
Comprehensive Evaluation of 1H-Isoindole-1,3(2H)-Dione Derivatives: Pharmacokinetic Studies and Analgesic Potential in Various Pain Models

 , , , and
, , , and
Abstract

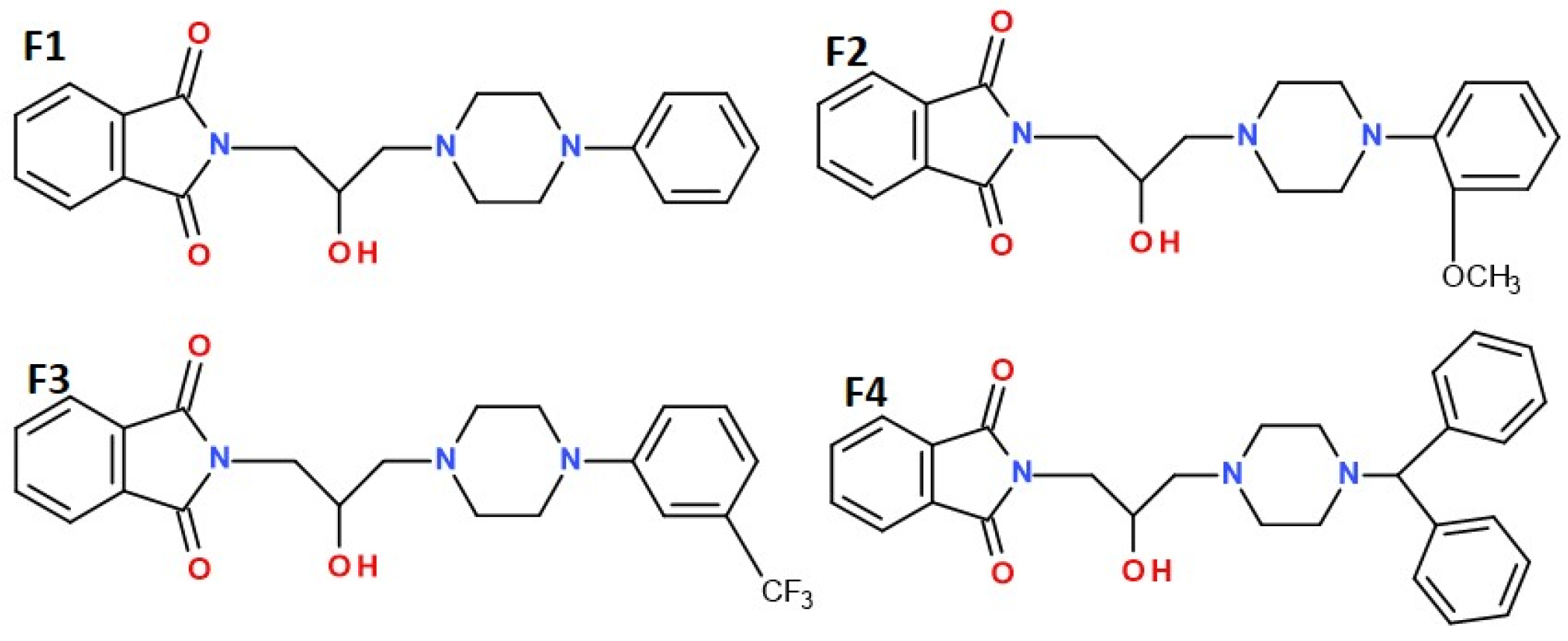
1. Introduction
2. Results
2.1. Pharmacokinetic Results
2.2. In Vivo Pharmacological Study Results
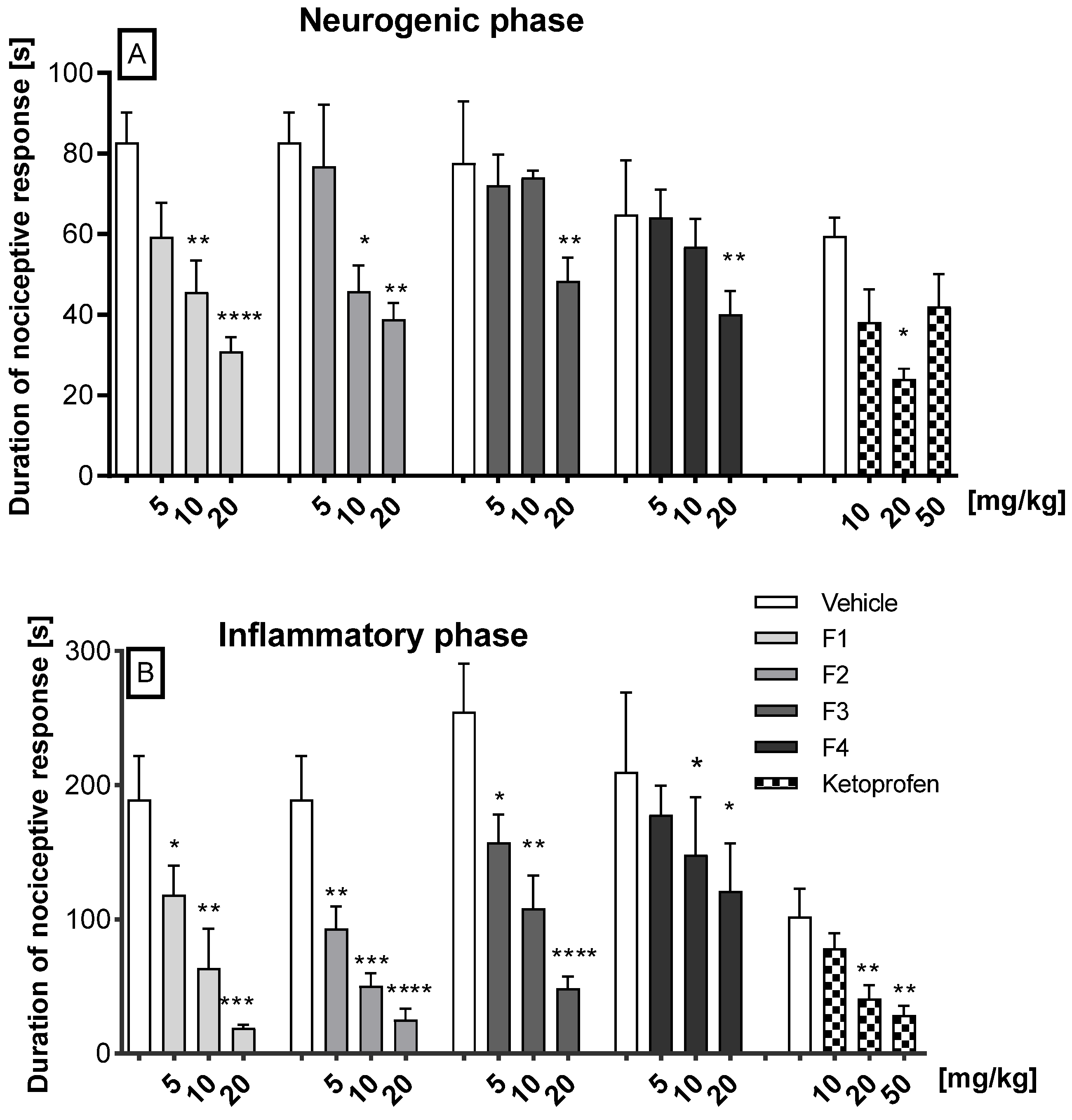
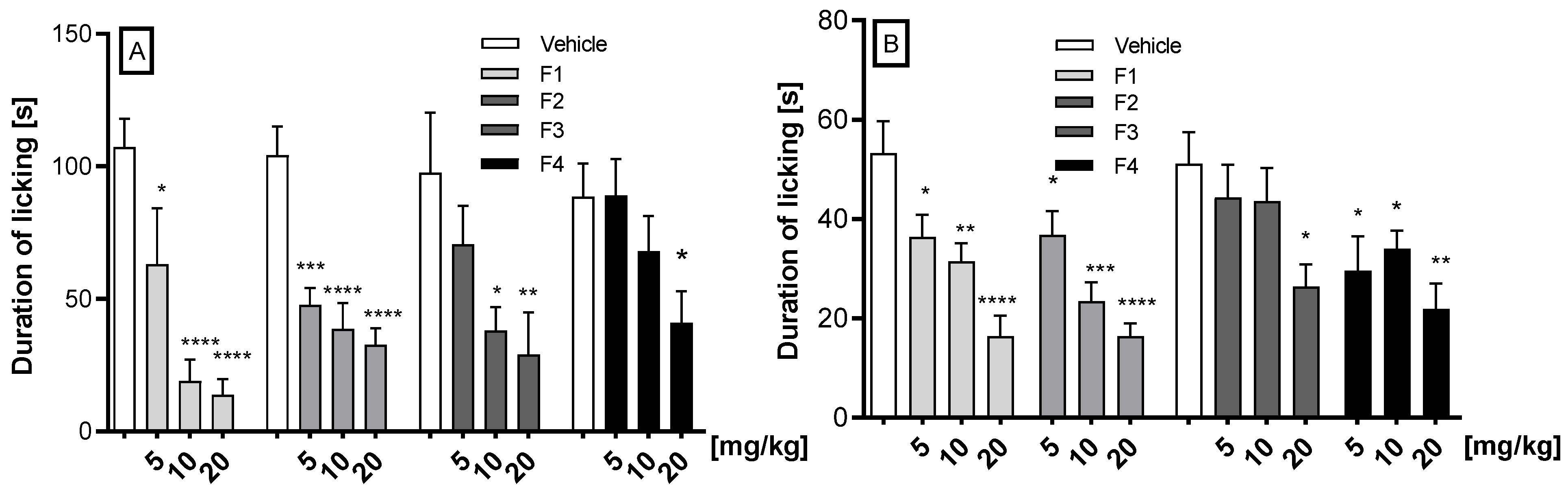
2.2.1. Antinociceptive Activity in the Formalin Test
2.2.2. Antinociceptive Activity in the Glutamate- and Capsaicin-Induced Pain Tests
2.2.3. Carrageenan-Induced Inflammatory Edema and Hyperalgesia
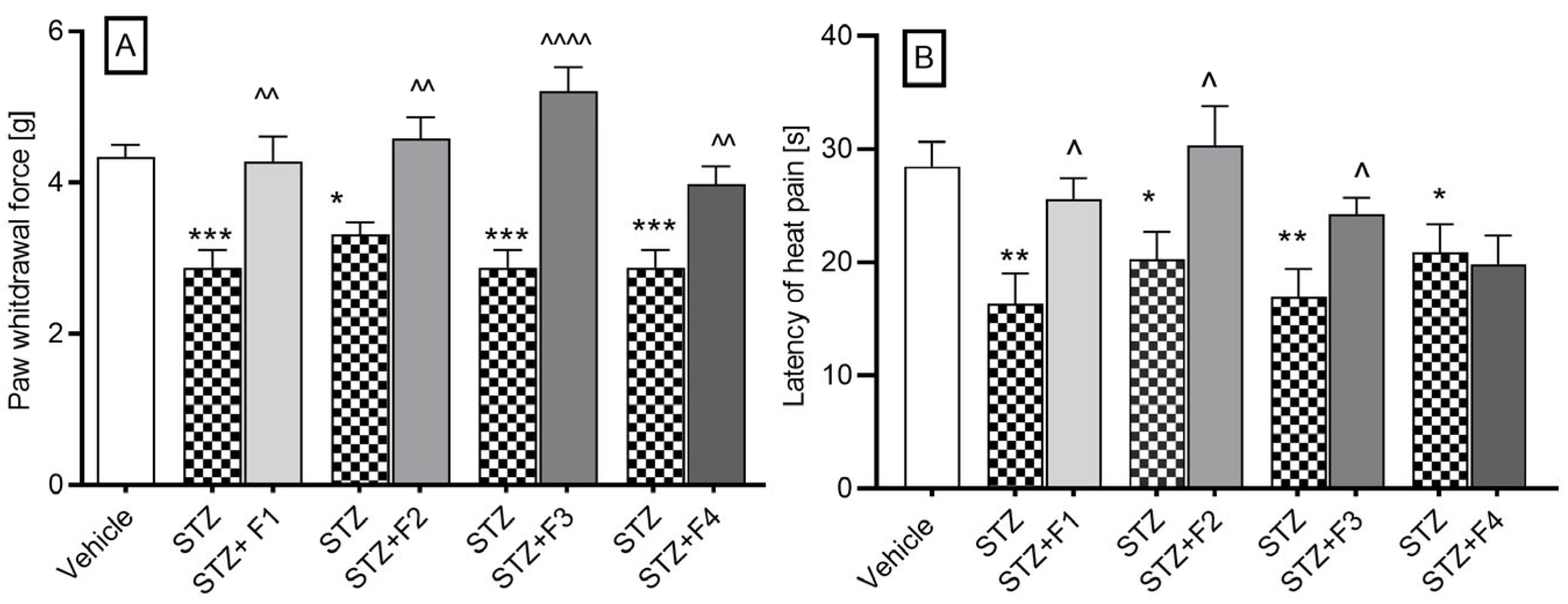
2.2.4. Streptozotocin-Induced Diabetic Neuropathy
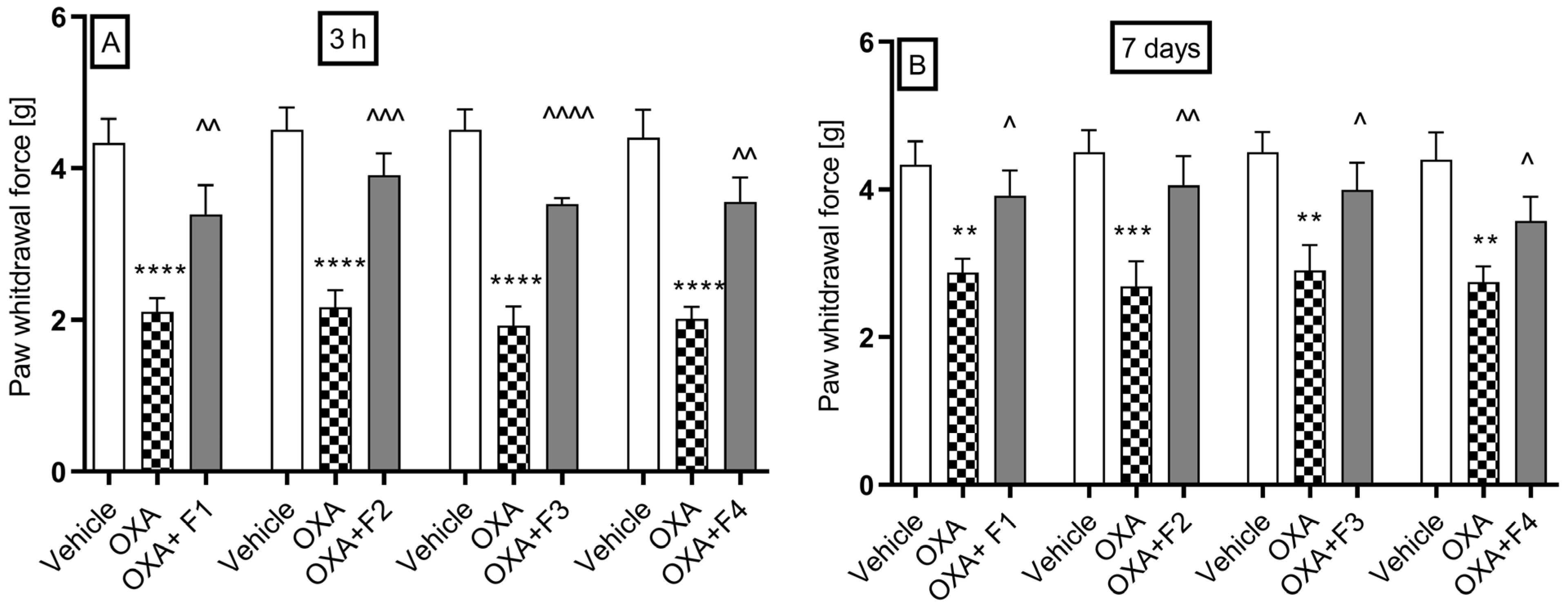
2.2.5. Oxaliplatin-Induced Neuropathic Pain
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs and Doses
4.3. Pharmacokinetic Study
4.3.1. Study Design
4.3.2. Sample Preparation and Analytical Method
4.4. In Vivo Pharmacological Studies
4.4.1. Formalin Test
4.4.2. Glutamate Test
4.4.3. Capsaicin-Induced Neurogenic Pain
4.4.4. Carrageenan-Induced Inflammatory Pain and Edema
4.4.5. Streptozotocin-Induced Diabetic Neuropathy
4.4.6. Oxaliplatin-Induced Neuropathic Pain
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fernandes, G.F.S.; Lopes, J.R.; Dos Santos, J.L.; Scarim, C.B. Phthalimide as a versatile pharmacophore scaffold: Unlocking its diverse biological activities. Drug Dev. Res. 2023, 84, 1346–1375. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.J.; McCoy, C. Thalidomide: A review of approved and investigational uses. Clin. Ther. 2003, 25, 342–395. [Google Scholar] [CrossRef] [PubMed]
- Fouquet, G.; Bories, C.; Guidez, S.; Renaud, L.; Herbaux, C.; Javed, S.; Facon, T.; Leleu, X. Pomalidomide for multiple myeloma. Expert Rev. Hematol. 2014, 7, 719–731. [Google Scholar] [CrossRef]
- Richardson, P.G.; Mitsiades, C.; Hideshima, T.; Anderson, K.C. Lenalidomide in multiple myeloma. Expert Rev. Anticancer Ther. 2006, 8, 1165–1173. [Google Scholar] [CrossRef]
- Schafer, P.H.; Parton, A.; Capone, L.; Cedzik, D.; Brady, H.; Evans, J.F.; Man, H.W.; Muller, G.W.; Stirling, D.I.; Chopra, R. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell Signal. 2014, 26, 2016–2029. [Google Scholar] [CrossRef]
- Dziubina, A.; Szkatuła, D.; Gdula-Argasińska, J.; Sapa, J. Synthesis and antinociceptive activity of four 1H-isoindolo-1,3(2H)-diones. J. Arch. Pharm. 2022, 355, e2100423. [Google Scholar] [CrossRef]
