
Inhibition of CD38 by 78c Enhanced NAD+, Alleviated Inflammation, and Decreased Oxidative Stress in Old Murine Macrophages Induced by Oral Pathogens


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
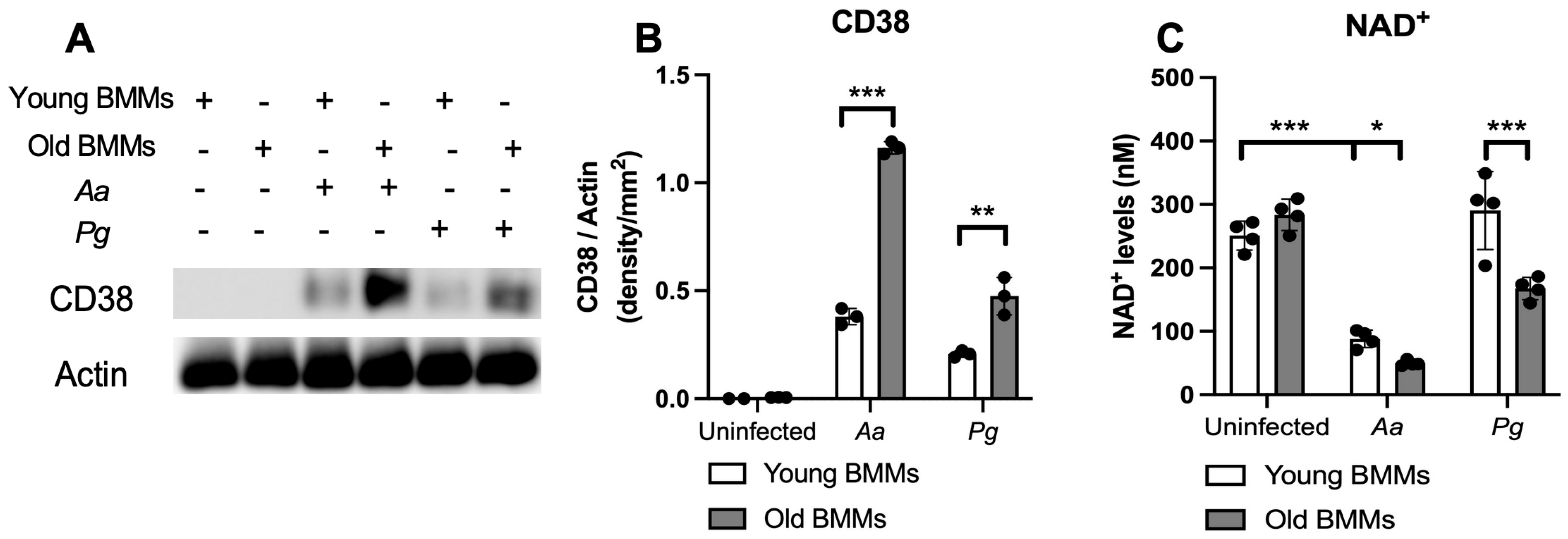
2.1. Old Murine BMMs Exhibited Significantly Higher CD38 Protein and Lower NAD+ Expressions After Infection with Oral Pathogens Aa or Pg Compared with Young Controls
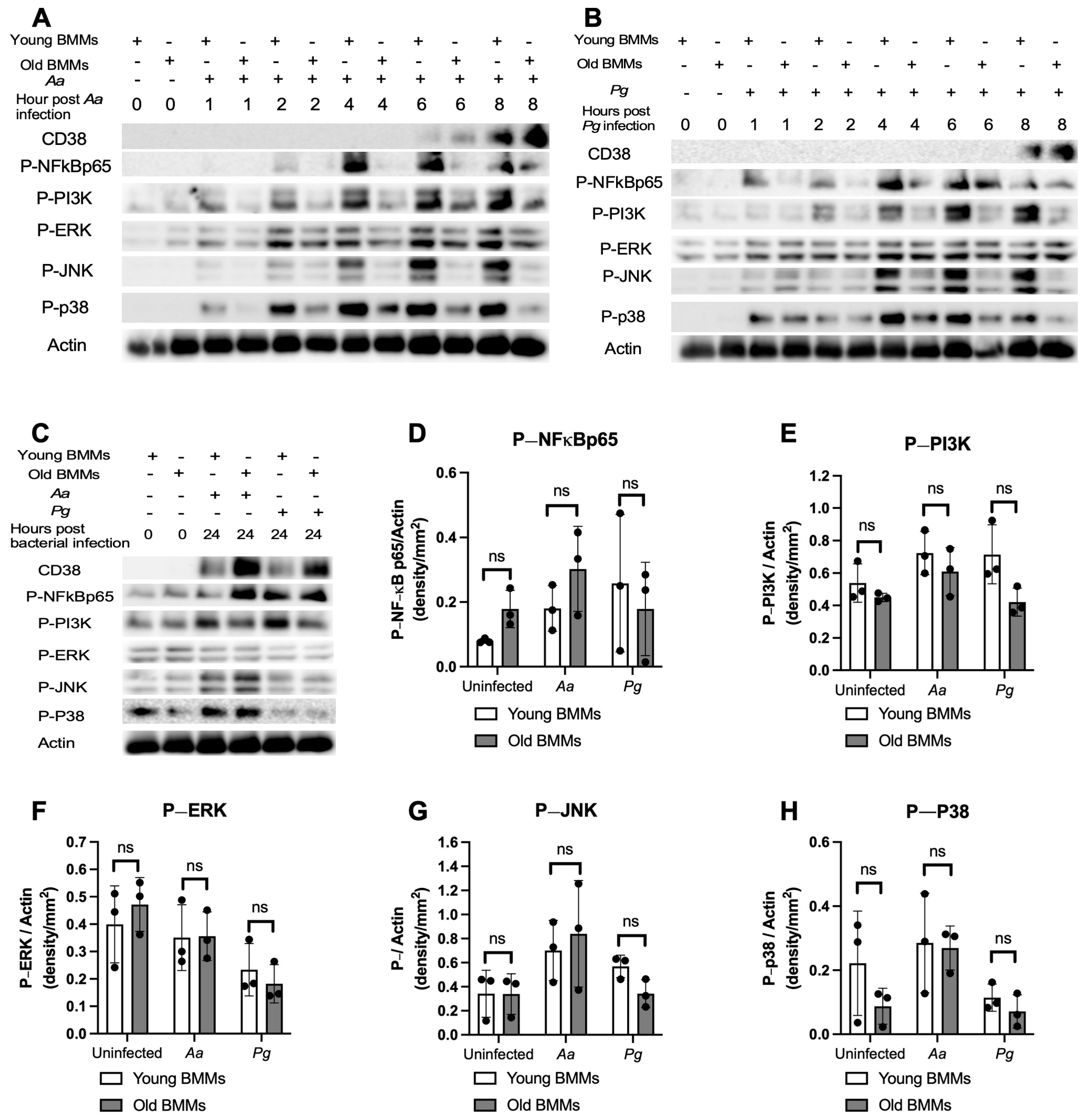
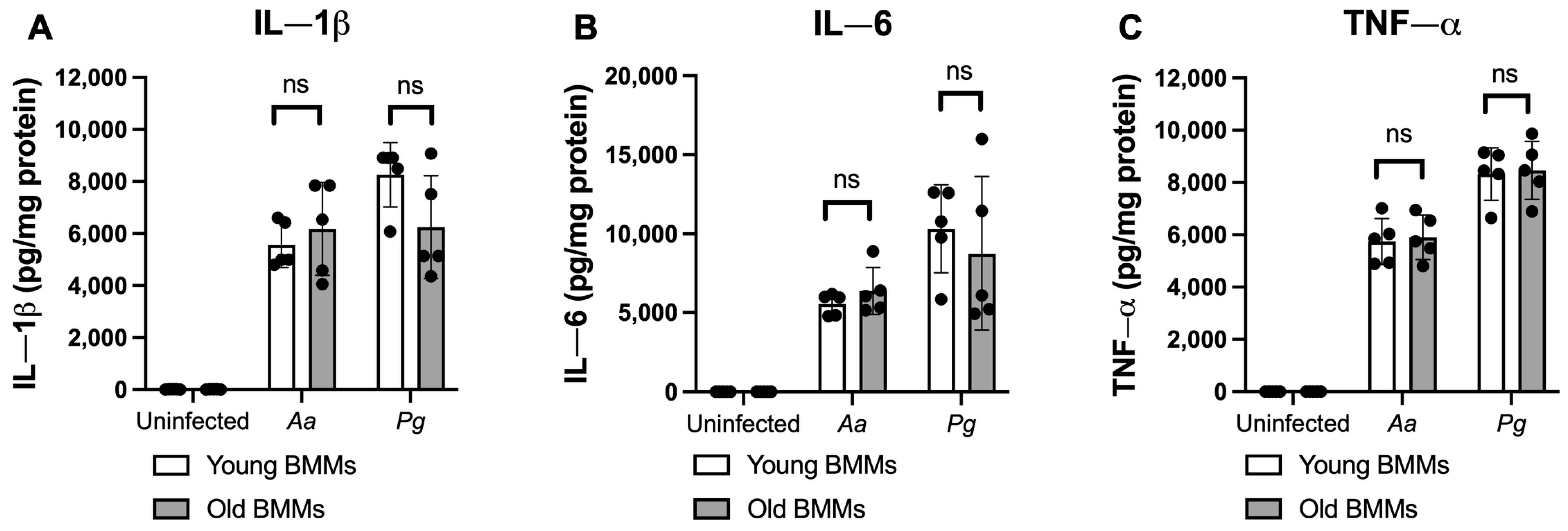
2.2. The Abnormal High CD38 Protein Level in Old Murine BMMs After Infection with the Oral Pathogens Aa or Pg Was Not Directly Correlated with the Level of Immune Responses in Old Murine BMMs Compared with Young Controls
2.3. Inhibition of CD38 by 78c Suppressed CD38, NF-κB, PI3K, and MAPK Protein Kinases, Enhanced NAD+, and Attenuated IL-1β, IL-6, and TNF-α Pro-Inflammatory Cytokine Levels in Old Murine BMMs Infected with Oral Pathogens Aa or Pg
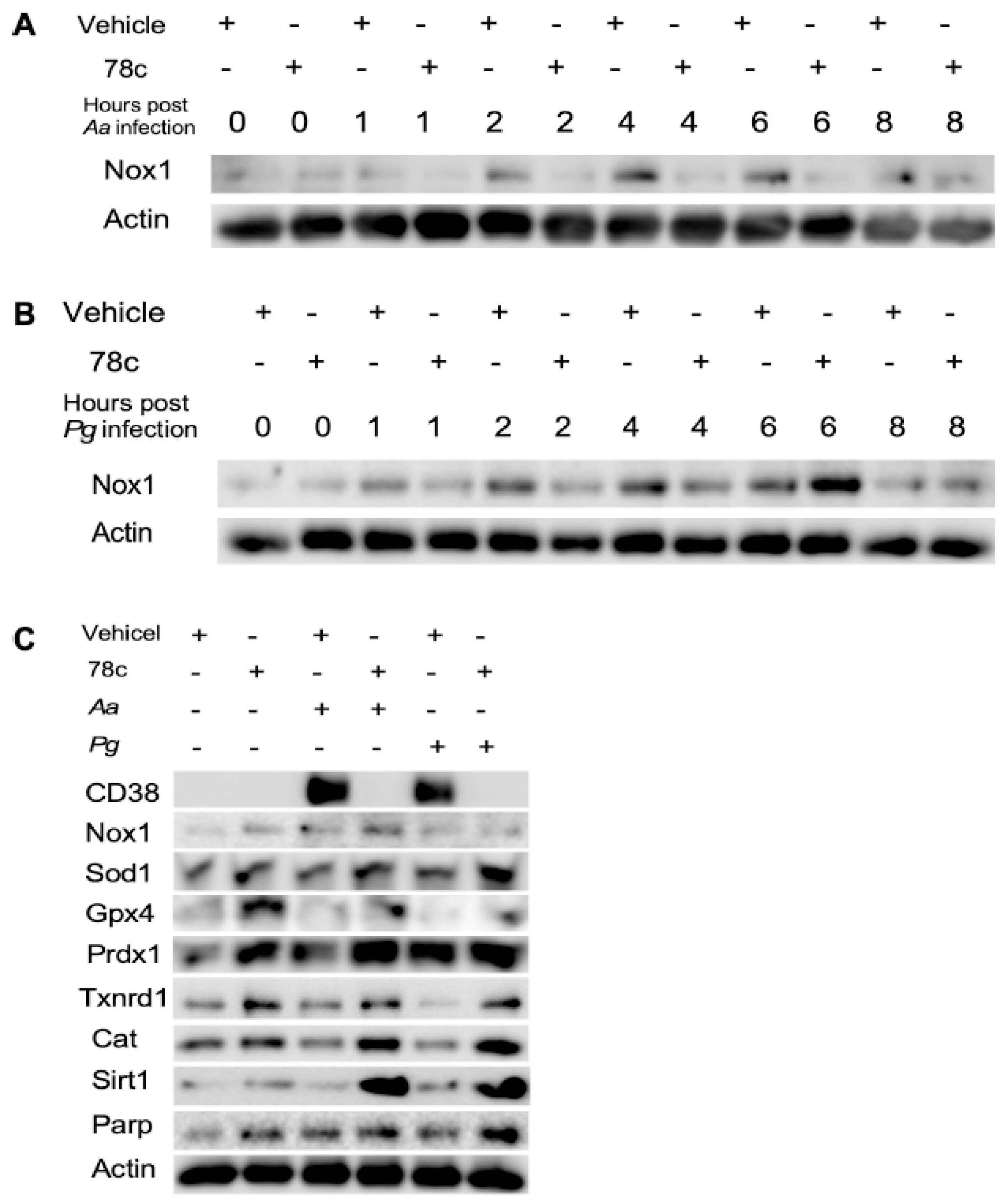
2.4. Inhibition of CD38 by 78c Reduced Oxidative Stress in Old Murine BMMs Infected with Oral Pathogens
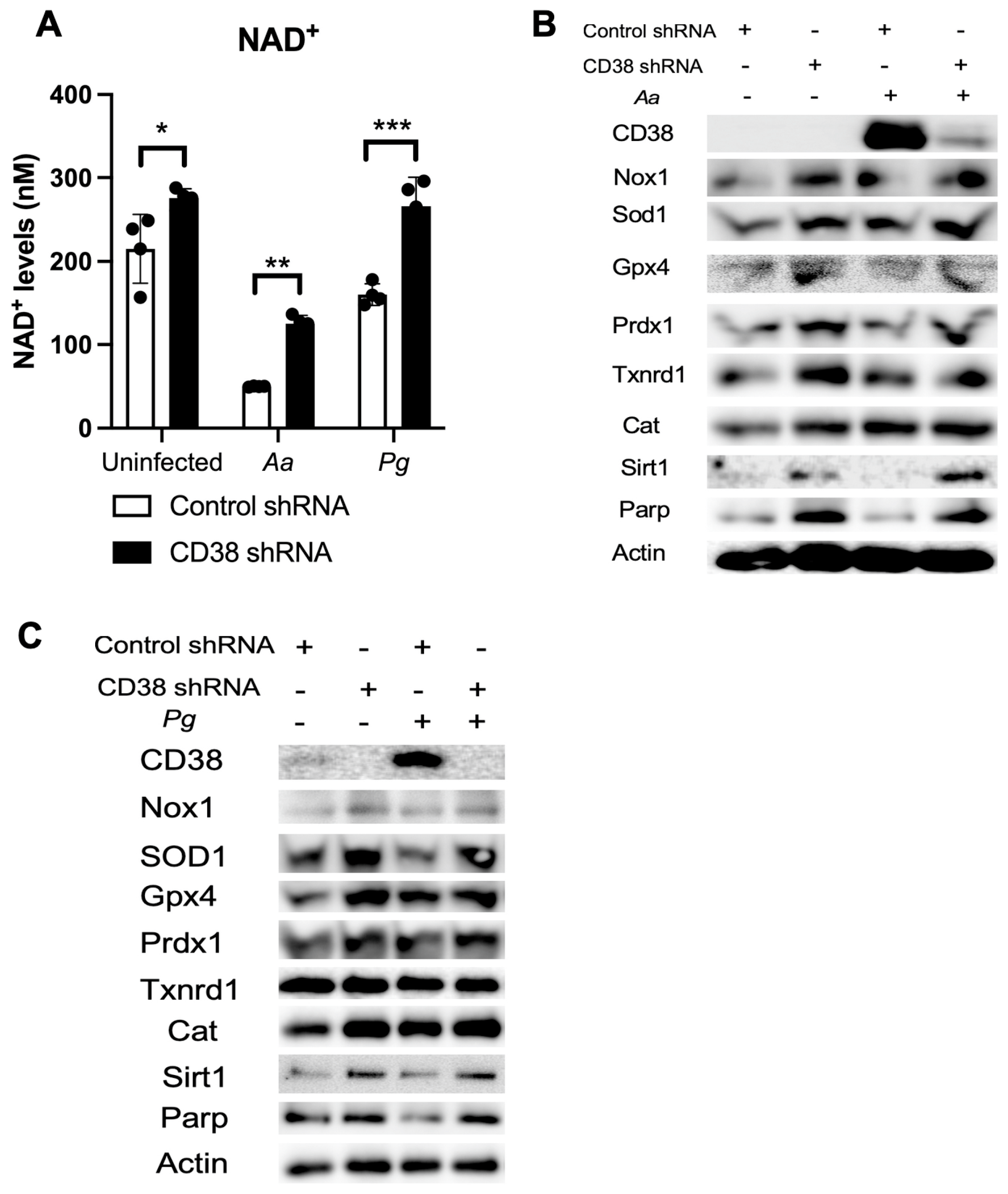
2.5. Knockdown of CD38 by CD38 shRNA Increased NAD+ and Reduced Oxidative Stress in Old Murine BMMs Infected with Oral Pathogens
3. Discussion
4. Materials and Methods
4.1. Animals and Reagents
4.2. Generation of Bone Marrow-Derived Monocytes and Macrophages (BMMs)
