
Gut Microbial Postbiotics as Potential Therapeutics for Lymphoma: Proteomics Insights of the Synergistic Effects of Nisin and Urolithin B Against Human Lymphoma Cells
 ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
2.1. Antiproliferative Activity of the Seven Postbiotics Against the HKB-11 (BL) Human Cell Line
2.2. The Synergy of N with UB Against HKB-11 Lymphoma Cells
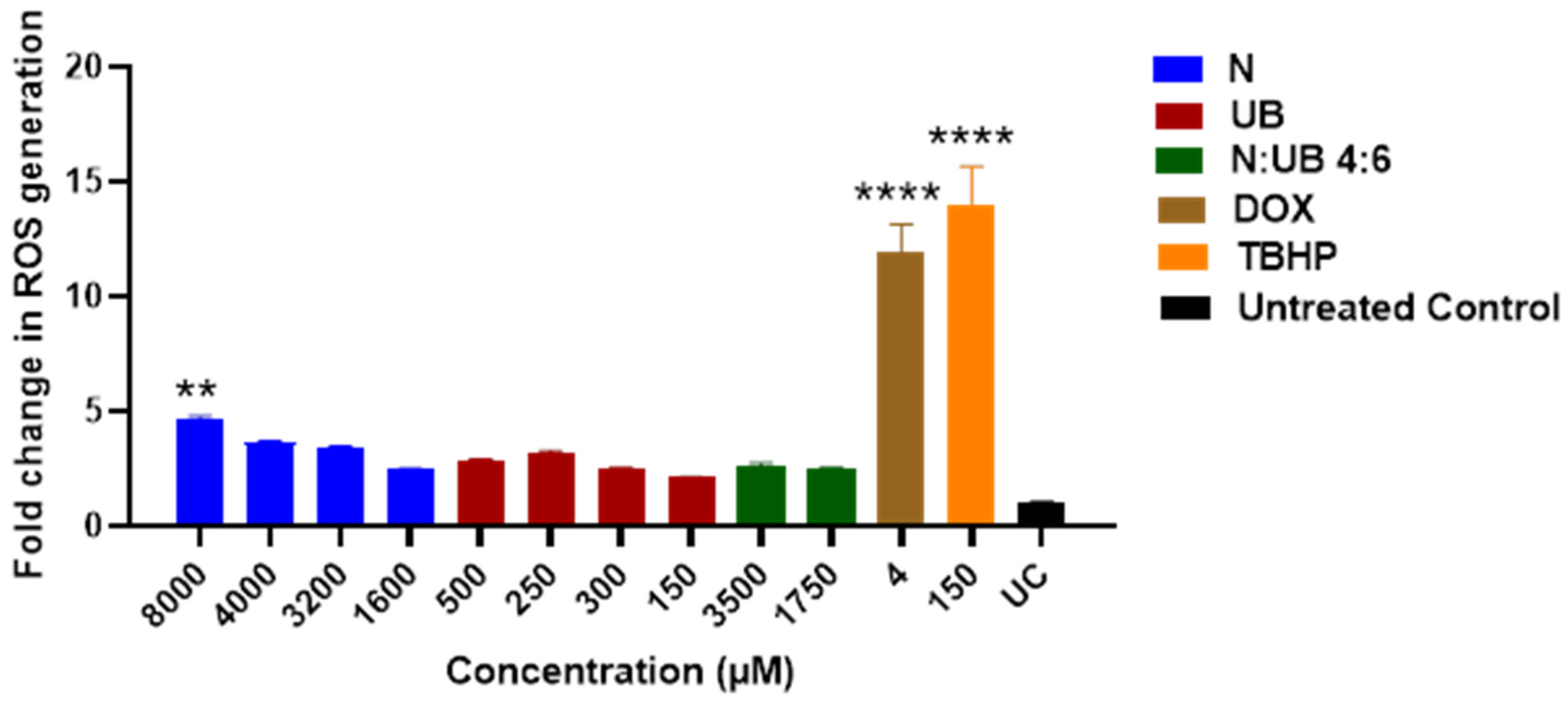
2.3. ROS Production in HKB-11 Lymphoma Cells After Treatment with Different Concentrations of N, UB, and N/UB (4:6)
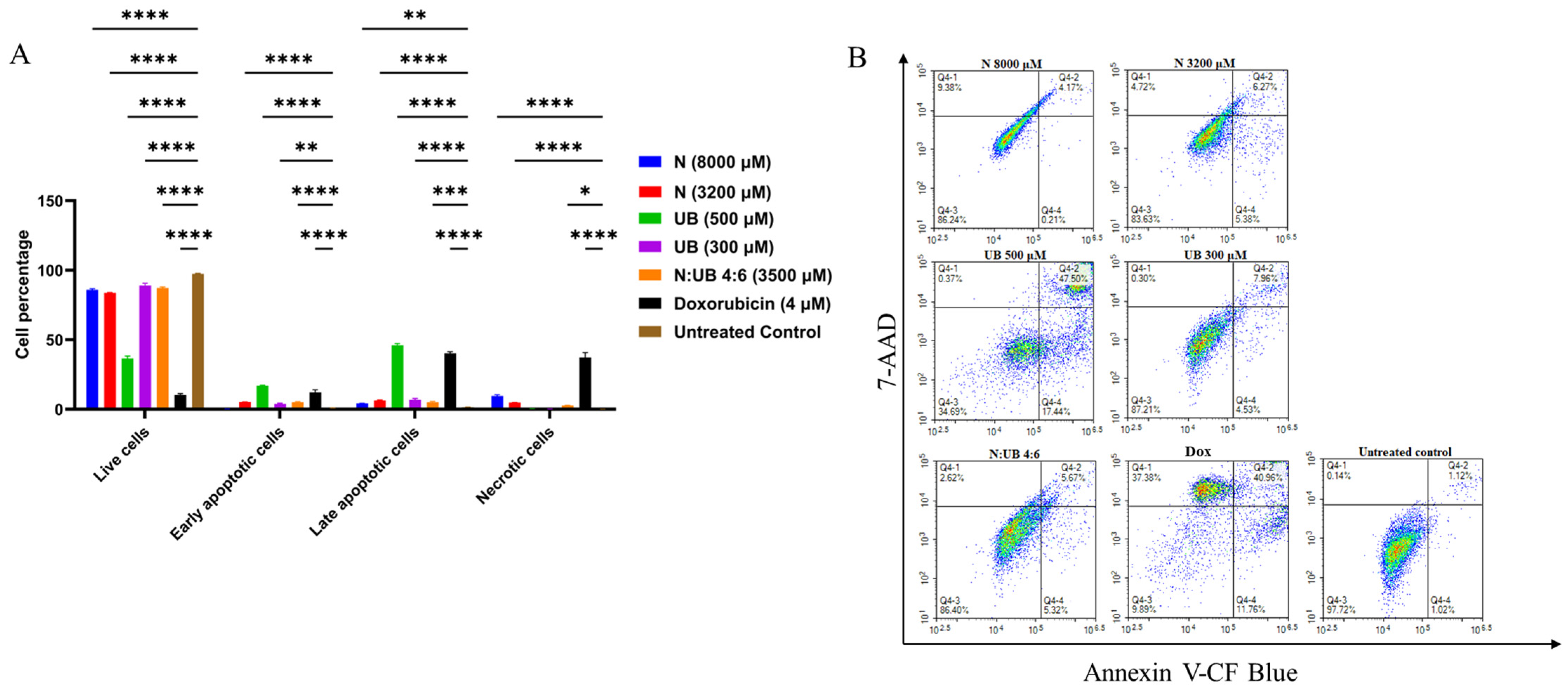
2.4. Flow Cytometric Analyses of Apoptotic Profiles of HKB-11 Lymphoma Cells After Treatment with Different Concentrations of N, UB, and N/UB (4:6)
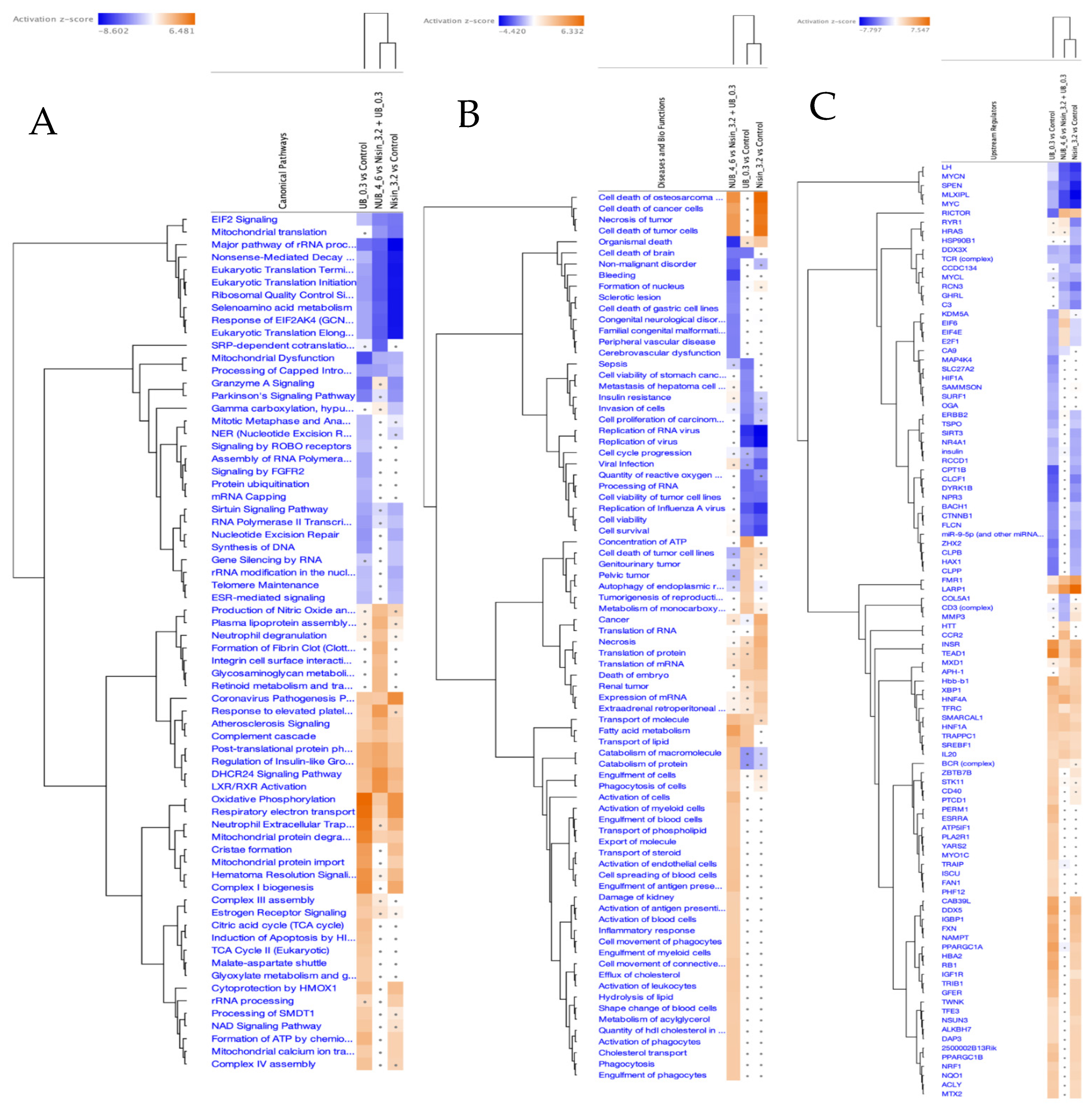
2.5. Proteomics Study of the HKB-11 Lymphoma Cells Treated with the Synergistic Combination vs. Monotreatments
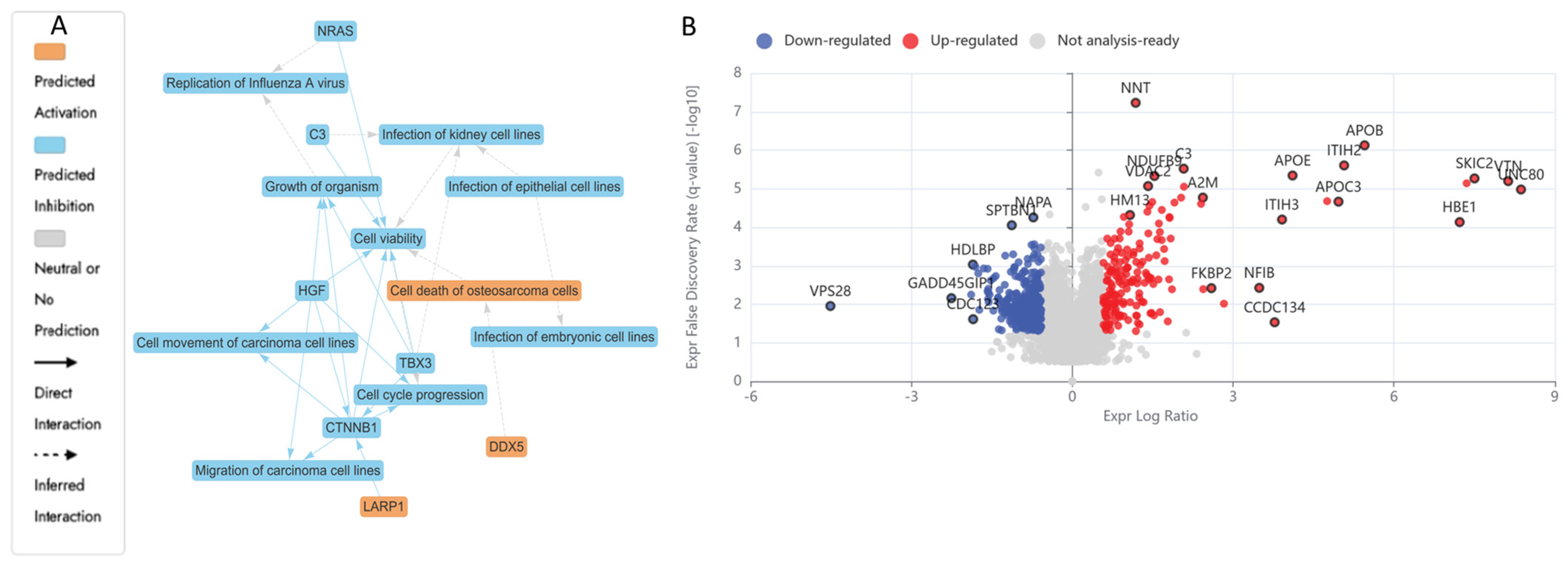
2.5.1. Differentially Expressed Proteins (DEPs) in N (3200 µM)-Treated HKB-11 Lymphoma Cells Compared to Untreated Control (Abs Log2FC ≥ 0.58 and Q ≤ 0.05)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Log2FC | Gene ID | Protein Descriptions | Molecular Pathway | Mechanism of Action | Reference |
|---|---|---|---|---|---|---|
| N 3200 μM | −1.27 | LARP1 | La-related protein 1 | mTOR signalling and RNA binding | Regulates mRNA stability and translation of survival genes | [80,89] |
| −1.54 | TYMS | Thymidylate synthase | Nucleotide synthesis and DNA replication | Catalyses thymidylate synthesis; target of 5-FU chemotherapy | [78,79] | |
| −0.80 | MAPK14 | Mitogen-activated protein kinase 14 | MAPK/p38 signalling pathway | Mediates cellular response to stress, inflammation, and proliferation | [81] | |
| −0.72 | PRC1 | Protein regulator of cytokinesis 1 | Cell cycle progression and mitosis | Regulates cytokinesis and mitotic spindle formation | [90] | |
| −0.70 | SRPK2 | SRSF protein kinase 2 | RNA splicing and nuclear mRNA processing | Works with SRPK1 in regulating alternative splicing | [91,92] | |
| −1.26 | CDK4 | Cyclin-dependent kinase 4 | Cell cycle (G1/S transition) | Phosphorylates RB1, promoting E2F release and progression through G1 phase | [82] | |
| −0.62 | POLR2E | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | Transcription (RNA Polymerase II complex) | Essential subunit for RNA Polymerase II assembly and mRNA transcription | [93] | |
| −0.63 | POLR2G | DNA-directed RNA Polymerase II subunit RPB7 | Transcription (RNA Polymerase II complex) | Structural component maintaining Polymerase II processivity | [94,95] | |
| −0.59 | POLR2H | DNA-directed RNA polymerases I, II, and III subunit RPABC3 | Transcription (RNA Polymerase II complex) | Stabilises RNA Pol II structure; shared across all RNA polymerases | [95,96] | |
| −0.85 | RFC1 | Replication factor C subunit 1 | DNA replication (clamp loader complex) | Loads PCNA onto DNA, facilitating DNA polymerase binding during replication | [97] | |
| −0.59 | RFC2 | Replication factor C subunit 2 | DNA replication and repair | Binds RFC1 to form RFC complex; essential for DNA synthesis fidelity | [83] | |
| −0.60 | NUP62 | Nuclear pore glycoprotein p62 | Nucleocytoplasmic transport (nuclear pore complex) | Central channel component; regulates import/export of macromolecules | [98] | |
| −0.93 | PPP1R14B | Protein phosphatase 1 regulatory subunit 14B | Actin cytoskeleton regulation | Inhibits protein phosphatase 1, affecting the cytoskeleton and cell motility | [99,100] | |
| −0.97 | CTNNA1 | Catenin alpha-1 | Cell adhesion (cadherin complex) | Links cadherins to actin cytoskeleton; maintains epithelial integrity | [101] | |
| −0.67 | HDAC1 | Histone deacetylase 1 | Epigenetic regulation (histone deacetylation) | Removes acetyl groups from histones, repressing transcription | [85] | |
| UB 300 μM | 2.04 | APOC3 | Apolipoprotein C-III | Triglyceride metabolism | Alters lipid signalling; enhances inflammatory microenvironment | [102] |
| 4.39 | C4BPA | C4b-binding protein alpha chain | Complement pathway | Inhibits complement-mediated lysis; immune evasion | [103] | |
| 3.06 | CLEC11A | C-type lectin domain family 11 member A | Cytokine signalling | Promotes endothelial and haematopoietic support in TME | [104] | |
| 2.07 | COX6C | Cytochrome c oxidase subunit 6C | Respiratory chain complex IV | Boosts mitochondrial respiration | [105] | |