- Marciniak, A.; Kotynia, A.; Szkatuła, D.; Krzyżak, E. The 2-hydroxy-3-(4-aryl-1-piperazinyl)propyl phthalimide derivatives as prodrugs-Spectroscopic and theoretical binding studies with plasma proteins. Int. J. Mol. Sci. 2022, 23, 7003. [Google Scholar] [CrossRef]
- Muley, M.M.; Krustev, E.; McDougall, J.J. Preclinical Assessment of Inflammatory Pain. CNS Neurosci. Ther. 2015, 22, 88–101. [Google Scholar] [CrossRef]
- Torres-Lopez, J.E.; Ortiz, M.I.; Castaneda-Hernandez, G.; Alonso-Lopez, R.; Asomoza-Espinosa, R.; Granados-Soto, V. Comparison of the antinociceptive effect of celecoxib, diclofenac and resveratrol in the formalin test. Life Sci. 2002, 70, 1669–1676. [Google Scholar] [CrossRef]
- López-Cano, M.; Fernández-Dueñas, V.; Llebaria, A.; Ciruela, F. Formalin murine model of pain. Bio Protoc. 2017, 7, e2628. [Google Scholar] [CrossRef]
- McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M. TRPA1 agonist mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA 2007, 104, 13525–13530. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Zhao, J.; Liu, S.-B.; Yang, C.-J.; Wang, Y.-Q.; Wu, G.-C.; Xu, D.-M.; Mi, W.-L. Interleukin-33 mediates formalin-induced inflammatory pain in mice. Neuroscience 2013, 241, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nozaki-Taguchi, N. The role of cyclooxygenase-1 and -2 in the rat formalin test. Anesth. Analg. 2002, 94, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Dickenson, A.H.; Sullivan, A.F. Peripheral origins and central modulation of subcutaneous formalin-induced activity of rat dorsal horn neurones. Neurosci. Lett. 1987, 83, 207–211. [Google Scholar] [CrossRef]
- Batista, C.R.A.; Godin, A.M.; Melo, I.S.F.; Coura, G.M.E.; Matsui, T.C.; Dutra, M.M.G.B.; Brito, A.M.S.; Canhestro, W.G.; Alves, R.J.; Araújo, D.P.; et al. The phthalimide analogues N-3-hydroxypropylphthalimide and N-carboxymethyl-3-nitrophthalimide exhibit activity in experimental models of inflammatory and neuropathic pain. Pharmacol Rep. 2019, 71, 1177–1183. [Google Scholar] [CrossRef]
- Frias, B.; Merighi, A. Capsaicin, nociception and pain. Molecules 2016, 21, 797. [Google Scholar] [CrossRef]
- Jin, Y.H.; Yamaki, F.; Takemura, M.; Koike, Y.; Furuyama, A.; Yonehara, N. Capsaicin-induced glutamate release is implicated in nociceptive processing through activation of ionotropic glutamate receptors and group I metabotropic glutamate receptor in primary afferent fibers. J. Pharmacol. Sci. 2009, 109, 233–241. [Google Scholar] [CrossRef]