4.3. Treatment with shRNA Lentivirus Vectors
4.4. Bacterial Culture and Cellular Infection
4.5. NAD+ Assay
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. RNA Extraction and Real-Time PCR
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 597959. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.S.; Cordeiro, H.S.; Tran, N.L.K.; Chini, E.N. NAD metabolism: Role in senescence regulation and aging. Aging Cell 2024, 23, e13920. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Kar, A.; Mehrotra, S.; Chatterjee, S. CD38: T Cell Immuno-Metabolic Modulator. Cells 2020, 9, 1716. [Google Scholar] [CrossRef]
- Benzi, A.; Grozio, A.; Spinelli, S.; Sturla, L.; Guse, A.H.; De Flora, A.; Zocchi, E.; Heeren, J.; Bruzzone, S. Role of CD38 in Adipose Tissue: Tuning Coenzyme Availability? Nutrients 2021, 13, 3734. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid. Redox Signal. 2019, 30, 251–294. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Kanasi, E.; Ayilavarapu, S.; Jones, J. The aging population: Demographics and the biology of aging. Periodontol. 2000 2016, 72, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarragó, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat. Metab. 2020, 2, 1284–1304. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Zapata-Pérez, R.; Wanders, R.J.A.; van Karnebeek, C.D.M.; Houtkooper, R.H. NAD+ homeostasis in human health and disease. EMBO Mol. Med. 2021, 13, e13943. [Google Scholar] [CrossRef] [PubMed]
