
Genome-Wide Association Study of COVID-19 Breakthrough Infections and Genetic Overlap with Other Diseases: A Study of the UK Biobank

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Results from SNP-Based Analysis
2.1.1. GWAS Results
2.1.2. Significant SNPs Mapped to Genes
2.2. Results from Gene-Based Analysis
2.3. Results from Analysis of Genetic Overlap
3. Discussion
3.1. Interpretation of Findings
3.1.1. Top Loci Identified via GWAS
3.1.2. Gene-Based Results
3.2. Pathway and GO Enrichment Analysis
3.3. Polygenic Score Analysis and Genetic Overlap with Other Disorders
3.4. Other Related Studies
3.5. Strengths and Limitations
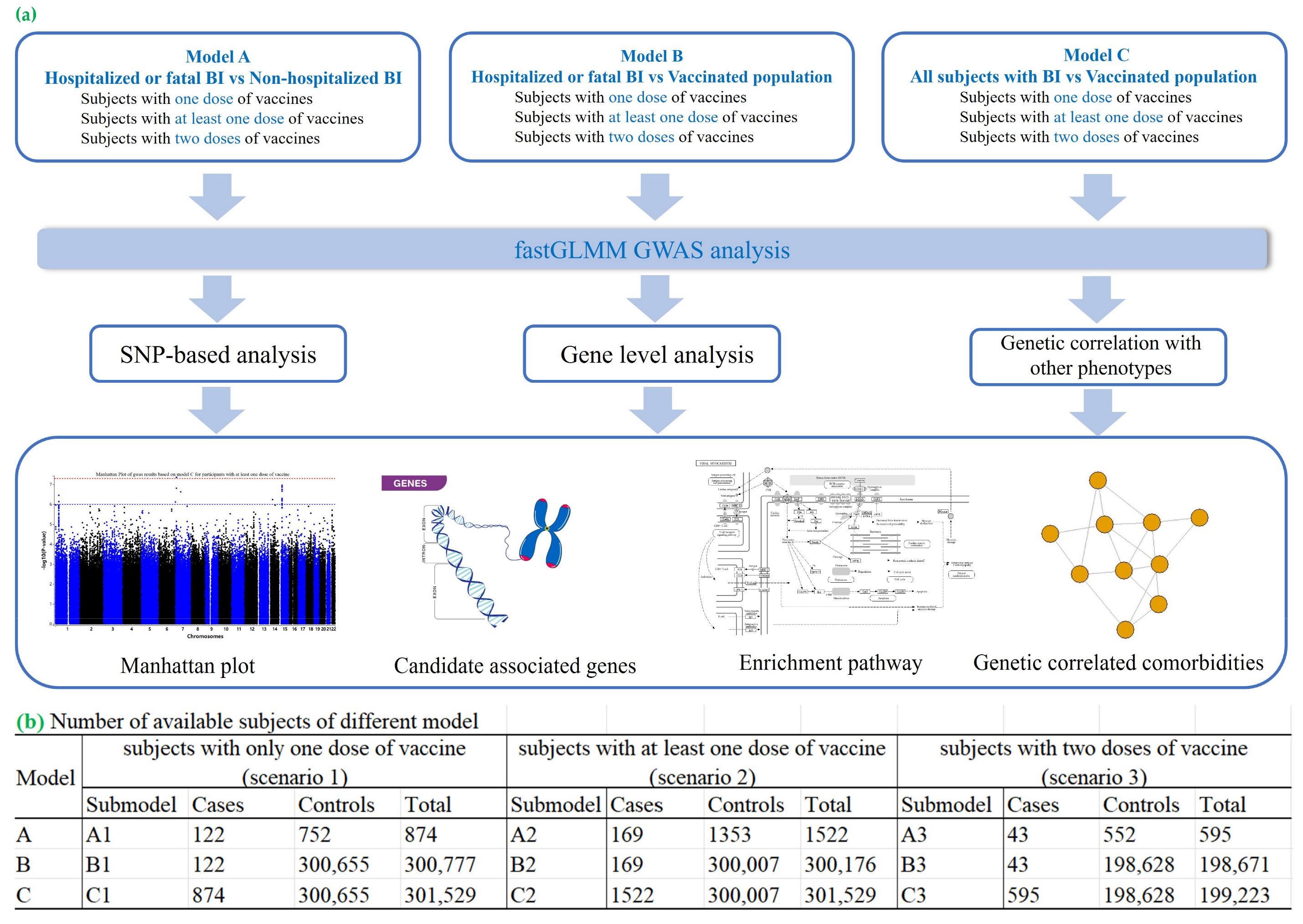
4. Materials and Methods
4.1. Data Source
4.2. COVID-19 Infection Status
4.3. Vaccination Status
4.4. Inclusion and Exclusion Criteria
4.5. Phenotype Definition
4.6. Genotyping and Quality Control (QC)
4.7. Genome-Wide Association Study (GWAS)
4.8. SNP-Based Analysis
4.9. Gene Set and Pathway Analyses
4.10. Transcriptome-Wide Association Studies (TWASs)
4.11. Phenome-Wide Association Studies (PheWASs)
4.12. Polygenic Risk Score (PRS) Analysis
4.13. Genetic Dependence Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guidotti, E.; Ardia, D. COVID-19 data hub. J. Open Source Softw. 2020, 5, 2376. [Google Scholar] [CrossRef]
- Lau, J.J.; Cheng, S.M.; Leung, K.; Lee, C.K.; Hachim, A.; Tsang, L.C.; Yam, K.W.; Chaothai, S.; Kwan, K.K.; Chai, Z.Y. Real-world COVID-19 vaccine effectiveness against the Omicron BA. 2 variant in a SARS-CoV-2 infection-naive population. Nat. Med. 2023, 29, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zheng, Q.; Madhira, V.; Olex, A.L.; Anzalone, A.J.; Vinson, A.; Singh, J.A.; French, E.; Abraham, A.G.; Mathew, J. Association between immune dysfunction and COVID-19 breakthrough infection after SARS-CoV-2 vaccination in the US. JAMA Intern. Med. 2022, 182, 153–162. [Google Scholar] [CrossRef]
- Bergwerk, M.; Gonen, T.; Lustig, Y.; Amit, S.; Lipsitch, M.; Cohen, C.; Mandelboim, M.; Levin, E.G.; Rubin, C.; Indenbaum, V. COVID-19 breakthrough infections in vaccinated health care workers. N. Engl. J. Med. 2021, 385, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Martin-Sancho, L.; Lewinski, M.K.; Pache, L.; Stoneham, C.A.; Yin, X.; Becker, M.E.; Pratt, D.; Churas, C.; Rosenthal, S.B.; Liu, S. Functional landscape of SARS-CoV-2 cellular restriction. Mol. Cell 2021, 81, 2656–2668.e8. [Google Scholar] [CrossRef]
- Mei, Z.W.; van Wijk, X.M.; Pham, H.P.; Marin, M.J. Role of von Willebrand factor in COVID-19 associated coagulopathy. J. Appl. Lab. Med. 2021, 6, 1305–1315. [Google Scholar] [CrossRef]
