
Metabolic Profiling of Distinct TP53-Mutant Esophageal Adenocarcinoma Models Reveals Different Bioenergetic Dependencies

 , , , , , ,
, , , , , ,  and
and 
Abstract
1. Introduction
2. Results
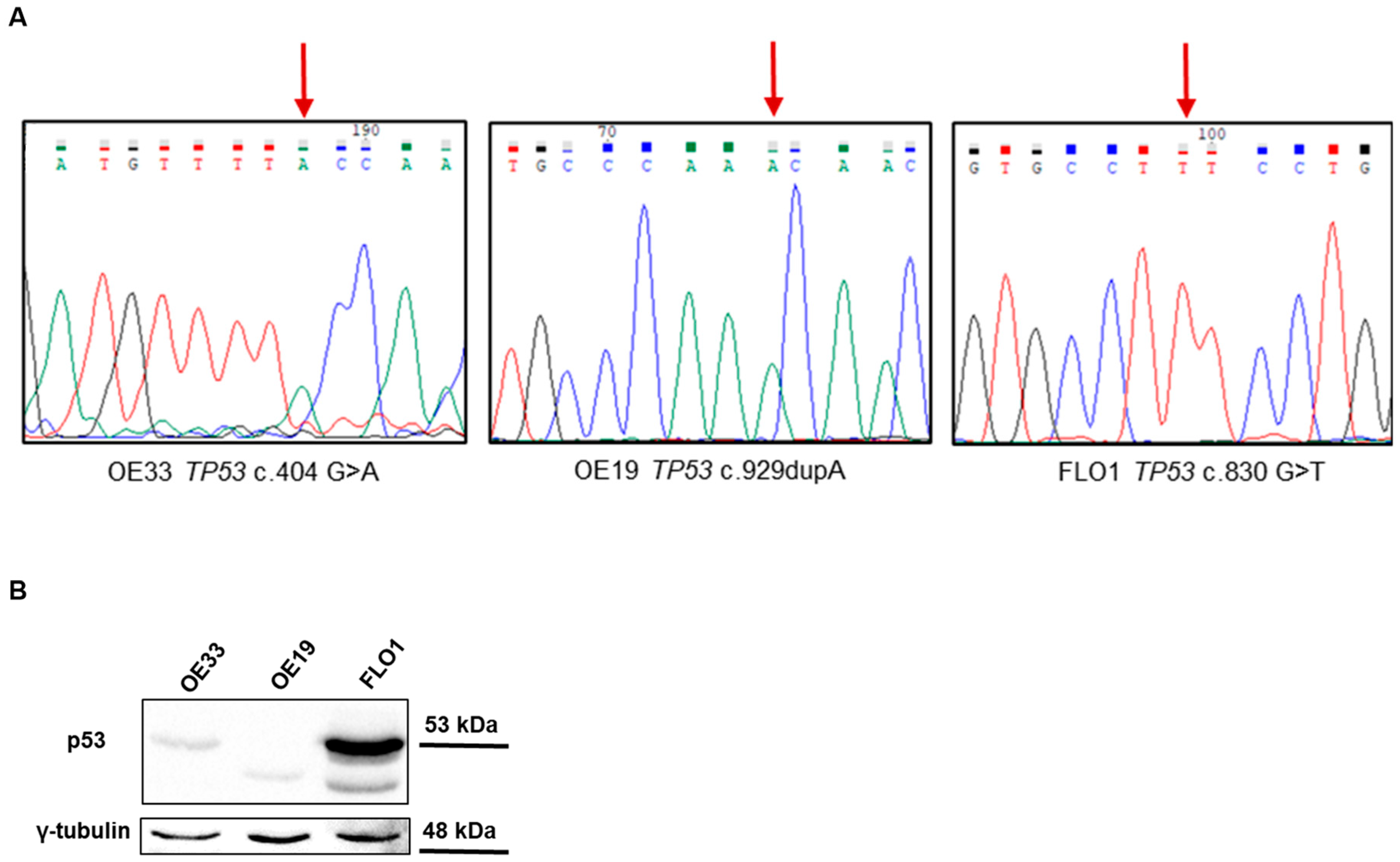
2.1. TP53 Variants in EAC Cell Lines
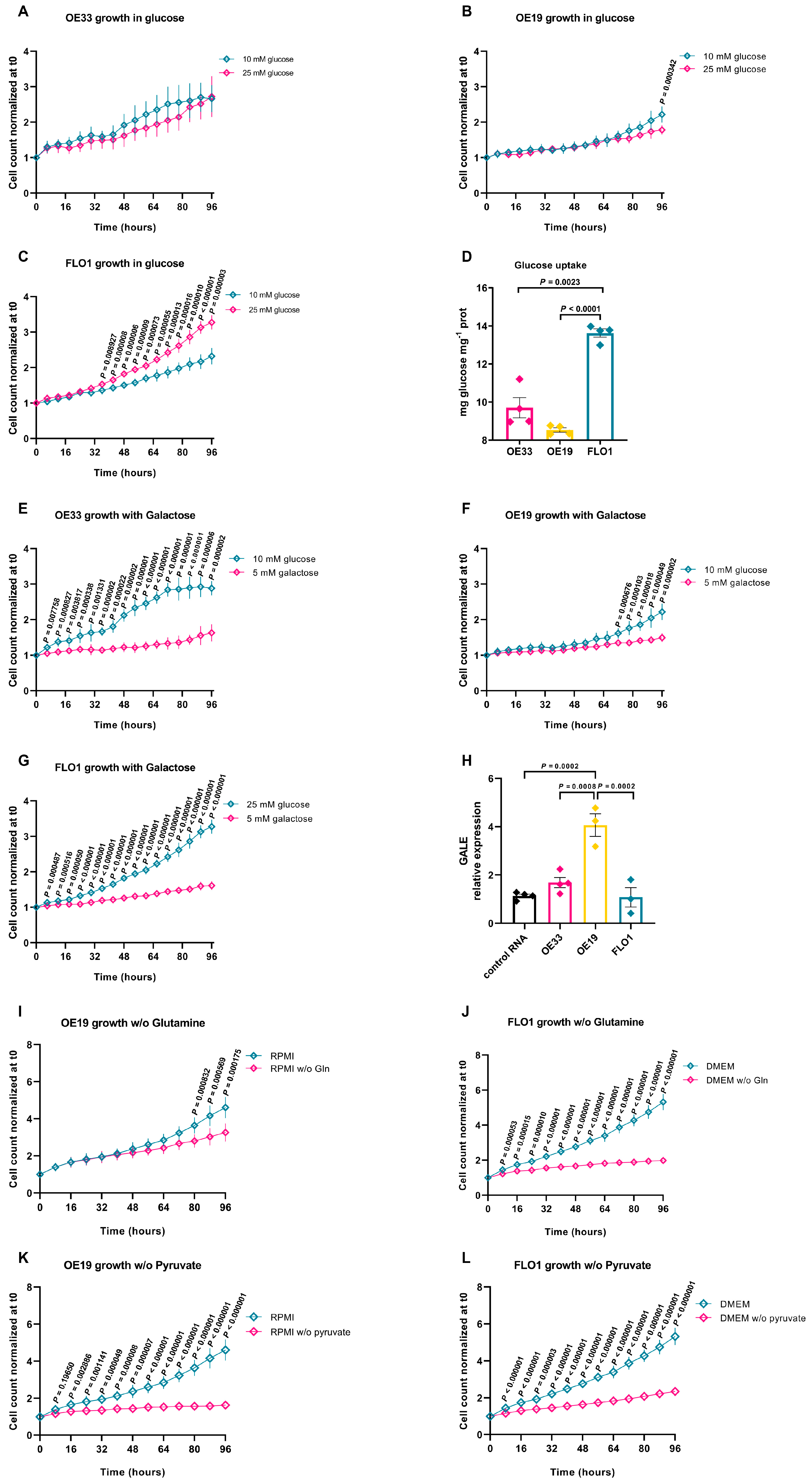
2.2. Metabolic Adaptations of EAC Cell Lines to Different Growth Environments
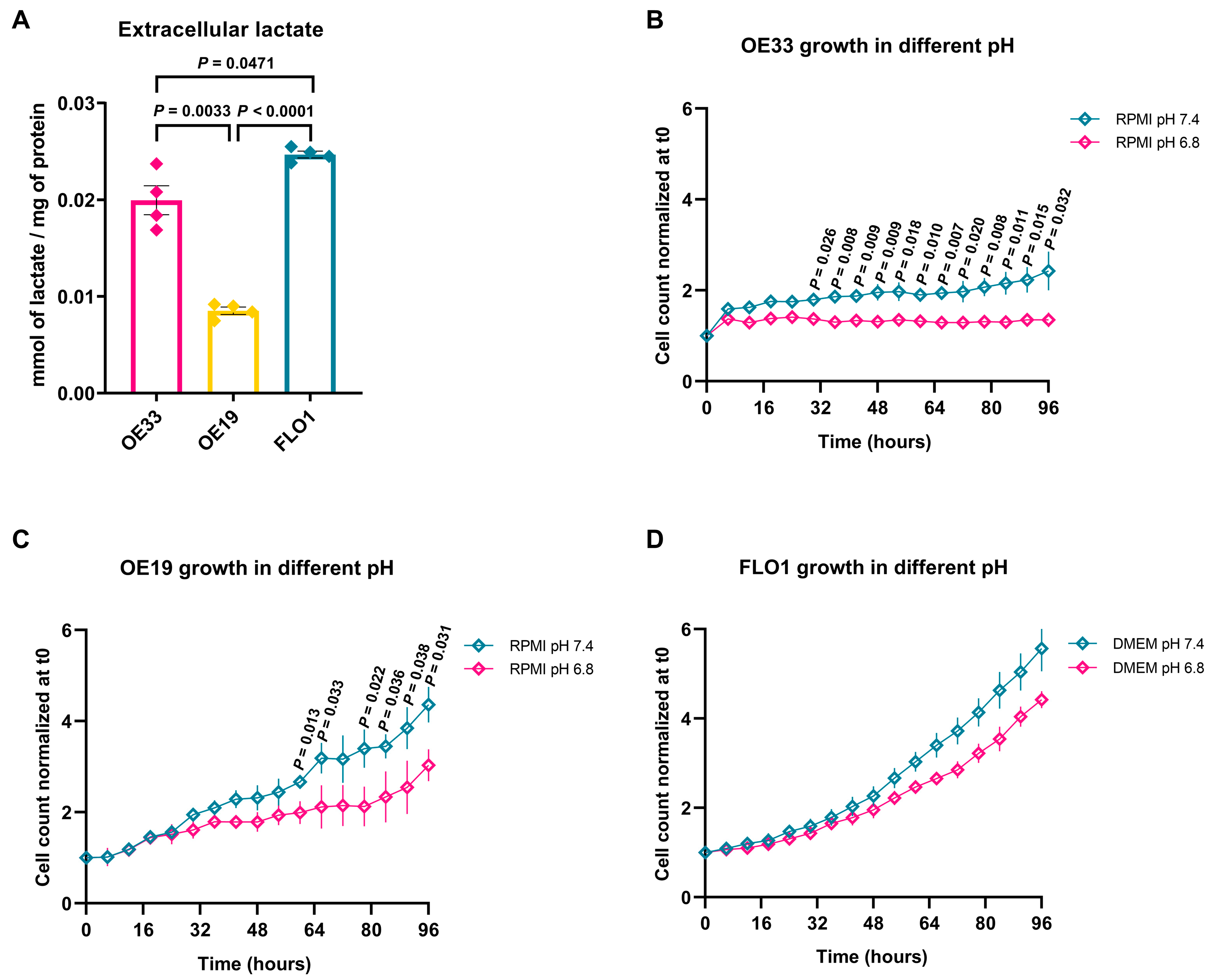
2.3. EAC Cell Lines Exhibit Different Levels of Lactate Production and Oxygen Consumption
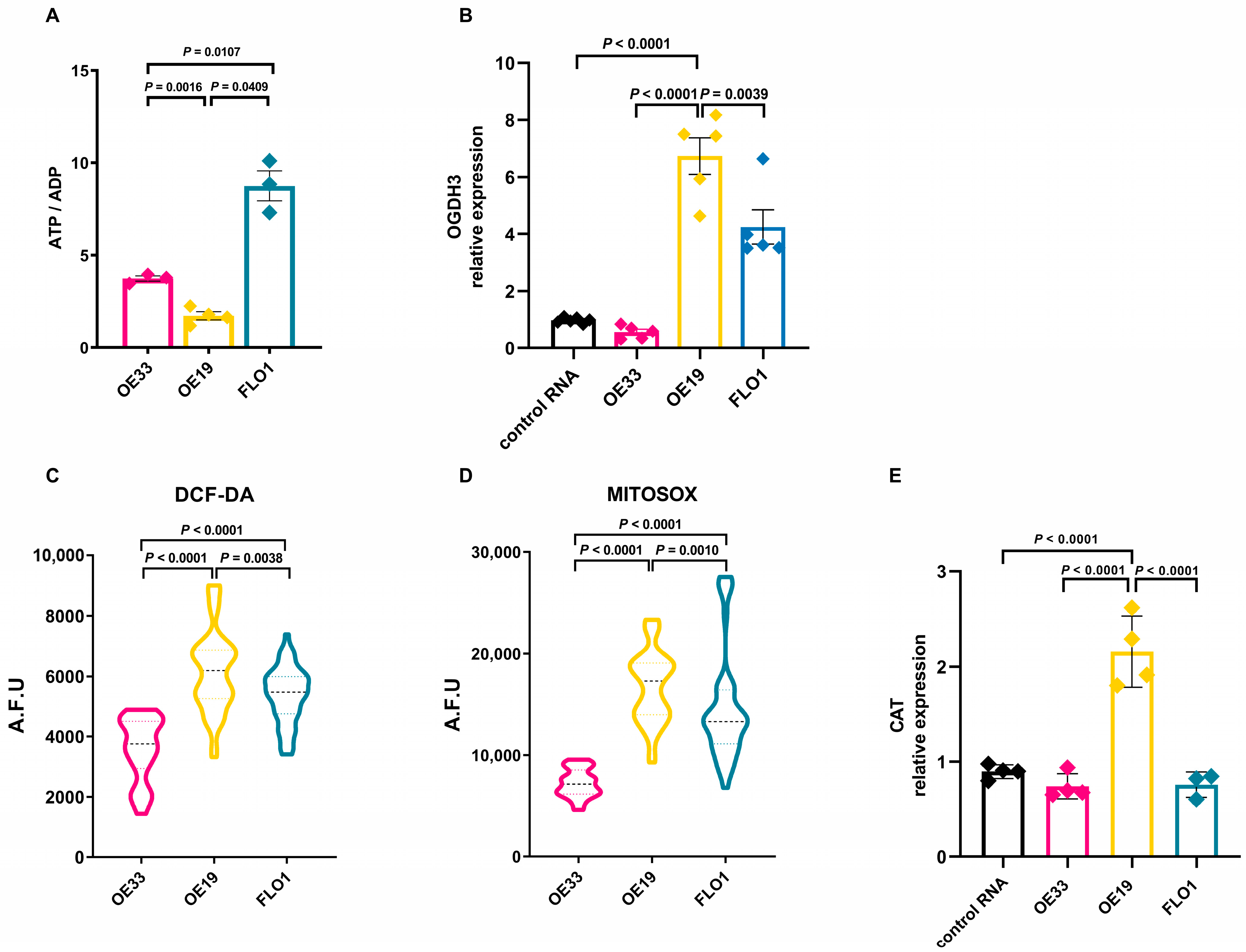
2.4. OE19 Cells Reveal an Energy Imbalance with Increased Reactive Oxygen Species Production
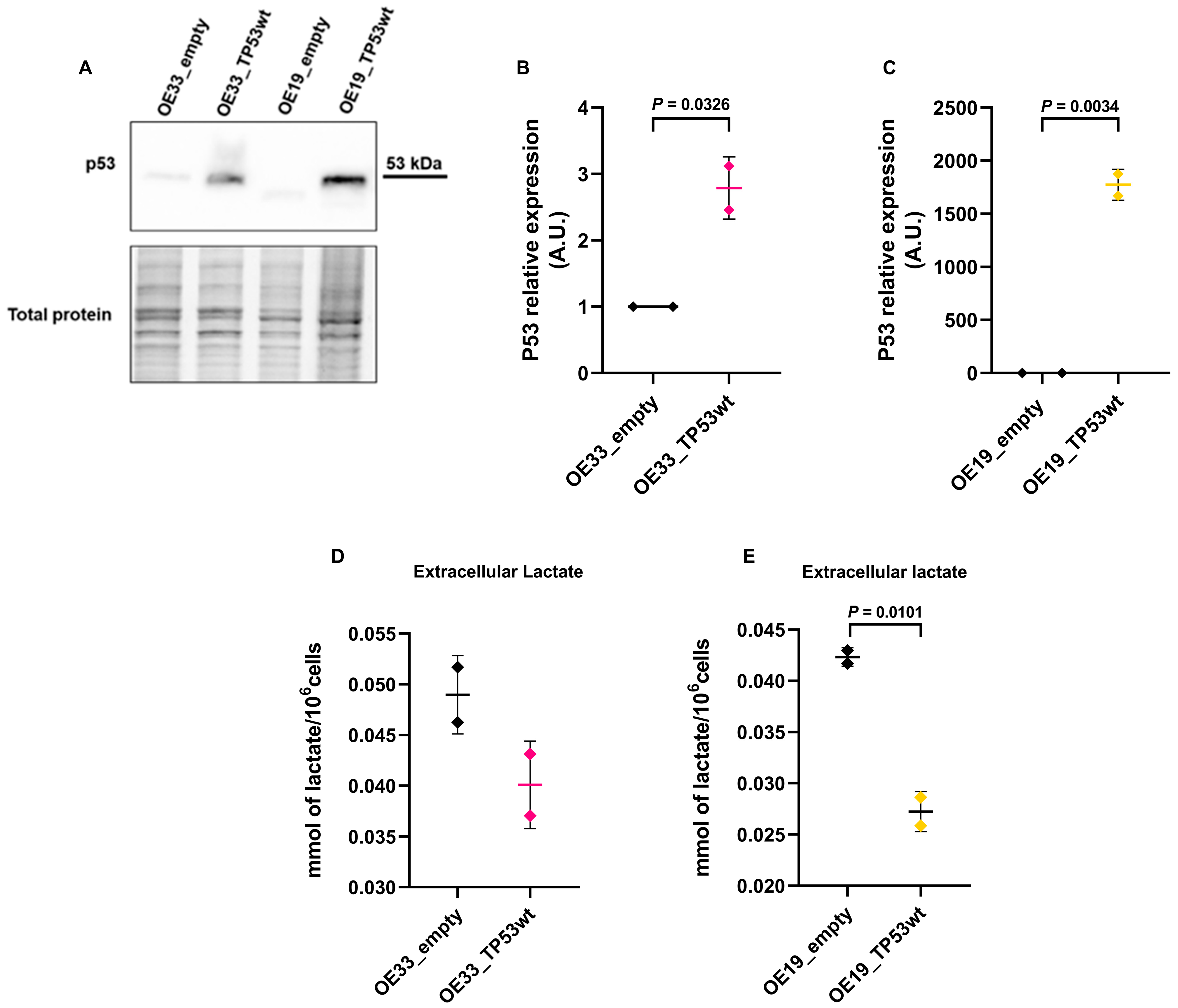
2.5. TP53 Restoration Reduces Extracellular Lactate Levels
2.6. Transcriptomic Analysis Reveals TP53 Mutation-Dependent Metabolic Reprogramming in EAC
3. Discussion
4. Materials and Methods
4.1. PCR and Sanger Sequencing
4.2. Western Blot Analysis
4.3. Cell Cultures and Experimental Conditions
4.4. Glucose Oxidase Assay
4.5. Quantitative Reverse Transcriptase Real-Time PCR (qRT-PCR)
4.6. Transient Transfection of Wild-Type TP53
4.7. Lactate Quantification
4.8. ATP and ADP Determination
4.9. Radical Oxygen Species and Mitochondrial Anion Superoxide Measurement
4.10. In Silico Analysis of TCGA Transcriptomic Data
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-DG | 2-deoxyglucose |
| ACTB | Human actin-beta |
| ATO | Arsenic trioxide |
| BFB | Breakage–fusion–bridge |
| CADD | Combined annotation-dependent depletion |
| CAT | Catalase |
| EAC | Esophageal Adenocarcinoma |
| EACSGE | Esophageal Adenocarcinoma Study Group Europe |
| GALE | UDP-galactose 4-epimerase |
| GC-MS | Gas chromatography mass spectrometry |
| GLS | Glutaminase |
| GOX | Glucose Oxidase |
| HPLC | High-Performance Liquid Chromatography |
| MAF | Minor allele frequency |
| NMR | Nuclear Magnetic Resonance |
| OGDH | Oxoglutarate dehydrogenase |
| OXPHOS | Oxidative phosphorylation |
| ROS | Reactive Oxygen Species |
| SOD1 | Superoxide Dismutase 1 |
| SOD2 | Superoxide Dismutase 2 |
| TCA | Tricarboxylic acid |
| TCGA | The Cancer Genome Atlas |