| 2.08 | COX7C | Cytochrome c oxidase subunit 7C, mitochondrial | Cytochrome c oxidase | Increases mitochondrial adaptability in tumours | [106] | |
| 2.14 | FGB | Fibrinogen beta chain | Coagulation cascade | Promotes vascularisation and fibrin scaffolding in tumours | [107] | |
| 2.26 | GSN | Gelsolin | Actin regulation | Modulates the actin cytoskeleton for migration and invasion | [108] | |
| 5.94 | HBD | Haemoglobin subunit delta; Haemoglobin subunit beta | Haemoglobin complex | Facilitates oxygen delivery; modulates redox status | [109] | |
| 5.63 | HBE1 | Haemoglobin subunit epsilon; Haemoglobin subunit gam1; Haemoglobin subunit gamma-2 | Foetal haemoglobin pathway | Reactivation may aid hypoxic survival in tumours | [110] | |
| 2.76 | ITIH2 | Inter-alpha-trypsin inhibitor heavy chain H2 | Matrix stability | Regulates hyaluronic acid and ECM stiffness | [111] | |
| 2.34 | MT-CO2 | Cytochrome c oxidase subunit 2 | Mitochondrial respiration | Supports tumour ATP production and ROS balance | [112] | |
| 2.24 | MT-ND4 | NADH-ubiquinone oxidoreductase chain 4 | Complex I, OXPHOS | Enhances mitochondrial respiration and survival under stress | [113] | |
| 2.18 | NDUFA11 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 11 | OXPHOS complex I | Maintains mitochondrial metabolism in cancer cells | [114] | |
| 2.01 | SAMM50 | Sorting and assembly machinery component 50 homolog | Protein import | Preserves outer membrane; supports anti-apoptotic signals | [115] | |
| −1.45 | MAD2L1 | Mitotic spindle assembly checkpoint protein MAD2A | Mitotic checkpoint (spindle assembly checkpoint, SAC) | Ensures proper chromosome segregation, disrupts mitosis, causing mitotic arrest or apoptotic cell death | [116] | |
| −0.59 | PSME3 | Proteasome activator complex subunit 3 | Proteasome activation, p53 degradation | Degrades tumour suppressor proteins. Its suppression stabilises p53, enhancing apoptosis and cell cycle arrest | [117] | |
| −1.21 | UBE2S | Ubiquitin-conjugating enzyme E2 S | Ubiquitination, mitotic exit | Ubiquitinates mitotic inhibitors (APC/C complex co-activator); downregulation leads to mitotic arrest, promoting senescence | [118] | |
| −0.63 | PCNA | Proliferating cell nuclear antigen | DNA replication and repair | Sliding clamp for DNA polymerases; loss of PCNA function causes replication stress and apoptosis | [84] | |
| −1.69 | UBE2E1 | Ubiquitin-conjugating enzyme E2 E1; Ubiquitin-conjugating enzyme E2 E3; Ubiquitin-conjugating enzyme E2 E2 | Ubiquitin-conjugating enzyme | Supports proteostasis, DNA repair; suppression disrupts protein quality control, leading to cell death | [119] | |
| N/UB 4:6 (3200:300 μM) | 2.30 | A2M | Alpha-2-macroglobulin | Protease inhibition, complement cascade | Regulates proteolysis; potentially restricts tumour invasion | [120] |
| 0.67 | CDC27 | Cell division cycle protein 27 homolog | Mitotic checkpoint (Anaphase-Promoting Complex, APC/C) | Regulates ubiquitination of mitotic regulators | [121] | |
| 2.45 | AFP | Alpha-fetoprotein | Oncofoetal protein, MAPK signalling | Supports tumour proliferation, angiogenesis; marker in hepatic and haematological cancers | [122] | |
| 2.56 | AHSG | Alpha-2-HS-glycoprotein | TGF-β inhibition | Inhibits calcification and regulates inflammation in tumours | [123] | |
| 2.32 | ALB | Albumin | Plasma transport | High levels may reflect cancer cachexia or liver activity during tumour burden | [124] | |
| 6.55 | ALG6 | Dolichyl pyrophosphate Man9GlcNAc2 alpha-1,3-glucosyltransferase | N-glycosylation | Promotes ER glycoprotein processing; linked to tumour cell survival | [125] | |
| 2.64 | IGLL5 | Immunoglobulin lambda-like polypeptide 5 | B-cell development | Overexpressed in some B-cell lymphomas; immune receptor surrogate | [126] | |
| 2.36 | LCAT | Phosphatidylcholine-sterol acyltransferase | HDL metabolism | Alters cholesterol availability in tumours | [127] | |
| 2.46 | NNMT | Nicotinamide N-methyltransferase | Nicotinamide methylation | Reprograms NAD+ metabolism; supports proliferation | [128] | |
| 2.38 | SERPINA7 | Thyroxine-binding globulin | Thyroid hormone transport | Affects hormone signalling relevant to cancer cell growth | [129] | |
| 2.27 | SERPINF1 | Pigment epithelium-derived factor | Anti-angiogenic | Inhibits neovascularisation; tumour-suppressive in some contexts | [130] | |