- Banerjee, S.; Cui, H.; Xie, N.; Tan, Z.; Yang, S.; Icyuz, M.; Thannickal, V.J.; Abraham, E.; Liu, G. miR-125a-5p regulates differential activation of macrophages and inflammation. J. Biol. Chem. 2013, 288, 35428–35436. [Google Scholar] [CrossRef]
- Boël, P.; Wildmann, C.; Sensi, M.L.; Brasseur, R.; Renauld, J.; Coulie, P.; Boon, T.; van der Bruggen, P. BAGE: A new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity 1995, 2, 167–175. [Google Scholar] [CrossRef]
- Vastrad, B.; Vastrad, C.; Tengli, A. Bioinformatics analyses of significant genes, related pathways, and candidate diagnostic biomarkers and molecular targets in SARS-CoV-2/COVID-19. Gene Rep. 2020, 21, 100956. [Google Scholar] [CrossRef]
- Barda, N.; Dagan, N.; Ben-Shlomo, Y.; Kepten, E.; Waxman, J.; Ohana, R.; Hernán, M.A.; Lipsitch, M.; Kohane, I.; Netzer, D. Safety of the BNT162b2 mRNA COVID-19 vaccine in a nationwide setting. N. Engl. J. Med. 2021, 385, 1078–1090. [Google Scholar] [CrossRef]
- Voleti, N.; Reddy, S.P.; Ssentongo, P. Myocarditis in SARS-CoV-2 infection vs. COVID-19 vaccination: A systematic review and meta-analysis. Front. Cardiovasc. Med. 2022, 9, 951314. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.; Okamoto, T.; John, A.E. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.J.; Guillen-Guio, B.; Croot, E.; Kraven, L.M.; Moss, S.; Stewart, I.; Jenkins, R.G.; Wain, L.V. Genetic overlap between idiopathic pulmonary fibrosis and COVID-19. Eur. Respir. J. 2022, 60, 2103132. [Google Scholar] [CrossRef]
- Shibolet, O.; Giallourakis, C.; Rosenberg, I.; Mueller, T.; Xavier, R.J.; Podolsky, D.K. AKAP13, a RhoA GTPase-specific guanine exchange factor, is a novel regulator of TLR2 signaling. J. Biol. Chem. 2007, 282, 35308–35317. [Google Scholar] [CrossRef] [PubMed]
- Abu-Farha, M.; Thanaraj, T.A.; Qaddoumi, M.G.; Hashem, A.; Abubaker, J.; Al-Mulla, F. The role of lipid metabolism in COVID-19 virus infection and as a drug target. Int. J. Mol. Sci. 2020, 21, 3544. [Google Scholar] [CrossRef]
- Maurya, R.; Sebastian, P.; Namdeo, M.; Devender, M.; Gertler, A. COVID-19 severity in obesity: Leptin and inflammatory cytokine interplay in the link between high morbidity and mortality. Front. Immunol. 2021, 12, 649359. [Google Scholar] [CrossRef]
- Bekele, Y.; Sui, Y.; Berzofsky, J.A. IL-7 in SARS-CoV-2 infection and as a potential vaccine adjuvant. Front. Immunol. 2021, 12, 737406. [Google Scholar] [CrossRef]