- Sałat, K.; Moniczewski, A.; Librowski, T. Transient receptor potential channels—Emerging novel drug targets for the treatment of pain. Curr. Med. Chem. 2013, 20, 1409–1436. [Google Scholar] [CrossRef]
- Song, T.; Wang, L.; Gu, K.; Yang, Y.; Yang, L.; Ma, P.; Ma, X.; Zhao, J.; Yan, R.; Guan, J.; et al. Involvement of peripheral TRPV1 channels in the analgesic effects of thalidomide. Neurochem. Int. 2015, 85–86, 40–45. [Google Scholar] [CrossRef]
- dos Santos, J.L.; Lanaro, C.; Lima, L.M.; Gambero, S.; Franco-Penteado, C.F.; Alexandre-Moreira, M.S.; Wade, M.; Yerigenahally, S.; Kutlar, A.; Meiler, S.E.; et al. Design, synthesis, and pharmacological evaluation of novel hybrid compounds to treat sickle cell disease symptoms. J. Med. Chem. 2011, 54, 5811–5819. [Google Scholar] [CrossRef]
- Beirith, A.; Santos, A.R.S.; Calixto, J.B. The role of neuropeptides and capsaicin-sensitive fibres in glutamate-induced nociception and paw oedema in mice. Brain Res. 2003, 969, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; White, T.D.; Sawynok, J. Intraplantar injection of glutamate evokes peripheral adenosine release in the rat hind paw: Involvement of peripheral ionotropic glutamate receptors and capsaicin-sensitive sensory afferents. J. Neurochem. 2002, 80, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Hoffman, E.M.; Sutharshan, M.; Schechter, R. Glutamate pharmacology and metabolism in peripheral primary afferents: Physiological and pathophysiological mechanisms. Pharmacol. Ther. 2011, 130, 283–309. [Google Scholar] [CrossRef]
- Meotti, F.C.; dos Santos Coelho, I.; Santos, A.R.S. The Nociception induced by glutamate in mice is potentiated by protons released into the solution. J. Pain 2010, 11, 570–578. [Google Scholar] [CrossRef]
- Dziubina, A.; Szkatuła, D.; Gdula-Argasińska, J.; Kotańska, M.; Filipek, B. Antinociceptive, antiedematous, and antiallodynic activity of 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-dione derivatives in experimental models of pain. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 813–827. [Google Scholar] [CrossRef]
- Wahnou, H.; Chgari, O.; Ndayambaje, M.; Hba, S.; Ouadghiri, Z.; Limami, Y.; Oudghiri, M. Carrageenan and TLR4 cross talk: A comprehensive review of inflammatory responses in animal models. Recent Adv. Inflamm. Allergy Drug Discov. 2025, 19, 5–17. [Google Scholar] [CrossRef]
- Cunha, T.M.; Verri, W.A.; Silva, J.S.; Poole, S.; Cunha, F.Q.; Ferreira, F.H. A cascade of cytokine mediates mechanical inflammatory hypernociception in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 1755–1760. [Google Scholar] [CrossRef]