- Peclat, T.R.; Shi, B.; Varga, J.; Chini, E.N. The NADase enzyme CD38: An emerging pharmacological target for systemic sclerosis, systemic lupus erythematosus and rheumatoid arthritis. Curr. Opin. Rheumatol. 2020, 32, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.; Kotronia, E.; Ramsay, S.E. Frailty, aging, and periodontal disease: Basic biologic considerations. Periodontol. 2000 2021, 87, 143–156. [Google Scholar] [CrossRef]
- Clark, D.; Halpern, B.; Miclau, T.; Nakamura, M.; Kapila, Y.; Marcucio, R. The Contribution of Macrophages in Old Mice to Periodontal Disease. J. Dent. Res. 2021, 100, 1397–1404. [Google Scholar] [CrossRef]
- Raja, M.; Ummer, F.; Dhivakar, C.P. Aggregatibacter actinomycetemcomitans—A tooth killer? J. Clin. Diagn. Res. 2014, 8, ZE13–ZE16. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef]
- Roberts, J.S.; Atanasova, K.R.; Lee, J.; Diamond, G.; Deguzman, J.; Hee Choi, C.; Yilmaz, Ö. Opportunistic Pathogen Porphyromonas gingivalis Modulates Danger Signal ATP-Mediated Antibacterial NOX2 Pathways in Primary Epithelial Cells. Front. Cell. Infect. Microbiol. 2017, 7, 291. [Google Scholar] [CrossRef]