- Chang, X.; Li, Y.; Nguyen, K.; Qu, H.; Liu, Y.; Glessner, J.; Sleiman, P.M.; Hakonarson, H. Genetic correlations between COVID-19 and a variety of traits and diseases. Innovation 2021, 2, 100112. [Google Scholar] [CrossRef]
- Marfella, R.; Sardu, C.; D’Onofrio, N.; Prattichizzo, F.; Scisciola, L.; Messina, V.; La Grotta, R.; Balestrieri, M.L.; Maggi, P.; Napoli, C. Glycaemic control is associated with SARS-CoV-2 breakthrough infections in vaccinated patients with type 2 diabetes. Nat. Commun. 2022, 13, 2318. [Google Scholar] [CrossRef]
- Boroumand, A.B.; Forouhi, M.; Karimi, F.; Moghadam, A.S.; Naeini, L.G.; Kokabian, P.; Naderi, D. Immunogenicity of COVID-19 vaccines in patients with diabetes mellitus: A systematic review. Front. Immunol. 2022, 13, 940357. [Google Scholar] [CrossRef]
- Alcalde-Herraiz, M.; Català, M.; Prats-Uribe, A.; Paredes, R.; Xie, J.; Prieto-Alhambra, D. Genome-wide association studies of COVID-19 vaccine seroconversion and breakthrough outcomes in UK Biobank. Nat. Commun. 2024, 15, 8739. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wong, K.C.; Tsui, W.K.; Zhang, R.; Xiang, Y.; So, H. Genome-wide association study of COVID-19 Breakthrough Infections and genetic overlap with other diseases: A study of the UK Biobank. medRxiv 2024. [Google Scholar] [CrossRef]
- Bian, S.; Guo, X.; Yang, X.; Wei, Y.; Yang, Z.; Cheng, S.; Yan, J.; Chen, Y.; Chen, G.; Du, X. Genetic determinants of IgG antibody response to COVID-19 vaccination. Am. J. Hum. Genet. 2024, 111, 181–199. [Google Scholar] [CrossRef]
- Mentzer, A.J.; O’connor, D.; Bibi, S.; Chelysheva, I.; Clutterbuck, E.A.; Demissie, T.; Dinesh, T.; Edwards, N.J.; Felle, S.; Feng, S. Human leukocyte antigen alleles associate with COVID-19 vaccine immunogenicity and risk of breakthrough infection. Nat. Med. 2023, 29, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, R.W.; Yavlinsky, A.; Nguyen, V.; Eyre, M.T.; Shrotri, M.; Navaratnam, A.M.; Beale, S.; Braithwaite, I.; Byrne, T.; Kovar, J. SARS-CoV-2 antibodies and breakthrough infections in the Virus Watch cohort. Nat. Commun. 2022, 13, 4869. [Google Scholar] [CrossRef]
- Tartof, S.Y.; Slezak, J.M.; Fischer, H.; Hong, V.; Ackerson, B.K.; Ranasinghe, O.N.; Frankland, T.B.; Ogun, O.A.; Zamparo, J.M.; Gray, S. Effectiveness of mRNA BNT162b2 COVID-19 vaccine up to 6 months in a large integrated health system in the USA: A retrospective cohort study. Lancet 2021, 398, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Jiang, L.; Zheng, Z.; Fang, H.; Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 2021, 53, 1616–1621. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef]