- Melo, I.S.F.; Ziviani, V.P.; Barbosa, B.C.M.; Rodrigues, F.F.; Silva, R.R.L.; da Silva Neto, L.; de Fátima, Â.; César, I.C.; Machado, R.R.; Coelho, M.M. Synthesis of 2-(2-(4-thioxo-3H-1,2-dithiole-5-yl) phenoxy)ethyl)isoindole-1,3-thione, a novel hydrogen sulfide-releasing phthalimide hybrid, and evaluation of its activity in models of inflammatory pain. Eur. J. Pharmacol. 2023, 938, 175409. [Google Scholar] [CrossRef]
- Eid, S.; Rumora, A.; Beirowski, E.; Bennett, D.L.; Hur, J.; Savelieff, M.G.; Feldman, E.L. New perspectives in diabetic neuropathy. Neuron 2023, 111, 2623–2641. [Google Scholar] [CrossRef]
- Pham, V.M.; Matsumura, S.; Katano, T.; Funatsu, N.; Ito, S. Diabetic neuropathy research: From mouse models to targets for treatment. Neural Regen Res. 2019, 14, 1870–1879. [Google Scholar] [CrossRef]
- Sandireddy, R.; Yerra, V.G.; Areti, A.; Komirishetty, P.; Kumar, A. Neuroinflammation and oxidative stress in diabetic neuropathy: Futuristic strategies based on these targets. Int. J. Endocrinol. 2014, 2014, 674987. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Abarca, A.B.; Avila-Rojas, S.H.; Barragán-Iglesias, P.; Pineda-Farias, J.B.; Granados-Soto, V. Formalin injection produces long-lasting hypersensitivity with characteristics of neuropathic pain. Eur. J. Pharmacol. 2017, 797, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Vissers, K.C.; Geenen, F.; Biermans, R.; Meert, T.F. Pharmacological correlation between the formalin test and the neuropathic pain behavior in different species with chronic constriction injury. Pharmacol. Biochem. Behav. 2006, 84, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zheng, Z.M. Animal models of diabetic neuropathic pain. Exp. Clin. Endocrinol. Diabetes 2014, 122, 100–106. [Google Scholar] [CrossRef]
- Obrosova, I.G. Diabetic painful and insensate neuropathy: Pathogenesis and potential treatments. Neurotherapeutics 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Ribeiro, R.A.; Vale, M.L.; Ferreira, S.H.; Cunha, F.Q. Analgesic effect of thalidomide on inflammatory pain. Eur. J. Pharmacol. 2000, 391, 97–103. [Google Scholar] [CrossRef]
- Taliyan, R.; Sharma, P.L. Possible mechanism of protective effect of thalidomide in STZ-induced-neuropathic pain behavior in rats. Inflammopharmacology 2012, 20, 89–97. [Google Scholar] [CrossRef]
- Xu, H.; Dang, S.J.; Cui, Y.Y.; Wu, Z.Y.; Zhang, J.F.; Mei, X.P.; Feng, Y.P.; Li, Y.Q. Systemic injection of thalidomide prevent and attenuate neuropathic pain and alleviate neuroinflammatory response in the spinal dorsal horn. J. Pain. Res. 2019, 12, 3221–3230. [Google Scholar] [CrossRef]