| 3.41 | TF | Serotransferrin | Iron transport | Modulates iron availability and oxidative stress in tumour cells | [131] | |
| 1.78 | F10 | Coagulation factor X | Coagulation cascade | Activation of prothrombin to thrombin; can affect tumour vascularization | [132] | |
| 0.71 | FGB | Fibrinogen beta chain | Extracellular matrix (ECM) interaction | Participates in clot formation and tissue remodelling | [107] | |
| 0.65 | ITGB1 | Integrin beta-1 | Cell adhesion, survival signalling | Binds ECM; activates FAK, PI3K pathways | [133] | |
| 0.77 | RANGAP1 | Ran GTPase-activating protein 1 | Nuclear transport and cell cycle regulation | Controls Ran GTPase cycle; vital for nuclear envelope reformation during mitosis | [134] | |
| −0.75 | BUB3 | Mitotic checkpoint protein BUB3 | Spindle assembly checkpoint (SAC) | Ensures correct chromosomal segregation; loss promotes chromosomal instability but can also trigger catastrophic cell death in tumours | [135] | |
| −1.18 | CCNB1 | G2/mitotic-specific cyclin-B1 | Cell cycle control (G2/M checkpoint) | Complexes with CDK1 to trigger mitosis; downregulation leads to G2/M arrest and apoptosis | [136] | |
| −0.60 | CDCA8 | Borealin | Chromosome passenger complex (CPC) | Regulates mitosis and cytokinesis; loss disrupts chromosomal stability, causing mitotic catastrophe | [137] | |
| −0.66 | CDK1 | Cyclin-dependent kinase 1 | Master G2/M checkpoint kinase | Phosphorylates downstream mitotic proteins; inhibition causes G2/M phase arrest, senescence, or apoptosis | [138] | |
| −0.59 | MAP2K4 | Dual-specificity mitogen-activated protein kinase kinase 4 | JNK/p38 MAPK stress pathway | Activates pro-apoptotic MAPK cascades; blocks survival signals, sensitizing cells to apoptosis | [139,140] |
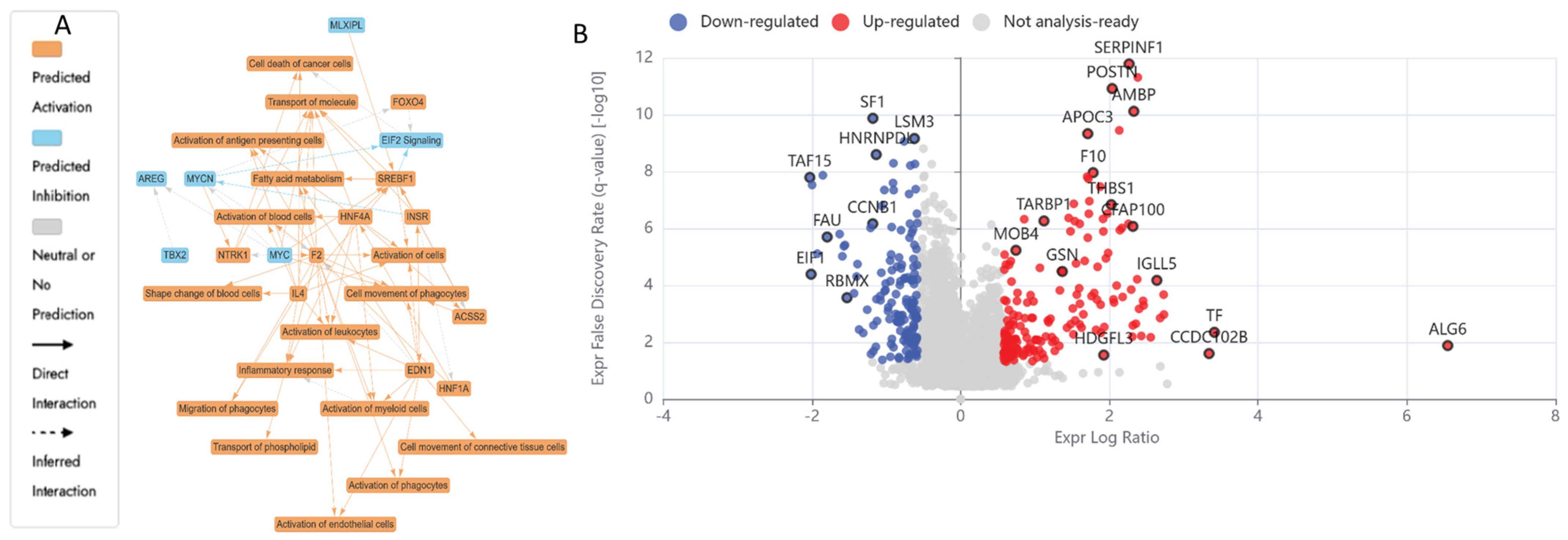
2.5.2. Differentially Expressed Proteins (DEPs) in UB (300 µM)-Treated HKB-11 Lymphoma Cells Compared to Untreated Control (Abs Log2FC ≥ 0.58 and Q ≤ 0.05)
2.5.3. Differentially Expressed Proteins (DEPs) in Combo N/UB (4:6)-Treated HBK-11 Cells vs. Monotreatments (N 3200 µM and UB 300 µM) (Abs Log2FC ≥ 0.58 and Q ≤ 0.05)
3. Materials and Methods
3.1. Chemicals and Drug Preparation
3.2. Cell Culture
3.3. Cell Viability Assays
3.4. Synergy Analysis
3.5. Analysis of ROS Production
3.6. Flow Cytometry Analyses of the Apoptotic Profiles
3.7. Liquid Chromatography–Mass Spectrometry (LC–MS)-Driven Bottom-Up Proteomics Analysis
3.7.1. Cell Culture, Treatment, and Protein Extraction
3.7.2. Protein Quantification
3.7.3. Peptide Preparation and Clean-Up
3.7.4. Label-Free Quantitative Proteomics Using Micro High-Performance Liquid Chromatography Coupled with Quadrupole Time-of-Flight Mass Spectrometry (Micro-HPLC-QTOF-MS)
Liquid Chromatography and Mass Spectrometry Setup
Mass Spectrometry Acquisition Parameters
Mass Calibration and Library Generation
Data Processing and Statistical Analysis
Data Availability
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; Cancer Tomorrow 2021; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Ansell, S.M. Non-Hodgkin lymphoma: Diagnosis and treatment. Mayo Clin. Proc. 2015, 90, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood J. Am. Soc. Hematol. 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Costa e Silva, L.; dos Santos, M.H.; Heluy, T.R.; Biaggio, R.T.; Ferreira, A.R.; Milhomens, J.; Kashima, S.; El Nemer, W.; El Hoss, S.; Picanço-Castro, V. Serum-free suspension cultured human cells can produce a high-level of recombinant human erythropoietin. Eng. Rep. 2020, 2, e12172. [Google Scholar] [CrossRef]
- Mei, B.; Chen, Y.; Chen, J.; Pan, C.Q.; Murphy, J.E. Expression of human coagulation factor VIII in a human hybrid cell line, HKB11. Mol. Biotechnol. 2006, 34, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Yee, H.; Chan, S. Establishment of a human somatic hybrid cell line for recombinant protein production. J. Biomed. Sci. 2002, 9, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Kho, Z.Y.; Lal, S.K. The human gut microbiome—A potential controller of wellness and disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Daillère, R.; Roberti, M.P.; Routy, B.; Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 2017, 15, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dwidar, M.; Nemet, I.; Buffa, J.A.; Sangwan, N.; Li, X.S.; Anderson, J.T.; Romano, K.A.; Fu, X.; Funabashi, M. Two distinct gut microbial pathways contribute to meta-organismal production of phenylacetylglutamine with links to cardiovascular disease. Cell Host Microbe 2023, 31, 18–32.e9. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zeevi, D.; Levy, M.; Segal, E.; Elinav, E. A day in the life of the meta-organism: Diurnal rhythms of the intestinal microbiome and its host. Gut Microbes 2015, 6, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Rafique, N.; Jan, S.Y.; Dar, A.H.; Dash, K.K.; Sarkar, A.; Shams, R.; Pandey, V.K.; Khan, S.A.; Amin, Q.A.; Hussain, S.Z. Promising bioactivities of postbiotics: A comprehensive review. J. Agric. Food Res. 2023, 14, 100708. [Google Scholar] [CrossRef]
- Peluzio, M.d.C.G.; Martinez, J.A.; Milagro, F.I. Postbiotics: Metabolites and mechanisms involved in microbiota-host interactions. Trends Food Sci. Technol. 2021, 108, 11–26. [Google Scholar] [CrossRef]
- O’keefe, S.J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Lewies, A.; Du Plessis, L.H.; Wentzel, J.F. The cytotoxic, antimicrobial and anticancer properties of the antimicrobial peptide nisin Z alone and in combination with conventional treatments. Cytotoxicity 2018, 25, 21. [Google Scholar]
- Niamah, A.K.; Al-Sahlany, S.T.G.; Verma, D.K.; Shukla, R.M.; Patel, A.R.; Tripathy, S.; Singh, S.; Baranwal, D.; Singh, A.K.; Utama, G.L. Emerging lactic acid bacteria bacteriocins as anti-cancer and anti-tumor agents for human health. Heliyon 2024, 10, e37054. [Google Scholar] [CrossRef] [PubMed]
- Al-Khazaleh, A.K.; Chang, D.; Münch, G.W.; Bhuyan, D.J. The Gut Connection: Exploring the Possibility of Implementing Gut Microbial Metabolites in Lymphoma Treatment. Cancers 2024, 16, 1464. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, G.R.; Antony, P.J.; Ceasar, S.A.; Vasconcelos, A.B.S.; Montalvão, M.M.; Farias de Franca, M.N.; Resende, A.d.S.; Sharanya, C.S.; Liu, Y.; Hariharan, G. Health functions and related molecular mechanisms of ellagitannin-derived urolithins. Crit. Rev. Food Sci. Nutr. 2024, 64, 280–310. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.S.; Shetty, S.; Kumari, N.S. Therapeutic efficacy of gut microbiota-derived polyphenol metabolite Urolithin A. Beni-Suef Univ. J. Basic Appl. Sci. 2024, 13, 31. [Google Scholar] [CrossRef]
- Hasheminezhad, S.H.; Boozari, M.; Iranshahi, M.; Yazarlu, O.; Sahebkar, A.; Hasanpour, M.; Iranshahy, M. A mechanistic insight into the biological activities of urolithins as gut microbial metabolites of ellagitannins. Phytother. Res. 2022, 36, 112–146. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.I.; Kapila, R. Dietary metabolites derived from gut microbiota: Critical modulators of epigenetic changes in mammals. Nutr. Rev. 2017, 75, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Lian, Y.; Zhao, C.; Du, H.; Han, Y.; Gao, W.; Xiao, H.; Zheng, J. Dietary fibers from fruits and vegetables and their health benefits via modulation of gut microbiota. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1514–1532. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.K.; Macia, L.; Mackay, C.R. Dietary fiber and SCFAs in the regulation of mucosal immunity. J. Allergy Clin. Immunol. 2023, 151, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Eladwy, R.A.; Alsherbiny, M.A.; Chang, D.; Fares, M.; Li, C.-G.; Bhuyan, D.J. The postbiotic sodium butyrate synergizes the antiproliferative effects of dexamethasone against the AGS gastric adenocarcinoma cells. Front. Nutr. 2024, 11, 1372982. [Google Scholar] [CrossRef] [PubMed]
- Jaye, K.; Alsherbiny, M.A.; Chang, D.; Li, C.-G.; Bhuyan, D.J. Mechanistic insights into the anti-proliferative action of gut microbial metabolites against breast adenocarcinoma cells. Int. J. Mol. Sci. 2023, 24, 15053. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Su, H.; Zhou, B.; Liu, S. The function of natural compounds in important anticancer mechanisms. Front. Oncol. 2023, 12, 1049888. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Li, S.; Wang, L.; Han, M.; Fan, H.; Tang, H.; Peng, C.; Du, J.; Zhou, Z. Combination of Hdac Inhibitor Sodium Butyrate and Immunotherapy in Glioma: Regulation of Immunologically Hot and Cold Tumors via Gut Microbiota and Metabolites. Available online: https://ssrn.com/abstract=4942877 (accessed on 21 May 2025).