- Carvalho-Silva, D.; Pierleoni, A.; Pignatelli, M.; Ong, C.; Fumis, L.; Karamanis, N.; Carmona, M.; Faulconbridge, A.; Hercules, A.; McAuley, E. Open Targets Platform: New developments and updates two years on. Nucleic Acids Res. 2019, 47, D1056–D1065. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, A.; Zhu, Z.; Vinkhuyzen, A.A.; Hill, W.D.; McRae, A.F.; Visscher, P.M.; Yang, J. Fast set-based association analysis using summary data from GWAS identifies novel gene loci for human complex traits. Sci. Rep. 2016, 6, 32894. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; VandeHaar, P.; Fritsche, L.G.; Zöllner, S.; Boehnke, M.; Scott, L.J.; Lee, S. A powerful subset-based method identifies gene set associations and improves interpretation in UK Biobank. Am. J. Hum. Genet. 2021, 108, 669–681. [Google Scholar] [CrossRef]
- Barbeira, A.N.; Dickinson, S.P.; Bonazzola, R.; Zheng, J.; Wheeler, H.E.; Torres, J.M.; Torstenson, E.S.; Shah, K.P.; Garcia, T.; Edwards, T.L. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018, 9, 1825. [Google Scholar] [CrossRef]
- Barbeira, A.N.; Pividori, M.; Zheng, J.; Wheeler, H.E.; Nicolae, D.L.; Im, H.K. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 2019, 15, e1007889. [Google Scholar] [CrossRef] [PubMed]
- Euesden, J.; Lewis, C.M.; O’reilly, P.F. PRSice: Polygenic risk score software. Bioinformatics 2015, 31, 1466–1468. [Google Scholar] [CrossRef]
- Willis, T.W.; Wallace, C. Accurate detection of shared genetic architecture from GWAS summary statistics in the small-sample context. PLoS Genet. 2023, 19, e1010852. [Google Scholar] [CrossRef]

{kind=link}
| Models | SNP | Chr. | Location (bp) | Effect Allele | Non-Effect Allele | Frequency of Effect Allele | BETA | SE | p | N | INFO | Gene Symbol | Gene Name | Total no. of SNPs from LD-Clumping | S0001 | Top Gene Prioritized by OpenTargets |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Participants with two doses of vaccine | rs28645263 | 5 | 16612885 | C | T | 0.416 | 0.347 | 0.06 | 9.46 × 10−9 | 199,223 | 0.964 | RETREG1 | reticulophagy regulator 1 | 3 | 3 | RETREG1 |
| rs4073656 | 2 | 48981646 | G | A | 0.502 | −0.288 | 0.059 | 9.89 × 10−7 | 199,223 | 0.988 | LHCGR | luteinizing hormone/choriogonadotropin receptor | 5 | 3 | STON1-GTF2A1L | |
| rs9661909 | 1 | 206714818 | T | C | 0.506 | −0.282 | 0.059 | 1.56 × 10−6 | 199,223 | 0.985 | RASSF5 | Ras association domain family member 5 | 11 | 6 | RASSF5 | |
| rs72718228 | 14 | 69475527 | T | C | 0.09 | 0.493 | 0.105 | 2.49 × 10−6 | 199,223 | 1 | 5 | 4 | ACTN1 | |||