- Tian, J.; Song, T.; Wang, H.; Wang, W.; Zhang, Z.; Yan, R. Thalidomide alleviates bone cancer pain by down-regulating expressions of NF-kappaB and GFAP in spinal astrocytes in a mouse model. Int. J. Neurosci. 2019, 129, 896–903. [Google Scholar] [CrossRef]
- Chaudhry, V.; Cornblath, D.R.; Corse, A.; Freimer, M.; Simmons-O’Brien, E.; Vogelsang, G. Thalidomide-induced neuropathy. Neurology 2002, 59, 1872–1875. [Google Scholar] [CrossRef]
- Wang, H.; Luo, J.; Yuan, Y.; Meng, L. Sural nerve injury in Behçet’s disease patients treated with thalidomide. Clin. Neuropathol. 2025, 44, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Gagne, J.J.; Landon, J.; Avorn, J.; Kesselheim, A.S. Comparative effectiveness and safety of thalidomide and lenalidomide in patients with multiple myeloma in the United States of America: A population-based cohort study. Eur. J. Cancer 2017, 70, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Leleu, X.; Palumbo, A.; Moreau, P.; Delforge, M.; Cavo, M.; Ludwig, H.; Morgan, G.J.; Davies, F.E.; Sonneveld, P.; et al. Expert panel consensus statement on the optimal use of pomalidomide in relapsed and refractory multiple myeloma. Leukemia 2014, 28, 1573–1585. [Google Scholar] [CrossRef] [PubMed]
- De Logu, F.; Trevisan, G.; Marone, I.M.; Coppi, E.; Dalenogare, D.P.; Titiz, M.; Marini, M.; Landini, L.; Souza Monteiro de Araujo, D.; Li Puma, S.; et al. Oxidative stress mediates thalidomide-induced pain by targeting peripheral TRPA1 and central TRPV4. BMC Biol. 2020, 18, 197. [Google Scholar] [CrossRef]
- Stino, A.M.; Bumma, N.; Smith, R.; Davalos, L.; Allen, J.; Ye, J.C.; Pianko, M.; Campagnaro, E.; Fierro, C.; Awad, A.; et al. Lenalidomide in the treatment of anti-myelin-associated glycoprotein neuropathy: A phase 1 study to identify the maximum tolerated dose. Eur. J. Neurol. 2024, 31, e16164. [Google Scholar] [CrossRef]
- Stino, A.M.; Efebera, Y. Lenalidomide-responsive anti-myelin-associated glycoprotein neuropathy. Muscle Nerve 2017, 56, e31–e32. [Google Scholar] [CrossRef]
- Moalem, G.; Tracey, D.J. Immune and inflammatory mechanisms in neuropathic pain. Brain. Res. Rev. 2006, 51, 240–264. [Google Scholar] [CrossRef]
- Ewertz, M.; Qvortrup, C.; Eckhoff, L. Chemotherapy-induced peripheral neuropathy in patients treated with taxanes and platinum derivatives. Acta Oncol. 2015, 54, 587–591. [Google Scholar] [CrossRef]