References
- Domper Arnal, M.J.; Ferrández Arenas, Á.; Lanas Arbeloa, Á. Esophageal Cancer: Risk Factors, Screening and Endoscopic Treatment in Western and Eastern Countries. World J. Gastroenterol. WJG 2015, 21, 7933–7943. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Off. J. Am. Coll. Gastroenterol. ACG 2017, 112, 1247. [Google Scholar] [CrossRef] [PubMed]
- Frankell, A.M.; Jammula, S.; Li, X.; Contino, G.; Killcoyne, S.; Abbas, S.; Perner, J.; Bower, L.; Devonshire, G.; Ococks, E.; et al. The Landscape of Selection in 551 Esophageal Adenocarcinomas Defines Genomic Biomarkers for the Clinic. Nat. Genet. 2019, 51, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Naeini, M.; Newell, F.; Aoude, L.G.; Bonazzi, V.F.; Patel, K.; Lampe, G.; Koufariotis, L.T.; Lakis, V.; Addala, V.; Kondrashova, O.; et al. Multi-Omic Features of Oesophageal Adenocarcinoma in Patients Treated with Preoperative Neoadjuvant Therapy. Nat. Commun. 2023, 14, 3155. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.; Armour, N.; Nancarrow, D.; Krause, L.; Hayward, N.; Lampe, G.; Smithers, B.M.; Barbour, A. Genome-Wide Analysis of Esophageal Adenocarcinoma Yields Specific Copy Number Aberrations That Correlate with Prognosis. Genes. Chromosomes Cancer 2014, 53, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.-M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic Catastrophes Frequently Arise in Esophageal Adenocarcinoma and Drive Tumorigenesis. Nat. Commun. 2014, 5, 5224. [Google Scholar] [CrossRef] [PubMed]
- Finley, J.C.; Reid, B.J.; Odze, R.D.; Sanchez, C.A.; Galipeau, P.; Li, X.; Self, S.G.; Gollahon, K.A.; Blount, P.L.; Rabinovitch, P.S. Chromosomal Instability in Barrett’s Esophagus Is Related to Telomere Shortening. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Mcneil, T.R.; Sikder, S.; Dalal, Y. Cancer cells’ chamber of secrets: The link between micronuclei, chromothripsis and malignancy. Open Biol. 2025, 15, 240388. [Google Scholar] [CrossRef] [PubMed]