| rs4991425 | 10 | 123485856 | T | C | 0.363 | −0.288 | 0.061 | 2.62 × 10−6 | 199,223 | 0.983 | 10 | 8 | FGFR2 | |||
| rs111692702 | 19 | 15651802 | A | G | 0.009 | 1.729 | 0.371 | 3.21 × 10−6 | 199,223 | 0.97 | CYP4F22 | cytochrome P450 family 4 subfamily F member 22 | 3 | 3 | CYP4F22 | |
| rs28718712 | 17 | 29882071 | T | G | 0.671 | −0.287 | 0.062 | 3.60 × 10−6 | 199,223 | 1 | 32 | 4 | RAB11FIP4 | |||
| rs4687124 | 3 | 189840935 | G | A | 0.232 | 0.319 | 0.07 | 4.86 × 10−6 | 199,223 | 0.998 | 22 | 22 | P3H2 | |||
| rs2874139 | 4 | 169751502 | C | G | 0.68 | −0.288 | 0.063 | 5.49 × 10−6 | 199,223 | 0.979 | PALLD | palladin, cytoskeletal associated protein | 43 | 15 | PALLD | |
| rs12466174 | 2 | 184802609 | T | G | 0.122 | 0.417 | 0.092 | 5.81 × 10−6 | 199,223 | 0.969 | 8 | 7 | NA | |||
| Participants with at least one dose of vaccine | rs36170929 | 7 | 12541187 | G | A | 0.64 | 0.21 | 0.038 | 4.39 × 10−8 | 301529 | 0.984254 | 11 | 5 | VWDE | ||
| rs56150535 | 15 | 31647722 | T | C | 0.359 | 0.203 | 0.038 | 1.09 × 10−7 | 301,529 | 0.996787 | KLF13 | Kruppel like factor 13 | 33 | 20 | KLF13 | |
| rs181987785 | 1 | 34977912 | G | A | 0.005 | 1.449 | 0.284 | 3.48 × 10−7 | 301,529 | 0.984316 | 30 | 30 | GJB5 | |||
| rs187268954 | 3 | 116529463 | C | T | 0.004 | 1.736 | 0.358 | 1.22 × 10−6 | 301,529 | 0.90264 | 4 | 3 | LSAMP | |||
| rs7590599 | 2 | 108915136 | C | T | 0.604 | 0.182 | 0.038 | 1.26 × 10−6 | 301,529 | 0.989482 | SULT1C2 | sulfotransferase family 1C member 2 | 8 | 5 | SULT1C2 | |
| rs3737328 | 13 | 110866065 | T | C | 0.246 | 0.198 | 0.042 | 3.05 × 10−6 | 301,529 | 1 | COL4A1 | collagen type IV alpha 1 chain | 4 | 4 | COL4A1 | |
| rs142193221 | 22 | 21166165 | A | G | 0.006 | 1.274 | 0.274 | 3.31 × 10−6 | 301,529 | 0.929992 | PI4KA | phosphatidylinositol 4-kinase alpha | 6 | 6 | PI4KA | |
| rs56070971 | 1 | 35025879 | T | C | 0.006 | 1.275 | 0.276 | 3.86 × 10−6 | 301,529 | 0.968667 | 29 | 29 | GJB5 | |||
| rs72664942 | 4 | 85808904 | G | A | 0.007 | 1.174 | 0.259 | 5.75 × 10−6 | 301,529 | 0.938787 | WDFY3 | WD repeat and FYVE domain containing 3 | 2 | 2 | WDFY3 | |
| rs79158353 | 10 | 78798475 | A | T | 0.082 | −0.304 | 0.067 | 6.48 × 10−6 | 301,529 | 0.995184 | KCNMA1 | potassium calcium-activated channel subfamily M alpha 1 | 23 | 13 | KCNMA1 |
| GeneSet | Length_GS | p-Value | Excluded | p_Adjust_BH | Model |
|---|---|---|---|---|---|
| KEGG_VIRAL_MYOCARDITIS | 41 | 9.05 × 10−6 | 22 | 5.69 × 10−2 | A1 |
| BIOCARTA_AKAP13_PATHWAY | 21 | 9.91 × 10−6 | 1 | 6.23 × 10−2 | B2 |