- Carozzi, V.A.; Canta, A.; Chiorazzi, A. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci. Lett. 2015, 596, 90–107. [Google Scholar] [CrossRef]
- Kang, L.; Tian, Y.; Xu, S.; Chen, H. Oxaliplatin-induced peripheral neuropathy: Clinical features, mechanisms, prevention and treatment. J. Neurol. 2021, 268, 3269–3282. [Google Scholar] [CrossRef]
- Szkatuła, D.; Krzyżak, E.; Mogilski, S.; Sapa, J.; Filipek, B.; Świątek, P. Bioresearch of New 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones. Molecules 2020, 25, 5883. [Google Scholar] [CrossRef] [PubMed]
- Hunskaar, S.; Hole, K. The formalin test in mice: Dissociation between inflammatory and non-inflammatory pain. Pain 1987, 30, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Tjølsen, A.; Berge, O.G.; Hunskaar, S.; Rosland, J.H.; Hole, K. The formalin test: An evaluation of the method. Pain 1992, 51, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Beirith, A.; Santos, A.R.S.; Calixto, J.B. Mechanisms underlying the nociception and paw oedema caused by injection of glutamate into the mouse paw. Brain Res. 2002, 924, 219–228. [Google Scholar] [CrossRef]
- Sałat, K.; Filipek, B. Antinociceptive activity of transient receptor potential channel TRPV1, TRPA1, and TRPM8 antagonists in neurogenic and neuropathic pain models in mice. J. Zhejiang Univ. Sci. B 2015, 16, 167–178. [Google Scholar] [CrossRef]
- Winter, C.; Risley, E.; Nuss, G. Carrageenin-induced edema in the hind paw of the rat as an assay for anti-inflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar] [CrossRef]
- Lence, P. A new device for plethysmoscopic measuring of small objects. Arch. Int. Pharmacodyn. Ther. 1962, 136, 237–240. [Google Scholar]
- Randall, L.; Selitto, J. A method for measurement of analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther. 1957, 111, 409–419. [Google Scholar]
- Tanabe, M.; Murakami, T.; Ono, H. Zonisamide Suppresses Pain Symptoms of Formalin-Induced Inflammatory and Streptozotocin-Induced Diabetic Neuropathy. J. Pharmacol. Sci. 2008, 107, 213–220. [Google Scholar] [CrossRef]
- Sałat, K.; Gawlik, K.; Witalis, J.; Pawlica-Gosiewska, D.; Filipek, B.; Solnica, B.; Więckowski, K.; Malawska, B. Evaluation of antinociceptive and antioxidant properties of 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]-dihydrofuran-2-one in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 493–505. [Google Scholar] [CrossRef]