- Díaz-Zúñiga, J.; Monasterio, G.; Alvarez, C.; Melgar-Rodríguez, S.; Benítez, A.; Ciuchi, P.; García, M.; Arias, J.; Sanz, M.; Vernal, R. Variability of the dendritic cell response triggered by different serotypes of Aggregatibacter actinomycetemcomitans or Porphyromonas gingivalis is toll-like receptor 2 (TLR2) or TLR4 dependent. J. Periodontol. 2015, 86, 108–119. [Google Scholar] [CrossRef]
- Cai, J.; Chen, J.; Guo, H.; Pan, Y.; Zhang, Y.; Zhao, W.; Li, X.; Li, Y. Recombinant fimbriae protein of Porphyromonas gingivalis induces an inflammatory response via the TLR4/NF-κB signaling pathway in human peripheral blood mononuclear cells. Int. J. Mol. Med. 2019, 43, 1430–1440. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Lory, W.; Chowdhury, N.; Wellslager, B.; Pandruvada, S.; Huang, Y.; Yilmaz, Ö.; Yu, H. CD38 Inhibitor 78c Attenuates Pro-Inflammatory Cytokine Expression and Osteoclastogenesis in Macrophages. Cells 2024, 13, 1971. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline. Biochem. Biophys. Res. Commun. 2019, 513, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Ogier-Denis, E.; Mkaddem, S.B.; Vandewalle, A. NOX enzymes and Toll-like receptor signaling. Semin. Immunopathol. 2008, 30, 291–300. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Y.; Zou, X.; Zhang, J.; Zheng, Z.; Wang, Z. Antioxidant Apigenin Relieves Age-Related Muscle Atrophy by Inhibiting Oxidative Stress and Hyperactive Mitophagy and Apoptosis in Skeletal Muscle of Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2081–2088. [Google Scholar] [CrossRef]