- Steliou, K.; Boosalis, M.S.; Perrine, S.P.; Sangerman, J.; Faller, D.V. Butyrate histone deacetylase inhibitors. BioResearch Open Access 2012, 1, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Berni Canani, R.; Di Costanzo, M.; Leone, L. The epigenetic effects of butyrate: Potential therapeutic implications for clinical practice. Clin. Epigenetics 2012, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, L.; Su, P.; Yu, T.; Ma, Z.; Liu, Y.; Yu, J. Urolithin A suppresses tumor progression and induces autophagy in gastric cancer via the PI3K/Akt/mTOR pathway. Drug Dev. Res. 2023, 84, 172–184. [Google Scholar] [CrossRef] [PubMed]
- El-Wetidy, M.S.; Ahmad, R.; Rady, I.; Helal, H.; Rady, M.I.; Vaali-Mohammed, M.-A.; Al-Khayal, K.; Traiki, T.B.; Abdulla, M.-H. Urolithin A induces cell cycle arrest and apoptosis by inhibiting Bcl-2, increasing p53-p21 proteins and reactive oxygen species production in colorectal cancer cells. Cell Stress Chaperones 2021, 26, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Gharsallaoui, A.; Oulahal, N.; Joly, C.; Degraeve, P. Nisin as a Food Preservative: Part 1: Physicochemical Properties, Antimicrobial Activity, and Main Uses. Crit. Rev. Food Sci. Nutr. 2016, 56, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS); Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, P.; Gundert-Remy, U.; et al. Extension of use of lycopene (E 160d) to certain meat preparations, meat products and fruit and vegetable preparations. EFSA J. 2017, 15, e05064. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.M.; Gwak, J.W.; Kamarajan, P.; Fenno, J.C.; Rickard, A.H.; Kapila, Y.L. Biomedical applications of nisin. J. Appl. Microbiol. 2016, 120, 1449–1465. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.Q.; Guerra-Librero, A.; Fernandez-Gil, B.I.; Florido, J.; García-López, S.; Martinez-Ruiz, L.; Mendivil-Perez, M.; Soto-Mercado, V.; Acuña-Castroviejo, D.; Ortega-Arellano, H. Combination of melatonin and rapamycin for head and neck cancer therapy: Suppression of AKT/mTOR pathway activation, and activation of mitophagy and apoptosis via mitochondrial function regulation. J. Pineal Res. 2018, 64, e12461. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.S.T.S.; Abdulrahman, Z.F.A.; Othman, R.T. Anticancer activity of cloned Nisin as an alternative therapy for MCF-7 brest cancer cell line. Cell. Mol. Biol. 2023, 69, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Preet, S.; Pandey, S.; Saini, N.; Koul, A.; Rishi, P. Nisin augments doxorubicin permeabilization and ameliorates signaling cascade during skin carcinogenesis. Transl. Med. 2015, 6, 1000161. [Google Scholar] [CrossRef]
- Stanisławska, I.; Piwowarski, J.; Granica, S.; Kiss, A. Urolithins, gut microbiota metabolites of ellagitannins, in prostate cancer chemoprevention. Planta Medica Int. Open 2017, 4, Mo-PO-16. [Google Scholar]
- Djedjibegovic, J.; Marjanovic, A.; Panieri, E.; Saso, L. Ellagic acid-derived urolithins as modulators of oxidative stress. Oxidative Med. Cell. Longev. 2020, 2020, 5194508. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Y.; De Zoete, M.R.; Van Putten, J.P.; Strijbis, K. Redirection of epithelial immune responses by short-chain fatty acids through inhibition of histone deacetylases. Front. Immunol. 2015, 6, 554. [Google Scholar] [CrossRef] [PubMed]
- Conaty, L.D. The Effects of Sodium Butyrate on Hox Gene Expression in a Human Colorectal Adenocarcinoma Cell Line, HT29. Master’s Thesis, East Carolina University, Greenville, NC, USA, 2013. [Google Scholar]
- Riester, D.; Hildmann, C.; Schwienhorst, A. Histone deacetylase inhibitors—Turning epigenic mechanisms of gene regulation into tools of therapeutic intervention in malignant and other diseases. Appl. Microbiol. Biotechnol. 2007, 75, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-J.; Huang, C.; Meng, X.-M.; Li, J. Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie 2015, 116, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Target for cancer therapy: Proliferating cells or stem cells. Leukemia 2006, 20, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, H. Biomarkers and targeted therapy for cancer stem cells. Trends Pharmacol. Sci. 2024, 45, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Du, F.-Y.; Zhou, Q.-F.; Sun, W.-J.; Chen, G.-L. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J. Stem Cells 2019, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. Chapter 74—Origin of Hodgkin Lymphoma. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1204–1211. [Google Scholar]
- Lombardi, V.R.; Carrera, I.; Cacabelos, R. In vitro and in vivo cytotoxic effect of AntiGan against tumor cells. Exp. Ther. Med. 2018, 15, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.; Ryan, A.; Hudson, S.P. Synergistic antimicrobial interactions of nisin A with biopolymers and solubilising agents for oral drug delivery. Eur. J. Pharm. Biopharm. 2022, 171, 29–38. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Giménez-Bastida, J.A.; Cortés-Martín, A.; Ávila-Gálvez, M.Á.; Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C.; González-Sarrías, A. Urolithins: A comprehensive update on their metabolism, bioactivity, and associated gut microbiota. Mol. Nutr. Food Res. 2022, 66, 2101019. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem.-Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Jäättelä, M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2001, 2, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- D’arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, P.; Kearney, C.J.; Martin, S.J. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol. Chem. 2014, 395, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Sheibani, M.; Azizi, Y.; Shayan, M.; Nezamoleslami, S.; Eslami, F.; Farjoo, M.H.; Dehpour, A.R. Doxorubicin-induced cardiotoxicity: An overview on pre-clinical therapeutic approaches. Cardiovasc. Toxicol. 2022, 22, 292–310. [Google Scholar] [CrossRef] [PubMed]
- Kuszczak, B.; Łacina, P.; Dratwa-Kuźmin, M.; Bogunia-Kubik, K.; Wróbel, T.; Rybka, J. Evaluation of polymorphism in BCL-2, PD-1, and PD-L1 genes in myelodysplastic neoplasms. Acta Haematol. Pol. 2025, 56, 222–230. [Google Scholar] [CrossRef]
- Radaic, A.; Malone, E.; Kamarajan, P.; Kapila, Y.L. Solid lipid nanoparticles loaded with nisin (sln-nisin) are more effective than free nisin as antimicrobial, antibiofilm, and anticancer agents. J. Biomed. Nanotechnol. 2022, 18, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Joo, N.E.; Ritchie, K.; Kamarajan, P.; Miao, D.; Kapila, Y.L. Nisin, an apoptogenic bacteriocin and food preservative, attenuates HNSCC tumorigenesis via CHAC1. Cancer Med. 2012, 1, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liu, Z.; Zhou, Y.; Hou, N.; Yan, W.; Qin, Y.; Ye, Q.; Cheng, X.; Xiao, Q.; Bao, Y. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol. Res. 2020, 153, 104655. [Google Scholar] [CrossRef] [PubMed]
- El-Wetidy, M.S.; Helal, H.; Rady, M.I. Urolithin A Targets Both PI3K/p-AKT/mTOR and pc-RAF/MEK/p-ERK Signaling Pathways in Colorectal Cancer. Egypt. Acad. J. Biol. Sci. C Physiol. Mol. Biol. 2024, 16, 387–401. [Google Scholar]
- Sadri, H.; Aghaei, M.; Akbari, V. Nisin induces apoptosis in cervical cancer cells via reactive oxygen species generation and mitochondrial membrane potential changes. Biochem. Cell Biol. 2022, 100, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K. Mitophagy, Diseases, and Aging. In Models, Molecules and Mechanisms in Biogerontology: Physiological Abnormalities, Diseases and Interventions; Springer: Berlin/Heidelberg, Germany, 2019; pp. 177–191. [Google Scholar]