- Dziubina, A.; Rapacz, A.; Czopek, A.; Góra, M.; Obniska, J.; Kamiński, K. Antinociceptive and Antiallodynic Activity of Some 3-(3-Methylthiophen-2- yl)pyrrolidine-2,5-dione derivatives in mouse models of tonic and neuropathic pain. Int. J. Mol. Sci. 2022, 23, 4057. [Google Scholar] [CrossRef] [PubMed]
- Eddy, N.B.; Leimbach, D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J. Pharmacol. Exp. Ther. 1953, 107, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Sałat, K.; Cios, A.; Wyska, E.; Sałat, R.; Mogilski, S.; Filipek, B.; Więckowski, K.; Malawska, B. Antiallodynic and antihyperalgesic activity of 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]-dihydrofuran-2-one compared to pregabalin in chemotherapy-induced neuropathic pain in mice. Pharmacol. Biochem. Behav. 2014, 122, 173–181. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetic Parameters | F1 | F2 | F3 | F4 | ||||
|---|---|---|---|---|---|---|---|---|
| Serum | Brain | Serum | Brain | Serum | Brain | Serum | Brain | |
| Cmax [µg/L(kg)] | 595.75 | 970 | 699.75 | 465.38 | 210.25 | 202.25 | 130.58 | 163.5 |
| tmax [h] | 0.083 | 0.083 | 0.083 | 0.083 | 0.083 | 0.25 | 0.083 | 0.25 |
| λz [h−1] | 0.73 | 0.76 | 0.92 | 0.77 | 0.55 | 0.53 | 0.86 | 0.89 |
| t1/2(λz) [h] | 0.95 | 0.91 | 0.75 | 0.90 | 1.26 | 1.31 | 0.80 | 0.78 |
| CL/F [L/h/kg] | 23.67 | - | 16.76 | - | 17.84 | - | 48.02 | - |
| AUC0–8h [µg∙h/L(kg)] | 205.95 | 368.04 | 292.78 | 230.83 | 258.81 | 520.05 | 101.99 | 164.50 |
| AUC0-inf [µg∙h/L(kg)] | 211.22 | 374.60 | 298.26 | 237.16 | 280.26 | 573.90 | 104.12 | 168.99 |
| Vz/F [L/kg] | 32.56 | - | 18.22 | - | 32.44 | - | 55.75 | - |
| MRT [h] | 1.32 | 1.36 | 1.14 | 1.41 | 2.8 | 3.16 | 1.47 | 1.63 |
| Kbrain/serum | - | 1.77 | - | 0.79 | - | 2.05 | - | 1.62 |
| Compounds [mg/kg] | Change in Edema Volume (mL) | |||
|---|---|---|---|---|
| 1 h | 2 h | 3 h | ||
| Control | 1.51 ± 0.04 | 1.83 ± 0.04 | 2.01 ± 0.05 | |
| F1 | 10 | 1.14 ± 0.05 **** | 1.51 ± 0.09 ** | 1.67± 0.09 * |
| 20 | 1.09 ± 0.02 **** | 1.15 ± 0.07 **** | 1.55 ± 0.09 ** | |
| Control | 1.57 ± 0.05 | 1.98 ± 0.02 | 2.12 ± 0.04 | |
| F2 | 10 | 1.51 ± 0.04 ns | 1.82 ± 0.03 ** | 1.99 ± 0.02 ns |
| 20 | 1.15 ± 0.08 *** | 1.63 ± 0.04 **** | 1.91± 0.03 ns | |
| Control | 1.37 ± 0.03 | 1.82 ± 0.02 | 2.09 ± 0.05 | |
| F3 | 10 | 1.37 ± 0.04 ns | 1.70 ± 0.04 ns | 1.89 ± 0.03 * |
| 20 | 1.14 ± 0.05 ** | 1.47 ± 0.07 *** | 1.67 ± 0.06 **** | |
| Control | 1.50 ± 0.04 | 1.88 ± 0.05 | 2.19 ± 0.05 | |
| F4 | 10 | 1.37 ± 0.04 * | 1.56 ± 0.06 *** | 1.85 ± 0.04 **** |
| 20 | 1.23 ± 0.01 *** | 1.47 ± 0.05 **** | 1.73 ± 0.02 **** | |
| Control | 1.51 ± 0.04 | 1.83 ± 0.04 | 2.01 ± 0.05 | |