- Tabibzadeh, S. Signaling pathways and effectors of aging. Front. Biosci. (Landmark Ed.) 2021, 26, 50–96. [Google Scholar] [CrossRef]
- Stumpferl, S.W.; Brand, S.E.; Jiang, J.C.; Korona, B.; Tiwari, A.; Dai, J.; Seo, J.G.; Jazwinski, S.M. Natural genetic variation in yeast longevity. Genome Res. 2012, 22, 1963–1973. [Google Scholar] [CrossRef]
- Ludewig, A.H.; Izrayelit, Y.; Park, D.; Malik, R.U.; Zimmermann, A.; Mahanti, P.; Fox, B.W.; Bethke, A.; Doering, F.; Riddle, D.L.; et al. Pheromone sensing regulates Caenorhabditis elegans lifespan and stress resistance via the deacetylase SIR-2.1. Proc. Natl. Acad. Sci. USA 2013, 110, 5522–5527. [Google Scholar] [CrossRef]
- Banerjee, K.K.; Ayyub, C.; Ali, S.Z.; Mandot, V.; Prasad, N.G.; Kolthur-Seetharam, U. dSir2 in the adult fat body, but not in muscles, regulates life span in a diet-dependent manner. Cell Rep. 2012, 2, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Chen, J.A.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 2015, 35, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dong, G.; Xiao, W.; Xiao, E.; Miao, F.; Syverson, A.; Missaghian, N.; Vafa, R.; Cabrera-Ortega, A.A.; Rossa, C., Jr.; et al. Effect of Aging on Periodontal Inflammation, Microbial Colonization, and Disease Susceptibility. J. Dent. Res. 2016, 95, 460–466. [Google Scholar] [CrossRef]