- Wang, C.; Wu, Y.; Gong, B.; Mou, J.; Cheng, X.; Zhang, L.; Wei, J. Agarwood Chromone Alleviates Gastric Ulcers by Inhibiting the NF-κB and Caspase Pathways Based on Network Pharmacology and Molecular Docking. Pharmaceuticals 2025, 18, 514. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Mazzara, S.; Salemi, D.; Zanetti, S.; Sapienza, M.R.; Orecchioni, S.; Talarico, G.; Falvo, P.; Davini, A.; Ceccarelli, C. Downregulation of rRNA synthesis by BCL-2 induces chemoresistance in Diffuse Large B-cell Lymphoma. iScience 2025, 28, 112333. [Google Scholar] [CrossRef] [PubMed]
- Szilveszter, R.-M.; Muntean, M.; Florea, A. Molecular Mechanisms in Tumorigenesis of Hepatocellular Carcinoma and in Target Treatments—An Overview. Biomolecules 2024, 14, 656. [Google Scholar] [CrossRef] [PubMed]
- Santos, O.F.; Moura, L.A.; Rosen, E.M.; Nigam, S.K. Modulation of HGF-induced tubulogenesis and branching by multiple phosphorylation mechanisms. Dev. Biol. 1993, 159, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.F.; Damerell, V.; Omar, R.; Du Toit, M.; Khan, M.; Maranyane, H.M.; Mlaza, M.; Bleloch, J.; Bellis, C.; Sahm, B.D. The roles and regulation of TBX3 in development and disease. Gene 2020, 726, 144223. [Google Scholar] [CrossRef] [PubMed]
- Matuszyk, J. MALAT1-miRNAs network regulate thymidylate synthase and affect 5FU-based chemotherapy. Mol. Med. 2022, 28, 89. [Google Scholar] [CrossRef] [PubMed]
- Peplow, P.V. Reprogramming T cells as an emerging treatment to slow human age-related decline in health. Front. Med. Technol. 2024, 6, 1384648. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Hopkins, T.G.; Michael, T.; Abd-Latip, N.; Weir, J.; Aboagye, E.; Mauri, F.; Jameson, C.; Sturge, J.; Gabra, H. LARP1 post-transcriptionally regulates mTOR and contributes to cancer progression. Oncogene 2015, 34, 5025–5036. [Google Scholar] [CrossRef] [PubMed]
- García-Hernández, L.; García-Ortega, M.B.; Ruiz-Alcalá, G.; Carrillo, E.; Marchal, J.A.; García, M.Á. The p38 MAPK components and modulators as biomarkers and molecular targets in cancer. Int. J. Mol. Sci. 2021, 23, 370. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Orr Brent, A.; Toprak Umut, H.; Hovestadt, V.; Jones David, T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott Paul, A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.L.G.; Gomes, X.V.; Burgers, P.M. ATP utilization by yeast replication factor C: III. The ATP-binding domains of Rfc2, Rfc3, and Rfc4 are essential for DNA recognition and clamp loading. J. Biol. Chem. 2001, 276, 34784–34791. [Google Scholar] [CrossRef] [PubMed]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2011, 107, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Gallinari, P.; Marco, S.D.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Dey, A.; Nandi, S.; Majumder, R.; Das, S.; Mandal, M. CTGF (CCN2): A multifaceted mediator in breast cancer progression and therapeutic targeting. Cancer Metastasis Rev. 2025, 44, 32. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-H.; Hsu, T.-I.; Chang, Y.-C.; Chan, M.-H.; Lu, P.-J.; Hsiao, M. Stationed or relocating: The seesawing emt/met determinants from embryonic development to cancer metastasis. Biomedicines 2021, 9, 1265. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Regulation of cell cycle progression by growth factor-induced cell signaling. Cells 2021, 10, 3327. [Google Scholar] [CrossRef] [PubMed]
- Oquendo, C.J.; Parker, H.; Oscier, D.; Ennis, S.; Gibson, J.; Strefford, J.C. The (epi)genomic landscape of splenic marginal zone lymphoma, biological implications, clinical utility, and future questions. J. Transl. Genet. Genom. 2021, 5, 89–111. [Google Scholar] [CrossRef]
- Li, J.; Dallmayer, M.; Kirchner, T.; Musa, J.; Grünewald, T.G. PRC1: Linking cytokinesis, chromosomal instability, and cancer evolution. Trends Cancer 2018, 4, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Gout, S.; Brambilla, E.; Boudria, A.; Drissi, R.; Lantuejoul, S.; Gazzeri, S.; Eymin, B. Abnormal expression of the pre-mRNA splicing regulators SRSF1, SRSF2, SRPK1 and SRPK2 in non small cell lung carcinoma. PLoS ONE 2012, 7, e46539. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, V.; Pereira, J.F.; Jordan, P. Signaling pathways driving aberrant splicing in cancer cells. Genes. 2017, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, H.; Hanada, A.; Kuzuyama, T.; Takagi, M.; Kamiya, Y.; Yamaguchi, S. Contribution of the Mevalonate and Methylerythritol Phosphate Pathways to the Biosynthesis of Gibberellins in Arabidopsis. J. Biol. Chem. 2002, 277, 45188–45194. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P. Structure and function of RNA polymerase II. Adv. Protein Chem. 2004, 67, 1–42. [Google Scholar] [PubMed]
- Osman, S.; Cramer, P. Structural biology of RNA polymerase II transcription: 20 years on. Annu. Rev. Cell Dev. Biol. 2020, 36, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Brandão, S.R.; Carvalho, F.; Amado, F.; Ferreira, R.; Costa, V.M. Insights on the molecular targets of cardiotoxicity induced by anticancer drugs: A systematic review based on proteomic findings. Metabolism 2022, 134, 155250. [Google Scholar] [CrossRef] [PubMed]
- Majka, J.; Burgers, P.M. The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid. Res. Mol. Biol. 2004, 78, 227–260. [Google Scholar] [PubMed]
- Bano, D.; Dinsdale, D.; Cabrera-Socorro, A.; Maida, S.; Lambacher, N.; McColl, B.; Ferrando-May, E.; Hengartner, M.; Nicotera, P. Alteration of the nuclear pore complex in Ca2+-mediated cell death. Cell Death Differ. 2010, 17, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhang, Y.-L.; Deng, L.; Chen, C.; Ma, X.-Y.; Andriani, L.; Yang, S.-Y.; Hu, S.-Y.; Zhang, F.-L.; Shao, Z.-M. Protein phosphatase 1 subunit PPP1R14B stabilizes STMN1 to promote progression and paclitaxel resistance in triple-negative breast cancer. Cancer Res. 2023, 83, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Ni, Y.; Zhang, A.; Chen, J. PPP1R14B as a potential biomarker for the identification of diagnosis and prognosis affecting tumor immunity, proliferation and migration in prostate cancer. J. Cancer 2024, 15, 6545–6564. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, H.; Xu, Y.; Li, C.; Lv, X.; Han, X.; Chen, X.; Chen, Y.; Yu, Z. The role of CTNNA1 in malignancies: An updated review. J. Cancer 2023, 14, 219. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ding, S.; Lu, G.; Lin, Z.; Liao, L.; Xiao, W.; Ding, Y.; Zhang, Y.; Wang, Z.; Gong, W. Apolipoprotein C-III itself stimulates the Syk/cPLA2-induced inflammasome activation of macrophage to boost anti-tumor activity of CD8+ T cell. Cancer Immunol. Immunother. 2023, 72, 4123–4144. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.M.; Villoutreix, B.O.; Dahlbäck, B. Complement inhibitor C4b-binding protein—Friend or foe in the innate immune system? Mol. Immunol. 2004, 40, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Wan, D.; Yu, Y.; Zhang, L. CLEC11A expression as a prognostic biomarker in correlation to immune cells of gastric cancer. Biomol. Biomed. 2024, 24, 101. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-E.; Mori, R.; Komatsu, T.; Chiba, T.; Hayashi, H.; Park, S.; Sugawa, M.D.; Dencher, N.A.; Shimokawa, I. Upregulation of cytochrome c oxidase subunit 6b1 (Cox6b1) and formation of mitochondrial supercomplexes: Implication of Cox6b1 in the effect of calorie restriction. Age 2015, 37, 9787. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Bonuccelli, G.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P. Vitamin C and Doxycycline: A synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget 2017, 8, 67269. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yu, X.; Chen, C.; Chen, C.; Wang, Y.; Su, D.; Zhu, L. Fibrinogen and tumors. Front. Oncol. 2024, 14, 1393599. [Google Scholar] [CrossRef] [PubMed]