| KEGG_TIGHT_JUNCTION | 73 | 1.38 × 10−5 | 11 | 6.29 × 10−2 | A2 |
| REACTOME_TRANSLATION | 295 | 2.00 × 10−5 | 76 | 6.29 × 10−2 | B2 |
| REACTOME_MITOCHONDRIAL_TRANSLATION | 96 | 1.10 × 10−4 | 4 | 1.73 × 10−1 | A2 |
| REACTOME_PASSIVE_TRANSPORT_BY_AQUAPORINS | 13 | 8.00 × 10−5 | 0 | 5.03 × 10−1 | C3 |
| MYLLYKANGAS_AMPLIFICATION_HOT_SPOT_29 | 33 | 1.60 × 10−4 | 0 | 5.35 × 10−1 | C1 |
| YAMASHITA_LIVER_CANCER_WITH_EPCAM_DN | 53 | 1.70 × 10−4 | 0 | 5.35 × 10−1 | C1 |
| APRELIKOVA_BRCA1_TARGETS | 48 | 2.00 × 10−4 | 8 | 7.17 × 10−1 | C2 |
| WP_LEPTIN_INSULIN_OVERLAP | 30 | 2.50 × 10−4 | 1 | 7.17 × 10−1 | C2 |
| REACTOME_INTERLEUKIN_7_SIGNALING | 9 | 3.90 × 10−4 | 13 | 7.17 × 10−1 | C2 |
| WP_LIPID_METABOLISM_PATHWAY | 23 | 3.40 × 10−4 | 0 | 7.76 × 10−1 | B1 |
| WP_STEROL_REGULATORY_ELEMENTBINDING_PROTEINS_SREBP_SIGNALLING | 8 | 3.70 × 10−4 | 4 | 7.76 × 10−1 | B1 |
| REACTOME_PI3K_AKT_ACTIVATION | 9 | 2.90 × 10−4 | 1 | 7.97 × 10−1 | C3 |
| WP_STRIATED_MUSCLE_CONTRACTION_PATHWAY | 11 | 3.00 × 10−4 | 2 | 8.39 × 10−1 | A1 |
| Body System | Exposure | pval_PRS | p_adjust_BH | Coefficient | r2 | nsnps | exposure_p_filter | clump_r2 |
|---|---|---|---|---|---|---|---|---|
| cardiovascular system | Heart Failure | 1.33 × 10−4 | 1.82 × 10−3 | 0.030588 | 4.84 × 10−5 | 41,900 | 0.05 | 0.05 |
| endocrine system | Type 1 diabetes, strict (exclude type 2) | 1.00 × 10−3 | 1.22 × 10−2 | 0.028586 | 3.59 × 10−5 | 131 | 5.00 × 10−8 | 0.05 |
| endocrine system | Glycaemic_HbA1c | 1.96 × 10−3 | 2.18 × 10−2 | 0.704835 | 3.18 × 10−5 | 250 | 1.00 × 10−4 | 0.05 |
| endocrine system | Diabetes mellitus (type 1 and 2) | 1.61 × 10−2 | 1.30 × 10−1 | 0.097242 | 1.92 × 10−5 | 128 | 5.00 × 10−8 | 0.05 |
| endocrine system | Obesity | 3.63 × 10−2 | 2.37 × 10−1 | 0.004535 | 1.45 × 10−5 | 56,424 | 0.05 | 0.05 |
| endocrine system | BMI | 1.49 × 10−2 | 1.32 × 10−1 | 0.17485 | 1.97 × 10−5 | 1365 | 1.00 × 10−7 | 0.05 |
| immune system | Human immunodeficiency virus disease | 3.71 × 10−2 | 2.41 × 10−1 | 0.042006 | 1.44 × 10−5 | 17 | 1.00 × 10−5 | 0.05 |
| nervous system | Dementia | 2.95 × 10−2 | 2.00 × 10−1 | 0.003864 | 1.57 × 10−5 | 78,932 | 0.1 | 0.05 |
| respiratory system | COPD/asthma-related infections | 9.15 × 10−3 | 8.58 × 10−2 | 0.01321 | 2.25 × 10−5 | 54,680 | 0.05 | 0.05 |
| respiratory system | Asthma | 2.09 × 10−2 | 1.62 × 10−1 | −0.009499 | 1.77 × 10−5 | 20,426 | 0.01 | 0.05 |
| respiratory system | Smoking Cessation | 4.00 × 10−2 | 2.46 × 10−1 | 0.15793 | 1.40 × 10−5 | 2871 | 0.001 | 0.05 |