- Espejo Valle-Inclan, J.; De Noon, S.; Trevers, K.; Elrick, H.; van Belzen, I.A.E.M.; Zumalave, S.; Sauer, C.M.; Tanguy, M.; Butters, T.; Muyas, F.; et al. Ongoing chromothripsis underpins osteosarcoma genome complexity and clonal evolution. Cell 2025, 188, 352–370.e22. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, S.; Jonas, C.; Wenzel, M.C.; Velazquez Camacho, O.; Arolt, C.; Zhao, Y.; Büttner, R.; Quaas, A.; Plum, P.S.; Hillmer, A.M. Genomic and Transcriptomic Characteristics of Esophageal Adenocarcinoma. Cancers 2021, 13, 4300. [Google Scholar] [CrossRef] [PubMed]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational Signatures in Esophageal Adenocarcinoma Define Etiologically Distinct Subgroups with Therapeutic Relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Murugaesu, N.; Wilson, G.A.; Birkbak, N.J.; Watkins, T.; McGranahan, N.; Kumar, S.; Abbassi-Ghadi, N.; Salm, M.; Mitter, R.; Horswell, S.; et al. Tracking the Genomic Evolution of Esophageal Adenocarcinoma through Neoadjuvant Chemotherapy. Cancer Discov. 2015, 5, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Isidori, F.; Malvi, D.; Fittipaldi, S.; Forcato, C.; Bozzarelli, I.; Sala, C.; Raulli, G.; D’Errico, A.; Fiorentino, M.; Seri, M.; et al. Genomic Profiles of Primary and Metastatic Esophageal Adenocarcinoma Identified via Digital Sorting of Pure Cell Populations: Results from a Case Report. BMC Cancer 2018, 18, 889. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of P53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Orsini, A.; Mastracci, L.; Bozzarelli, I.; Ferrari, A.; Isidori, F.; Fiocca, R.; Lugaresi, M.; D’Errico, A.; Malvi, D.; Cataldi-Stagetti, E.; et al. Correlations between Molecular Alterations, Histopathological Characteristics, and Poor Prognosis in Esophageal Adenocarcinoma. Cancers 2023, 15, 1408. [Google Scholar] [CrossRef] [PubMed]
- Fiocca, R.; Mastracci, L.; Lugaresi, M.; Grillo, F.; D’Errico, A.; Malvi, D.; Spaggiari, P.; Tomezzoli, A.; Albarello, L.; Ristimäki, A.; et al. The Prognostic Impact of Histology in Esophageal and Esophago-Gastric Junction Adenocarcinoma. Cancers 2021, 13, 5211. [Google Scholar] [CrossRef] [PubMed]