- Alsofyani, A.A.; Nedjadi, T. Gelsolin, an actin-binding protein: Bioinformatic analysis and functional significance in urothelial bladder carcinoma. Int. J. Mol. Sci. 2023, 24, 15763. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.J. Redox and peroxidase activities of the hemoglobin superfamily: Relevance to health and disease. Antioxid. Redox Signal. 2017, 26, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Woodard, K.J. Genomic Characterization of Sickle Cell Mouse Models for Therapeutic Genome Editing Applications; The University of Tennessee Health Science Center: Memphis, TN, USA, 2021. [Google Scholar]
- Huth, S.; Huth, L.; Marquardt, Y.; Fietkau, K.; Dahl, E.; Esser, P.R.; Martin, S.F.; Heise, R.; Merk, H.F.; Baron, J.M. Inter-α-trypsin inhibitor heavy chain 5 (ITIH5) is a natural stabilizer of hyaluronan that modulates biological processes in the skin. Ski. Pharmacol. Physiol. 2020, 33, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Vikramdeo, K.S.; Sudan, S.K.; Singh, A.P.; Singh, S.; Dasgupta, S. Mitochondrial respiratory complexes: Significance in human mitochondrial disorders and cancers. J. Cell. Physiol. 2022, 237, 4049–4078. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-C.; Su, J.; Zhou, J.-J.; Yuan, Q.; Han, J.-S. Roles of MT-ND1 in Cancer. Curr. Med. Sci. 2023, 43, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Song, M.; Zeng, Z.-L.; Zhu, C.-F.; Lu, W.-H.; Yang, J.; Ma, M.-Z.; Huang, A.-M.; Hu, Y.; Huang, P. Identification of NDUFAF1 in mediating K-Ras induced mitochondrial dysfunction by a proteomic screening approach. Oncotarget 2015, 6, 3947. [Google Scholar] [CrossRef] [PubMed]
- Lefort, N.; Yi, Z.; Bowen, B.; Glancy, B.; De Filippis, E.A.; Mapes, R.; Hwang, H.; Flynn, C.R.; Willis, W.T.; Civitarese, A. Proteome profile of functional mitochondria from human skeletal muscle using one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. J. Proteom. 2009, 72, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.; Furlong, F.; Gallagher, M.F.; Spillane, C.D.; McCann, A.; O’Toole, S.; O’Leary, J.J. Too MAD or not MAD enough: The duplicitous role of the spindle assembly checkpoint protein MAD2 in cancer. Cancer Lett. 2020, 469, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Dong, X.; Jin, J.; He, Y. The expression patterns and prognostic value of the proteasome activator subunit gene family in gastric cancer based on integrated analysis. Front. Cell Dev. Biol. 2021, 9, 663001. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Mansfeld, J.; Godwin, C.; Matsusaka, T.; Wu, J.; Russell, P.; Pines, J.; Venkitaraman, A.R. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat. Cell Biol. 2009, 11, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Maffeo, B.; Cilloni, D. The ubiquitin-conjugating enzyme E2 O (UBE2O) and its therapeutic potential in human leukemias and solid tumors. Cancers 2024, 16, 3064. [Google Scholar] [CrossRef] [PubMed]
- Lagrange, J.; Lecompte, T.; Knopp, T.; Lacolley, P.; Regnault, V. Alpha-2-macroglobulin in hemostasis and thrombosis: An underestimated old double-edged sword. J. Thromb. Haemost. 2022, 20, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; Gorbsky, G.J. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Samban, S.S.; Hari, A.; Nair, B.; Kumar, A.R.; Meyer, B.S.; Valsan, A.; Vijayakurup, V.; Nath, L.R. An insight into the role of alpha-fetoprotein (AFP) in the development and progression of hepatocellular carcinoma. Mol. Biotechnol. 2024, 66, 2697–2709. [Google Scholar] [CrossRef] [PubMed]
- Odiase, P.; Ma, J.; Ranganathan, S.; Ogunkua, O.; Turner, W.B.; Marshall, D.; Ochieng, J. The Role of Fetuin-A in Tumor Cell Growth, Prognosis, and Dissemination. Int. J. Mol. Sci. 2024, 25, 12918. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-X.; Zhang, X.; Tang, M.; Zhang, Q.; Deng, L.; Song, C.-H.; Li, W.; Shi, H.-P.; Cong, M.-H. Comprehensive evaluation of serum hepatic proteins in predicting prognosis among cancer patients with cachexia: An observational cohort study. BMC Cancer 2024, 24, 293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhao, Y.C.; Liu, H.; Luo, S.; Amos, C.I.; Lee, J.E.; Li, X.; Nan, H.; Wei, Q. Novel genetic variants of ALG6 and GALNTL4 of the glycosylation pathway predict cutaneous melanoma-specific survival. Cancers 2020, 12, 288. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, H.; Tabata, S.; Kosako, H.; Hori, Y.; Okamura, T.; Yamashita, Y.; Fujimoto, K.; Kajioka, D.; Suzuki, K.; Osato, M. IGLL5 controlled by super-enhancer affects cell survival and MYC expression in mature B-cell lymphoma. Leuk. Res. Rep. 2024, 21, 100451. [Google Scholar] [CrossRef] [PubMed]
- D’Arca, D.; Severi, L.; Ferrari, S.; Dozza, L.; Marverti, G.; Magni, F.; Chinello, C.; Pagani, L.; Tagliazucchi, L.; Villani, M. Serum mass spectrometry proteomics and protein set identification in response to FOLFOX-4 in drug-resistant ovarian carcinoma. Cancers 2023, 15, 412. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-D.; Zhu, X.-J.; Li, J.-J.; Mei, Y.-Z.; Li, W.-S.; Li, J.-H. Nicotinamide N-methyltransferase (NNMT): A key enzyme in cancer metabolism and therapeutic target. Int. Immunopharmacol. 2024, 142, 113208. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Lima, C.J.; Maciel, A.A.F.L.; de Oliveira Andrade, M.; da Cunha, V.S.; Mazzeu, J.F.; Bleicher, L.; Neves, F.d.A.R.; Lofrano-Porto, A. Thyroxine-binding globulin deficiency due to a novel SERPINA7 mutation: Clinical characterization, analysis of X-chromosome inactivation pattern and protein structural modeling. Gene 2018, 666, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Pączek, S.; Zajkowska, M.; Mroczko, B. Pigment Epithelial-Derived Factor in Pancreatic and Liver Cancers—From Inflammation to Cancer. Biomedicines 2024, 12, 2260. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, P.; Duan, X.; Cheng, M.; Xu, L.X. Deferoxamine-induced high expression of TfR1 and DMT1 enhanced iron uptake in triple-negative breast cancer cells by activating IL-6/PI3K/AKT pathway. OncoTargets Ther. 2019, 12, 4359–4377. [Google Scholar] [CrossRef] [PubMed]
- Alexander, E.T.; Gilmour, S.K. Immunomodulatory role of thrombin in cancer progression. Mol. Carcinog. 2022, 61, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Mo, J.; Dong, S.; Liao, Z.; Zhang, B.; Zhu, P. Integrinβ-1 in disorders and cancers: Molecular mechanisms and therapeutic targets. Cell Commun. Signal. 2024, 22, 71. [Google Scholar] [CrossRef] [PubMed]
- Ciciarello, M.; Mangiacasale, R.; Lavia, P. Spatial control of mitosis by the GTPase Ran. Cell. Mol. Life Sci. 2007, 64, 1891–1914. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Sahoo, P.K.; Mahata, S.; Pal, R.; Ghosh, D.; Mistry, T.; Ghosh, S.; Bera, T.; Nasare, V.D. Mitotic checkpoint defects: En route to cancer and drug resistance. Chromosome Res. 2021, 29, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Role of FOXM1, GPX1, and CCNB1 in Cisplatin Resistance in Malignant Thymomas and Thymic Carcinomas. Ph.D. Thesis, Georg-August-Universität Göttingen, Göttingen, Germany, 2024. [Google Scholar]
- Zhang, C.; Zhao, L.; Leng, L.; Zhou, Q.; Zhang, S.; Gong, F.; Xie, P.; Lin, G. CDCA8 regulates meiotic spindle assembly and chromosome segregation during human oocyte meiosis. Gene 2020, 741, 144495. [Google Scholar] [CrossRef] [PubMed]