| Ketoprofen | 20 | 0.92 ± 0.05 **** | 0.97 ± 0.02 **** | 1.29 ± 0.05 **** |
| Compounds [mg/kg] | Pain Threshold (g) | ||||
|---|---|---|---|---|---|
| 0 h | 1 h | 2 h | 3 h | ||
| Control | 130.8 ± 2.4 | 119.2 ± 0.8 | 119.2 ± 1.5 | 117.5 ± 1.1 | |
| F1 | 10 | 128.3 ± 1.1 | 138.3 ± 2.1 **** | 142.5 ± 2.1 **** | 134.2 ± 2.1 **** |
| 20 | 130.0 ± 2.9 | 143.3 ± 2.5 **** | 148.3 ± 2.8 **** | 146.7 ± 1.1 **** | |
| Control | 137.5 ± 1.1 | 131.7 ± 1.7 | 124.2 ± 2 | 125 ± 1.8 | |
| F2 | 10 | 132.5 ± 2.1 | 135. ± 1.3 ns | 127.5 ± 1.5 ns | 130 ± 2.6 ns |
| 20 | 132.5 ± 2.8 | 147.5 ± 2.5 **** | 141.7± 1.7 **** | 139.2 ± 0.8 ** | |
| Control | 140.8 ± 2.0 | 132.5 ± 2.5 | 126.7 ± 1.05 | 125 ± 1.8 | |
| F3 | 10 | 134.2 ± 2.4 | 141.7 ± 1.7 * | 142.5 ± 2.1 ** | 137.5 ± 1.7 ** |
| 20 | 135.8 ± 1.5 | 148.3± 3.3 ** | 150.8 ± 3.9 **** | 148.3 ± 3.1 **** | |
| Control | 134.2 ± 0.8 | 126.0 ± 2.0 | 125.0 ± 1.8 | 123.3 ± 1.8 | |
| F4 | 10 | 131.7 ± 1.7 | 139.2 ± 2.0 *** | 141.7 ± 1.7 **** | 139.2 ± 1.2 **** |
| 20 | 134.2 ± 1.5 | 144.2 ± 0.8 **** | 145 ± 1.5 **** | 140.8 ± 0.8 **** | |
| Control | 159.2 ± 8.9 | 130 ± 2.9 | 135.8 ± 3.8 | 133.3 ± 5.3 | |
| Ketoprofen | 20 | 130 ± 2.9 | 155.8 ± 5.2 ** | 187.5 ± 9.0 *** | 178.3 ± 11.7 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziubina, A.; Szkatuła, D.; Szczukowski, Ł.; Szafarz, M.; Rapacz, A. Comprehensive Evaluation of 1H-Isoindole-1,3(2H)-Dione Derivatives: Pharmacokinetic Studies and Analgesic Potential in Various Pain Models. Int. J. Mol. Sci. 2025, 26, 6026. https://doi.org/10.3390/ijms26136026
Dziubina A, Szkatuła D, Szczukowski Ł, Szafarz M, Rapacz A. Comprehensive Evaluation of 1H-Isoindole-1,3(2H)-Dione Derivatives: Pharmacokinetic Studies and Analgesic Potential in Various Pain Models. International Journal of Molecular Sciences. 2025; 26(13):6026. https://doi.org/10.3390/ijms26136026
Chicago/Turabian StyleDziubina, Anna, Dominika Szkatuła, Łukasz Szczukowski, Małgorzata Szafarz, and Anna Rapacz. 2025. "Comprehensive Evaluation of 1H-Isoindole-1,3(2H)-Dione Derivatives: Pharmacokinetic Studies and Analgesic Potential in Various Pain Models" International Journal of Molecular Sciences 26, no. 13: 6026. https://doi.org/10.3390/ijms26136026
APA StyleDziubina, A., Szkatuła, D., Szczukowski, Ł., Szafarz, M., & Rapacz, A. (2025). Comprehensive Evaluation of 1H-Isoindole-1,3(2H)-Dione Derivatives: Pharmacokinetic Studies and Analgesic Potential in Various Pain Models. International Journal of Molecular Sciences, 26(13), 6026. https://doi.org/10.3390/ijms26136026