| renal system | Diabetic kidney disease in type 1 DM | 9.75 × 10−3 | 9.00 × 10−2 | −0.015166 | 2.22 × 10−5 | 1449 | 0.001 | 0.05 |
| renal system | Serum urate | 1.16 × 10−2 | 1.04 × 10−1 | 1.205126 | 2.11 × 10−5 | 33 | 0.05 | 0.05 |
| Exposure | Outcome | pthres | n | Dn | Scaled | p. Value | p.adj_pthres&traitB_Separate |
|---|---|---|---|---|---|---|---|
| Respiratory | |||||||
| Abnormal findings on diagnostic imaging of lung | A2 | 0.1 | 102,776 | 1.29 × 10−6 | 4.76 | 2.05 × 10−4 | 8.00 × 10−3 |
| Abnormal findings on diagnostic imaging of lung | B2 | 0.1 | 102,787 | 7.19 × 10−7 | 2.66 | 4.47 × 10−3 | 1.74 × 10−1 |
| Asthma (only as main diagnosis) | A2 | 0.5 | 372,099 | 1.94 × 10−7 | 2.6 | 4.88 × 10−3 | 1.43 × 10−1 |
| Asthma (only as main diagnosis) | B2 | 0.5 | 372,141 | 1.81 × 10−7 | 2.42 | 6.41 × 10−3 | 8.33 × 10−2 |
| Asthma (only as main diagnosis) | C2 | 0.05 | 68,429 | 1.55 × 10−6 | 3.82 | 8.08 × 10−4 | 2.69 × 10−2 |
| Asthma, hospital admissions, main diagnosis only | A2 | 0.5 | 371,828 | 1.63 × 10−7 | 2.19 | 9.11 × 10−3 | 1.43 × 10−1 |
| COPD/asthma-related infections | B2 | 1.00 × 10−5 | 44 | 8.24 × 10−4 | 1.28 | 3.77 × 10−2 | 2.56 × 10−1 |
| COPD/asthma-related pneumonia or pneumonia-derived septicaemia | A2 | 0.01 | 15,042 | 2.35 × 10−6 | 1.27 | 3.81 × 10−2 | 2.97 × 10−1 |
| Interstitial lung disease | A2 | 0.3 | 248,253 | 2.02 × 10−7 | 1.81 | 1.63 × 10−2 | 3.08 × 10−1 |
| Interstitial lung disease endpoints | C2 | 0.2 | 190,993 | 1.70 × 10−7 | 1.17 | 4.49 × 10−2 | 6.65 × 10−1 |
| Obesity-related asthma | A2 | 0.01 | 15,347 | 3.12 × 10−6 | 1.73 | 1.85 × 10−2 | 2.41 × 10−1 |
| Obesity-related asthma | B2 | 0.01 | 15,350 | 2.12 × 10−6 | 1.17 | 4.48 × 10−2 | 5.82 × 10−1 |
| Pulmonary embolism | B2 | 0.05 | 58,577 | 1.46 × 10−6 | 3.07 | 2.43 × 10−3 | 6.17 × 10−2 |
| Tuberculosis | A2 | 0.01 | 13,123 | 3.08 × 10−6 | 1.45 | 2.84 × 10−2 | 2.77 × 10−1 |
| Cardiovascular | |||||||
| Cardiomyopathy | C2 | 0.1 | 103,175 | 9.18 × 10−7 | 3.41 | 1.48 × 10−3 | 5.75 × 10−2 |
| Cardiomyopathy (excluding other) | B2 | 0.5 | 363,183 | 1.87 × 10−7 | 2.44 | 6.18 × 10−3 | 8.33 × 10−2 |
| Cardiomyopathy (no controls excluded) | A2 | 0.01 | 14,204 | 4.44 × 10−6 | 2.27 | 8.07 × 10−3 | 1.57 × 10−1 |
| Endocrine | |||||||
| Diabetes mellitus (type 1 and 2) | A2 | 0.3 | 275,918 | 1.17 × 10−7 | 1.16 | 4.52 × 10−2 | 3.52 × 10−1 |
| Diabetes mellitus (type 1 and 2) | C2 | 0.1 | 129,409 | 4.66 × 10−7 | 2.17 | 9.33 × 10−3 | 1.82 × 10−1 |
| Obesity | B2 | 0.4 | 319,934 | 1.34 × 10−7 | 1.54 | 2.49 × 10−2 | 3.95 × 10−1 |
| Type 1 diabetes, strict definition | A2 | 1.00 × 10−4 | 728 | 9.35 × 10−5 | 2.45 | 6.16 × 10−3 | 1.20 × 10−1 |