- Dahlberg, P.S.; Jacobson, B.A.; Dahal, G.; Fink, J.M.; Kratzke, R.A.; Maddaus, M.A.; Ferrin, L.J. ERBB2 Amplifications in Esophageal Adenocarcinoma. Ann. Thorac. Surg. 2004, 78, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.; Lowe, S. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Schmitt, E.M.; Smitht, S.W.; Osbornet, B.A.; Jacks, T. pS3 Is Required for Radiation-Induced Apoptosis in Mouse Thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of P53 Protein in the Cellular Response to DNA Damage1. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [PubMed]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate Metabolism in Human Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Meehan, J.; Gray, M.E.; Murray, A.F.; Argyle, D.J.; Kunkler, I.H.; Langdon, S.P. The Impact of Tumour pH on Cancer Progression: Strategies for Clinical Intervention. Explor. Target. Anti-Tumor Ther. 2020, 1, 71–100. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [PubMed]
- Uhlenhopp, D.J.; Then, E.O.; Sunkara, T.; Gaduputi, V. Epidemiology of Esophageal Cancer: Update in Global Trends, Etiology and Risk Factors. Clin. J. Gastroenterol. 2020, 13, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Fortuno, C.; Cipponi, A.; Ballinger, M.L.; Tavtigian, S.V.; Olivier, M.; Ruparel, V.; Haupt, Y.; Haupt, S.; Study, I.S.K.; Tucker, K.; et al. A Quantitative Model to Predict Pathogenicity of Missense Variants in the TP53 Gene. Hum. Mutat. 2019, 40, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Hilsenbeck, S.; Gazitt, Y. Arsenic Trioxide–Induced Apoptosis in Myeloma Cells: P53-Dependent G1 or G2/M Cell Cycle Arrest, Activation of Caspase-8 or Caspase-9, and Synergy with APO2/TRAIL: Presented in Preliminary Form at the 43rd Annual Meeting of the American Society of Hematology, Orlando, FL, December 8, 2001.42. Blood 2003, 101, 4078–4087. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qu, X.; Qu, J.; Zhang, Y.; Liu, J.; Teng, Y.; Hu, X.; Hou, K.; Liu, Y. Arsenic Trioxide Induces Apoptosis and G2/M Phase Arrest by Inducing Cbl to Inhibit PI3K/Akt Signaling and Thereby Regulate P53 Activation. Cancer Lett. 2009, 284, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Frey, P.A. The Leloir Pathway: A Mechanistic Imperative for Three Enzymes to Change the Stereochemical Configuration of a Single Carbon in Galactose. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 461–470. [Google Scholar] [CrossRef]
