insights Towards CRIB-Par6 Protein Allostery Through a Graph-Theoretic Analysis. Bachelor’s Thesis, Wesleyan University, Middletown, CT, USA, 2022. [Google Scholar]
- ElGhawalby, H.; Hancock, E.R. Heat Kernel Embeddings, Differential Geometry and Graph Structure. Axioms 2015, 4, 275–293. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % of Residues with EED Value ≥ Cuttoff | |||||||
| Residue EED Value Cutoff | |||||||
| p53-DBD Motif | Average α-carbon distance (Å) between Motif and Y220C | 0.0002 | 0.0003 | 0.0004 | 0.0005 | 0.001 | 0.002 |
| L1 | 29.1 | 54.5 | 45.5 | 36.4 | 18.2 | 9.1 | 0.0 |
| L2 | 25.5 | 38.5 | 7.7 | 3.8 | 0.0 | 0.0 | 0.0 |
| H1 | 29.1 | 66.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| S6-S7 | 19.4 | 85.7 | 71.4 | 71.4 | 71.4 | 28.6 | 14.3 |
| L6 | 15.2 | 90.0 | 70.0 | 70.0 | 70.0 | 30.0 | 10.0 |
| L3 | 30.5 | 28.6 | 21.4 | 14.3 | 14.3 | 14.3 | 0.0 |
| H2 | 36.7 | 70.0 | 46.2 | 30.8 | 15.4 | 15.4 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cowan, B.S.; Thayer, K.M. Network Theory Analysis of Allosteric Drug-Rescue Mechanisms in the Tumor Suppressor Protein p53 Y220C Mutant. Int. J. Mol. Sci. 2025, 26, 6884. https://doi.org/10.3390/ijms26146884
Cowan BS, Thayer KM. Network Theory Analysis of Allosteric Drug-Rescue Mechanisms in the Tumor Suppressor Protein p53 Y220C Mutant. International Journal of Molecular Sciences. 2025; 26(14):6884. https://doi.org/10.3390/ijms26146884
Chicago/Turabian StyleCowan, Benjamin S., and Kelly M. Thayer. 2025. "Network Theory Analysis of Allosteric Drug-Rescue Mechanisms in the Tumor Suppressor Protein p53 Y220C Mutant" International Journal of Molecular Sciences 26, no. 14: 6884. https://doi.org/10.3390/ijms26146884
APA StyleCowan, B. S., & Thayer, K. M. (2025). Network Theory Analysis of Allosteric Drug-Rescue Mechanisms in the Tumor Suppressor Protein p53 Y220C Mutant. International Journal of Molecular Sciences, 26(14), 6884. https://doi.org/10.3390/ijms26146884