| Type 1 diabetes, wide definition | B2 | 0.2 | 179,971 | 6.59 × 10−7 | 4.27 | 4.21 × 10−4 | 1.64 × 10−2 |
| Type 1 diabetes, wide definition | C2 | 0.05 | 56,637 | 6.89 × 10−7 | 1.41 | 3.07 × 10−2 | 3.99 × 10−1 |
| Neurological | |||||||
| Schizophrenia or delusion | C2 | 1.00 × 10−5 | 35 | 2.00 × 10−3 | 2.44 | 6.19 × 10−3 | 2.41 × 10−1 |
| Schizophrenia or delusion (more controls excluded) | A2 | 0.01 | 15,032 | 6.43 × 10−6 | 3.48 | 1.33 × 10−3 | 5.20 × 10−2 |
| Schizophrenia, schizotypal and delusional disorders | B2 | 1.00 × 10−5 | 43 | 3.09 × 10−3 | 4.68 | 2.33 × 10−4 | 9.09 × 10−3 |
| Any dementia | B2 | 1.00 × 10−5 | 109 | 4.62 × 10−4 | 1.8 | 1.66 × 10−2 | 2.56 × 10−1 |
| Any dementia (more controls excluded) | A2 | 0.001 | 1858 | 2.18 × 10−5 | 1.45 | 2.84 × 10−2 | 2.77 × 10−1 |
| Liver | |||||||
| Alcoholic liver disease | A2 | 0.001 | 1690 | 3.69 × 10−5 | 2.25 | 8.35 × 10−3 | 2.77 × 10−1 |
| Cirrhosis, broad definition | A2 | 1.00 × 10−4 | 202 | 3.92 × 10−4 | 2.84 | 3.43 × 10−3 | 1.20 × 10−1 |
| Cirrhosis, broad definition | C2 | 0.3 | 248,811 | 1.36 × 10−7 | 1.22 | 4.12 × 10−2 | 6.57 × 10−1 |
| Nonalcoholic fatty liver disease | B2 | 0.2 | 178,801 | 1.82 × 10−7 | 1.17 | 4.45 × 10−2 | 4.34 × 10−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Y.; Wong, K.C.-Y.; Tsui, W.K.; Zhang, R.; Xiang, Y.; So, H.-C. Genome-Wide Association Study of COVID-19 Breakthrough Infections and Genetic Overlap with Other Diseases: A Study of the UK Biobank. Int. J. Mol. Sci. 2025, 26, 6441. https://doi.org/10.3390/ijms26136441
Feng Y, Wong KC-Y, Tsui WK, Zhang R, Xiang Y, So H-C. Genome-Wide Association Study of COVID-19 Breakthrough Infections and Genetic Overlap with Other Diseases: A Study of the UK Biobank. International Journal of Molecular Sciences. 2025; 26(13):6441. https://doi.org/10.3390/ijms26136441
Chicago/Turabian StyleFeng, Yaning, Kenneth Chi-Yin Wong, Wai Kai Tsui, Ruoyu Zhang, Yong Xiang, and Hon-Cheong So. 2025. "Genome-Wide Association Study of COVID-19 Breakthrough Infections and Genetic Overlap with Other Diseases: A Study of the UK Biobank" International Journal of Molecular Sciences 26, no. 13: 6441. https://doi.org/10.3390/ijms26136441
APA StyleFeng, Y., Wong, K. C.-Y., Tsui, W. K., Zhang, R., Xiang, Y., & So, H.-C. (2025). Genome-Wide Association Study of COVID-19 Breakthrough Infections and Genetic Overlap with Other Diseases: A Study of the UK Biobank. International Journal of Molecular Sciences, 26(13), 6441. https://doi.org/10.3390/ijms26136441