- Dott, W.; Mistry, P.; Wright, J.; Cain, K.; Herbert, K.E. Modulation of Mitochondrial Bioenergetics in a Skeletal Muscle Cell Line Model of Mitochondrial Toxicity. Redox Biol. 2014, 2, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Gohil, V.M.; Sheth, S.A.; Nilsson, R.; Wojtovich, A.P.; Lee, J.H.; Perocchi, F.; Chen, W.; Clish, C.B.; Ayata, C.; Brookes, P.S.; et al. Nutrient-Sensitized Screening for Drugs That Shift Energy Metabolism from Mitochondrial Respiration to Glycolysis. Nat. Biotechnol. 2010, 28, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Byun, J.-K.; Choi, Y.-K.; Park, K.-G. Targeting Glutamine Metabolism as a Therapeutic Strategy for Cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Silwal-Pandit, L.; Vollan, H.K.M.; Chin, S.-F.; Rueda, O.M.; McKinney, S.; Osako, T.; Quigley, D.A.; Kristensen, V.N.; Aparicio, S.; Børresen-Dale, A.-L.; et al. TP53 Mutation Spectrum in Breast Cancer Is Subtype Specific and Has Distinct Prognostic Relevance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3569–3580. [Google Scholar] [CrossRef] [PubMed]
- Novotna, E.; Milosevic, M.; Prukova, D.; Magalhaes-Novais, S.; Dvorakova, S.; Dmytruk, K.; Gemperle, J.; Zudova, D.; Nickl, T.; Vrbacky, M.; et al. Mitochondrial HER2 Stimulates Respiration and Promotes Tumorigenicity. Eur. J. Clin. Investig. 2024, 54, e14174. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Lau, M.C.; Zhao, M.; Haruki, K.; Kosumi, K.; Mima, K.; Gu, M.; Väyrynen, J.P.; Twombly, T.S.; Baba, Y.; et al. Metabolic Profiling of Formalin-Fixed Paraffin-Embedded Tissues Discriminates Normal Colon from Colorectal Cancer. Mol. Cancer Res. MCR 2020, 18, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Santaliz-Casiano, A.; Mehta, D.; Danciu, O.C.; Patel, H.; Banks, L.; Zaidi, A.; Buckley, J.; Rauscher, G.H.; Schulte, L.; Weller, L.R.; et al. Identification of metabolic pathways contributing to ER+ breast cancer disparities using a machine-learning pipeline. Sci. Rep. 2023, 13, 12136. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.; Sarfaraz, M.O.; Farshidfar, F.; Bebb, D.G.; Lee, C.Y.; Card, C.M.; David, M.; Weljie, A.M. Temporal characterization of serum metabolite signatures in lung cancer patients undergoing treatment. Metabolomics 2016, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Boros, L.G.; D’Agostino, D.P.; Katz, H.E.; Roth, J.P.; Meuillet, E.J.; Somlyai, G. Submolecular regulation of cell transformation by deuterium depleting water exchange reactions in the tricarboxylic acid substrate cycle. Med. Hypotheses 2016, 87, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Boros, L.G.; Seneff, S.; Túri, M.; Palcsu, L.; Zubarev, R.A. Active involvement of compartmental, inter- and intramolecular deuterium disequilibrium in adaptive biology. Proc. Natl. Acad. Sci. USA 2024, 121, e2412390121. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.; Neugent, M.L.; Lee, S.Y.; Choe, J.H.; Choi, H.; Jenkins, D.M.R.; Ruthenborg, R.J.; Robinson, M.W.; Jeong, J.Y.; Wake, M.; et al. The Distinct Metabolic Phenotype of Lung Squamous Cell Carcinoma Defines Selective Vulnerability to Glycolytic Inhibition. Nat. Commun. 2017, 8, 15503. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zheng, M.; Yang, C.; Wang, K.; Lv, Z.; Liu, Z.; Tang, Z.; Chen, X. Azo-Based Hypoxic-Activated 6-Diazo-5-Oxo-L-Norleucine (DON) Prodrug Combined with Vascular Disrupting Agent Nanoparticles for Tumor-Selective Glutamine Metabolism Blockade. Chem. Eng. J. 2024, 481, 148281. [Google Scholar] [CrossRef]
- Yu, W.; Yang, X.; Zhang, Q.; Sun, L.; Yuan, S.; Xin, Y. Targeting GLS1 to Cancer Therapy through Glutamine Metabolism. Clin. Transl. Oncol. 2021, 23, 2253–2268. [Google Scholar] [CrossRef] [PubMed]
- Hoang, G.; Nguyen, K.; Le, A. Metabolic Intersection of Cancer and Cardiovascular Diseases: Opportunities for Cancer Therapy. Adv. Exp. Med. Biol. 2021, 1311, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Pei, J.; Xu, S.; Liu, J.; Yu, J. A glutamine tug-of-war between cancer and immune cells: Recent advances in unraveling the ongoing battle. J. Exp. Clin. Cancer Res. 2024, 43, 74. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Campos-Sandoval, J.A.; Santos-Jiménez, J.d.L.; Márquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I.; Mkrtchyan, G.; Grabarska, A.; Oppermann, H.; Daloso, D.; Araujo, W.L.; Juszczak, M.; Rzeski, W.; Bettendorff, L.; Fernie, A.R.; et al. Inhibition of Mitochondrial 2-Oxoglutarate Dehydrogenase Impairs Viability of Cancer Cells in a Cell-Specific Metabolism-Dependent Manner. Oncotarget 2016, 7, 26400–26421. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, B.P.; Nazlamova, L.A.; Tse, C.; Johnston, D.A.; Thomas, J.; Blyth, R.; Pickering, O.J.; Grace, B.; Harrington, J.; Rajak, R.; et al. Patient-Derived Tumor Organoid and Fibroblast Assembloid Models for Interrogation of the Tumor Microenvironment in Esophageal Adenocarcinoma. Cell Rep. Methods 2024, 4, 100909. [Google Scholar] [CrossRef] [PubMed]
- Diquigiovanni, C.; Rizzardi, N.; Kampmeier, A.; Liparulo, I.; Bianco, F.; De Nicolo, B.; Cataldi-Stagetti, E.; Cuna, E.; Severi, G.; Seri, M.; et al. Mutant SPART Causes Defects in Mitochondrial Protein Import and Bioenergetics Reversed by Coenzyme Q. Open Biol. 2023, 13, 230040. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.A.; Zámocká, M.; Majtán, J.; Bauerová-Hlinková, V. Glucose Oxidase, an Enzyme “Ferrari”: Its Structure, Function, Production and Properties in the Light of Various Industrial and Biotechnological Applications. Biomolecules 2022, 12, 472. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An Improvement of the 2−ΔΔCT Method for Quantitative Real-Time Polymerase Chain Reaction Data Analysis. Biostat. Bioinforma. Biomath. 2013, 3, 71–85. [Google Scholar] [PubMed]
- Liparulo, I.; Bergamini, C.; Bortolus, M.; Calonghi, N.; Gasparre, G.; Kurelac, I.; Masin, L.; Rizzardi, N.; Rugolo, M.; Wang, W.; et al. Coenzyme Q Biosynthesis Inhibition Induces HIF-1α Stabilization and Metabolic Switch toward Glycolysis. FEBS J. 2021, 288, 1956–1974. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Determination of Pyridine Dinucleotides in Cell Extracts by High-Performance Liquid Chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1981, 225, 446–449. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Mutation | Variant ID | Position (GRCh38) | CADD | MAF | IARC Classification | Functional Impact |