- Fatma, F.; Kumar, A. The Cell Cycle, Cyclins, Checkpoints and Cancer. Asian J. Res. Pharm. Sci. 2021, 11, 175–183. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, G.; Yang, Z.; Kang, X. Stress-Activated Protein Kinases in Intervertebral Disc Degeneration: Unraveling the Impact of JNK and p38 MAPK. Biomolecules 2024, 14, 393. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.-Y.; Zhang, J.; Zeng, F.-Y.; Zhu, Y.Z. Roles of the peroxisome proliferator-activated receptors (PPARs) in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Pharmacol. Res. 2023, 192, 106786. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bai, X.; Liu, G.; Li, X. A transcriptional regulatory network of HNF4α and HNF1α involved in human diseases and drug metabolism. Drug Metab. Rev. 2022, 54, 361–385. [Google Scholar] [CrossRef] [PubMed]
- Subbian, S.; Bandyopadhyay, N.; Tsenova, L.; O’Brien, P.; Khetani, V.; Kushner, N.L.; Peixoto, B.; Soteropoulos, P.; Bader, J.S.; Karakousis, P.C. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun. Signal. 2013, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Elsori, D.; Verma, M.; Pandey, S.; Obaidur Rab, S.; Siddiqui, S.; Alabdallah, N.M.; Saeed, M.; Pandey, P. Unraveling the intricate relationship between lipid metabolism and oncogenic signaling pathways. Front. Cell Dev. Biol. 2024, 12, 1399065. [Google Scholar] [CrossRef] [PubMed]
- Bionaz, M.; Thering, B.J.; Loor, J.J. Fine metabolic regulation in ruminants via nutrient–gene interactions: Saturated long-chain fatty acids increase expression of genes involved in lipid metabolism and immune response partly through PPAR-α activation. Br. J. Nutr. 2012, 107, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S. The Influence of Stromal Fibroblast on Antigen-Presenting Cell Function and Alteration of Their Biology. Doctoral Dissertation, University of Nottingham, Nottingham, UK, 2021. [Google Scholar]
- Parra-Medina, R.; Cardona, A.G.; Quintero-Ronderos, P.; Rodríguez, É.G. Cytokines, Chemokines and Growth Factors. In Cytokines, Chemokines and Growth Factors; El Rosario University Press: Bogota, Colombia, 2013. [Google Scholar]
- Errafii, K. Identification of the Long Non-Coding RNAs and the Signaling Pathways Involved in the Protective Effect of the Glucagon-Like Peptide-1 Receptor Agonist Exendin-4 on Hepatic Steatosis. Doctoral Dissertation, Hamad Bin Khalifa University (Qatar), Doha, Qatar, 2022. [Google Scholar]
- Alsherbiny, M.A.; Bhuyan, D.J.; Low, M.N.; Chang, D.; Li, C.G. Synergistic interactions of cannabidiol with chemotherapeutic drugs in mcf7 cells: Mode of interaction and proteomics analysis of mechanisms. Int. J. Mol. Sci. 2021, 22, 10103. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, I.H.; Alsherbiny, M.A.; Chang, D.; Li, C.G.; Bhuyan, D.J. Antiproliferative effects of Australian native plums against the MCF7 breast adenocarcinoma cells and UPLC-qTOF-IM-MS-driven identification of key metabolites. Food Biosci. 2023, 54, 102864. [Google Scholar] [CrossRef]
- Dang, V.B.; Alsherbiny, M.A.; Lin, R.; Gao, Y.; Li, C.; Bhuyan, D.J. Impact of a Functional Dairy Powder and Its Primary Component on the Growth of Pathogenic and Probiotic Gut Bacteria and Human Coronavirus 229E. Int. J. Mol. Sci. 2024, 25, 9353. [Google Scholar] [CrossRef] [PubMed]
- Pino, L.K.; Just, S.C.; MacCoss, M.J.; Searle, B.C. Acquiring and analyzing data independent acquisition proteomics experiments without spectrum libraries. Mol. Cell. Proteom. 2020, 19, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bandla, C.; Kundu, D.J.; Kamatchinathan, S.; Bai, J.; Hewapathirana, S.; John, N.S.; Prakash, A.; Walzer, M.; Wang, S. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 2025, 53, D543–D553. [Google Scholar] [CrossRef] [PubMed]

| Cell Growth Inhibition (%) of HKB-11 Lymphoma Cell Line | ||||||||
|---|---|---|---|---|---|---|---|---|
| Concentration (µM) | N | Sodium Butyrate | Sodium Propionate | Magnesium Acetate | Inosine | Concentration (µM) | UA | UB |
| 16,000 | NA | 63.83 ± 8.99 a | 51.45 ± 12.41 b | 18.23 ± 5.66 c | 35.2 ± 10.76 d | 500 | 61.51 ± 13.44 a | 81.81 ± 9.51 b |
| 8000 | 100.26 ± 0.09 a | 57.83 ± 9.95 b | 38.07 ± 10.39 c | 9.97 ± 3.12 d | 30.91 ± 6.27 e | 250 | 39.31 ± 12.18 a | 74.24 ± 4.91 b |
| 4000 | 100.21 ± 0.18 a | 53.18 ± 10.22 b | 16.31 ± 7.30 c | 9.86 ± 4.18 d | 27.02 ± 8.27 e | 125 | 18.22 ± 10.15 a | 55.51 ± 7.58 b |
| 2000 | 65.22 ± 1.35 a | 45.36 ± 9.08 b | 11.82 ± 2.51 c | 8.57 ± 4.41 d | 19.32 ± 7.15 e | 62.5 | 12.03 ± 5.16 a | 39.63 ± 7.48 b |
| 1000 | 34.84 ± 3.52 a | 23.33 ± 6.66 b | 10.26 ± 3.23 c | 7.85 ± 4.89 d | 14.41 ± 5.2 e | 31.25 | 10.77 ± 6.16 a | 36.16 ± 11.92 b |
| 500 | 15.18 ± 6.98 a | 18.58 ± 3.59 b | 10.99 ± 2.83 c | 7.61 ± 5.66 d | 13.54 ± 4.57 e | 15.625 | 8.16 ± 5.42 a | 30.96 ± 15.63 b |
| 250 | 2.64 ± 3.47 a | 15.31 ± 3.23 b | 9.47 ± 2.61 c | 6.82 ± 6.07 d | 12.42 ± 6.04 e | 7.8125 | 6.84 ± 4.75 a | 23.05 ± 2.71 b |
| 125 | ND | 13.38 ± 3.51 a | 7.75 ± 4.79 b | 6.66 ± 4.01 c | 9.45 ± 4.76 d | 3.90625 | 5.72 ± 5.12 a | 17.99 ± 3.26 b |
| 62.5 | ND | 11.64 ± 3.91 a | 5.7 ± 4.21 b | 5.47 ± 3.16 c | 7.69 ± 5.33 d | 1.953125 | 5.64 ± 5.61 a | 12.22 ± 2.73 b |
| IC50 | 1467 µM | 2022 µM | 14597.14 µM | NA | NA | IC50 | 384.41 µM | 87.56 µM |
| Combinations N/UB | IC50 | IC75 | IC90 | IC95 |
|---|---|---|---|---|
| 1:9 (800:450 μM) | 1.02 | 0.74 | 0.54 | 0.44 |
| 2:8 (1600:400 μM) | 1.03 | 0.88 | 0.76 | 0.69 |
| 3:7 (2400:350 μM) | 0.95 | 0.61 | 0.40 | 0.31 |
| 4:6 (3200:300 μM) | 0.77 | 0.33 | 0.15 | 0.09 |
| 5:5 (4000:250 μM) | 0.94 | 0.53 | 0.31 | 0.22 |
| 6:4 (4800:200 μM) | 1.09 | 0.66 | 0.42 | 0.32 |
| 7:3 (5600:150 μM) | 0.91 | 0.46 | 0.25 | 0.17 |
| 8:2 (6400:100 μM) | 0.61 | 0.41 | 0.30 | 0.26 |
| 9:1 (7200:50 μM) | 1.32 | 0.92 | 0.70 | 0.61 |
| Concentration (μM) N/UB 4:6 (3200:300) | Cell Growth Inhibition (%) | Cell Viability (%) | |
|---|---|---|---|
| HKB-11 | Hs 313.T | HS-5 | |
| 3500 | 98.49 ± 2.43 a | 100.55 ± 0.04 a | 10.08 ± 4.01 |
| 1750 | 65.77 ± 9.03 a | 90.26 ± 1.17 b | 17.92 ± 2.52 |
| 875 | 46.80 ± 1.32 a | 86.33 ± 1.59 b | 23.46 ± 3.96 |
| 437.5 | 30.04 ± 7.51 a | 76.06 ± 4.43 b | 66.37 ± 11.53 |
| 218.75 | 25.74 ± 17.07 a | 29.84 ± 7.04 a | 89.08 ± 9.20 |
| 109.375 | 21.86 ± 16.07 a | 9.99 ± 8.81 a | 92.32 ± 7.72 |
| IC50 | 1304 μM | 335.4 μM | 551.6 μM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Khazaleh, A.K.; Alsherbiny, M.A.; Münch, G.; Chang, D.; Bhuyan, D.J. Gut Microbial Postbiotics as Potential Therapeutics for Lymphoma: Proteomics Insights of the Synergistic Effects of Nisin and Urolithin B Against Human Lymphoma Cells. Int. J. Mol. Sci. 2025, 26, 6829. https://doi.org/10.3390/ijms26146829
Al-Khazaleh AK, Alsherbiny MA, Münch G, Chang D, Bhuyan DJ. Gut Microbial Postbiotics as Potential Therapeutics for Lymphoma: Proteomics Insights of the Synergistic Effects of Nisin and Urolithin B Against Human Lymphoma Cells. International Journal of Molecular Sciences. 2025; 26(14):6829. https://doi.org/10.3390/ijms26146829
Chicago/Turabian StyleAl-Khazaleh, Ahmad K., Muhammad A. Alsherbiny, Gerald Münch, Dennis Chang, and Deep Jyoti Bhuyan. 2025. "Gut Microbial Postbiotics as Potential Therapeutics for Lymphoma: Proteomics Insights of the Synergistic Effects of Nisin and Urolithin B Against Human Lymphoma Cells" International Journal of Molecular Sciences 26, no. 14: 6829. https://doi.org/10.3390/ijms26146829
APA StyleAl-Khazaleh, A. K., Alsherbiny, M. A., Münch, G., Chang, D., & Bhuyan, D. J. (2025). Gut Microbial Postbiotics as Potential Therapeutics for Lymphoma: Proteomics Insights of the Synergistic Effects of Nisin and Urolithin B Against Human Lymphoma Cells. International Journal of Molecular Sciences, 26(14), 6829. https://doi.org/10.3390/ijms26146829