- Peclat, T.R.; Thompson, K.L.; Warner, G.M.; Chini, C.C.S.; Tarragó, M.G.; Mazdeh, D.Z.; Zhang, C.; Zavala-Solorio, J.; Kolumam, G.; Liang Wong, Y.; et al. CD38 inhibitor 78c increases mice lifespan and healthspan in a model of chronological aging. Aging Cell 2022, 21, e13589. [Google Scholar] [CrossRef]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018, 27, 1081–1095.e1010. [Google Scholar] [CrossRef]
- Nagao, Y.; Tanigawa, T. Red complex periodontal pathogens are risk factors for liver cirrhosis. Biomed. Rep. 2019, 11, 199–206. [Google Scholar] [CrossRef]
- Martin, T.G.; Corzo, K.; Chiron, M.; Velde, H.V.; Abbadessa, G.; Campana, F.; Solanki, M.; Meng, R.; Lee, H.; Wiederschain, D.; et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells 2019, 8, 1522. [Google Scholar] [CrossRef]
- Wu, H.T.; Zhao, X.Y. Regulation of CD38 on Multiple Myeloma and NK Cells by Monoclonal Antibodies. Int. J. Biol. Sci. 2022, 18, 1974–1988. [Google Scholar] [CrossRef] [PubMed]
- Wellslager, B.; Roberts, J.; Chowdhury, N.; Madan, L.; Orellana, E.; Yilmaz, Ö. Porphyromonas gingivalis activates Heat-Shock-Protein 27 to drive a LC3C-specific probacterial form of select autophagy that is redox sensitive for intracellular bacterial survival in human gingival mucosa. bioRxiv 2024. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, K.; Chowdhury, N.; Wellslager, B.; Hill, W.D.; Yilmaz, Ö.; Yu, H. Inhibition of CD38 by 78c Enhanced NAD+, Alleviated Inflammation, and Decreased Oxidative Stress in Old Murine Macrophages Induced by Oral Pathogens. Int. J. Mol. Sci. 2025, 26, 6180. https://doi.org/10.3390/ijms26136180
Cao K, Chowdhury N, Wellslager B, Hill WD, Yilmaz Ö, Yu H. Inhibition of CD38 by 78c Enhanced NAD+, Alleviated Inflammation, and Decreased Oxidative Stress in Old Murine Macrophages Induced by Oral Pathogens. International Journal of Molecular Sciences. 2025; 26(13):6180. https://doi.org/10.3390/ijms26136180
Chicago/Turabian StyleCao, Kimberly, Nityananda Chowdhury, Bridgette Wellslager, William D. Hill, Özlem Yilmaz, and Hong Yu. 2025. "Inhibition of CD38 by 78c Enhanced NAD+, Alleviated Inflammation, and Decreased Oxidative Stress in Old Murine Macrophages Induced by Oral Pathogens" International Journal of Molecular Sciences 26, no. 13: 6180. https://doi.org/10.3390/ijms26136180
APA StyleCao, K., Chowdhury, N., Wellslager, B., Hill, W. D., Yilmaz, Ö., & Yu, H. (2025). Inhibition of CD38 by 78c Enhanced NAD+, Alleviated Inflammation, and Decreased Oxidative Stress in Old Murine Macrophages Induced by Oral Pathogens. International Journal of Molecular Sciences, 26(13), 6180. https://doi.org/10.3390/ijms26136180