|---|---|---|---|---|---|---|---|
| OE33 | TP53 c.404 G>A, p.Cys135Tyr | rs587781991 | chr17:7675208 | 28.2 | 1.2 × 10−6 | Damaging | DNE-LOF |
| OE19 | TP53 c.929dupA, p.Asn310Lysfs*27 | n.a. | chr17:7673598 | n.a. | n.a. | n.a. | n.a. |
| FLO1 | TP53 c.830 G>T, p.Cys277Phe | rs763098116 | chr17:7673790 | 27.1 | 6.2 × 10−7 | Damaging | DNE-LOF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cataldi-Stagetti, E.; Rizzardi, N.; Orsini, A.; De Nicolo, B.; Diquigiovanni, C.; Pincigher, L.; Moruzzi, N.; Fato, R.; Bergamini, C.; Bonora, E. Metabolic Profiling of Distinct TP53-Mutant Esophageal Adenocarcinoma Models Reveals Different Bioenergetic Dependencies. Int. J. Mol. Sci. 2025, 26, 6869. https://doi.org/10.3390/ijms26146869
Cataldi-Stagetti E, Rizzardi N, Orsini A, De Nicolo B, Diquigiovanni C, Pincigher L, Moruzzi N, Fato R, Bergamini C, Bonora E. Metabolic Profiling of Distinct TP53-Mutant Esophageal Adenocarcinoma Models Reveals Different Bioenergetic Dependencies. International Journal of Molecular Sciences. 2025; 26(14):6869. https://doi.org/10.3390/ijms26146869
Chicago/Turabian StyleCataldi-Stagetti, Erica, Nicola Rizzardi, Arianna Orsini, Bianca De Nicolo, Chiara Diquigiovanni, Luca Pincigher, Noah Moruzzi, Romana Fato, Christian Bergamini, and Elena Bonora. 2025. "Metabolic Profiling of Distinct TP53-Mutant Esophageal Adenocarcinoma Models Reveals Different Bioenergetic Dependencies" International Journal of Molecular Sciences 26, no. 14: 6869. https://doi.org/10.3390/ijms26146869
APA StyleCataldi-Stagetti, E., Rizzardi, N., Orsini, A., De Nicolo, B., Diquigiovanni, C., Pincigher, L., Moruzzi, N., Fato, R., Bergamini, C., & Bonora, E. (2025). Metabolic Profiling of Distinct TP53-Mutant Esophageal Adenocarcinoma Models Reveals Different Bioenergetic Dependencies. International Journal of Molecular Sciences, 26(14), 6869. https://doi.org/10.3390/ijms26146869