
A Pilot Metabolomic Study for Diagnosing Aspergillus Infection in Immunocompromised Pediatric Cancer Patients
 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Study Subjects
2.2. Sample Collection and Preparation
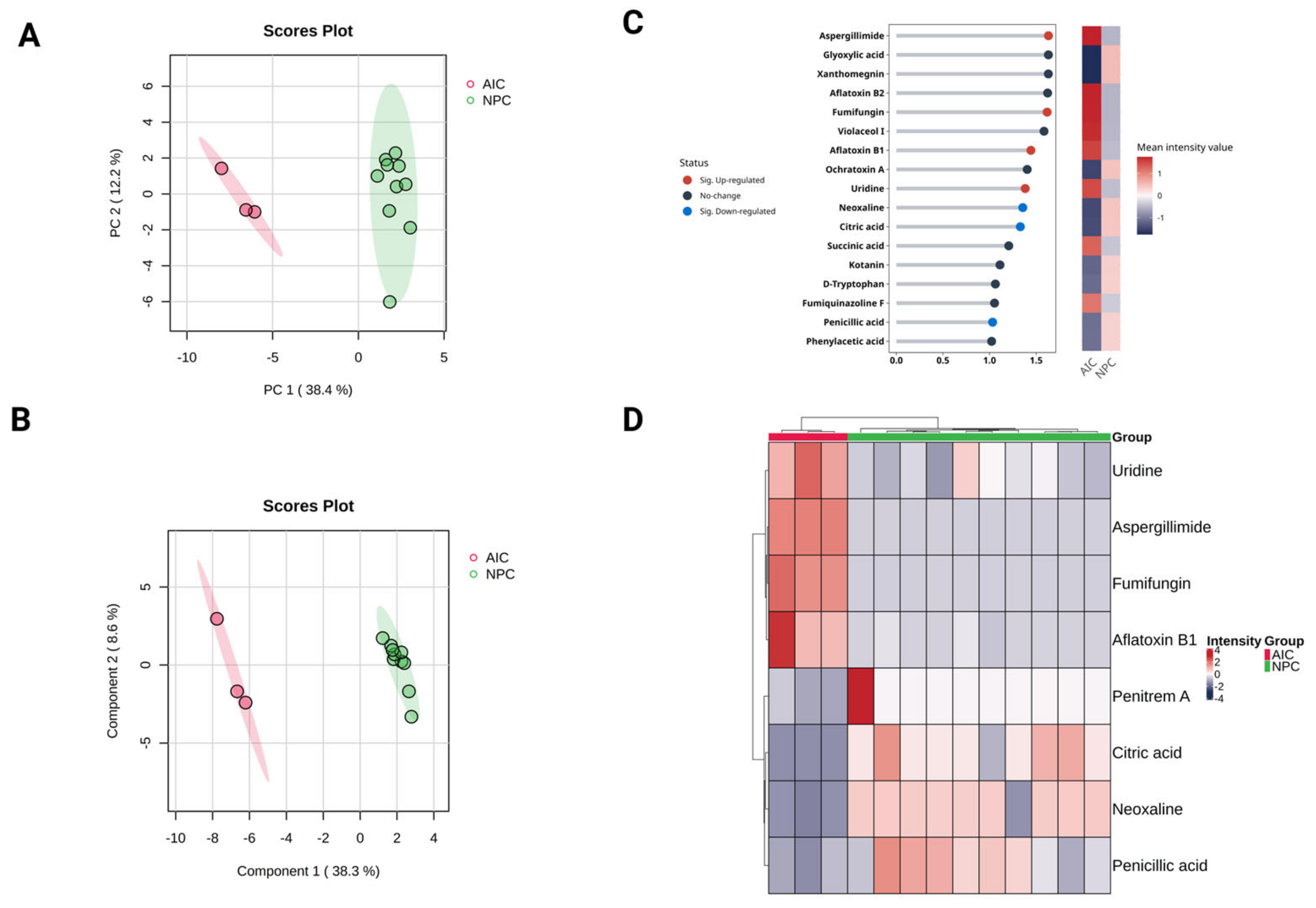
2.3. Univariate Statistical Analysis
2.4. Multivariate Statistical Analysis
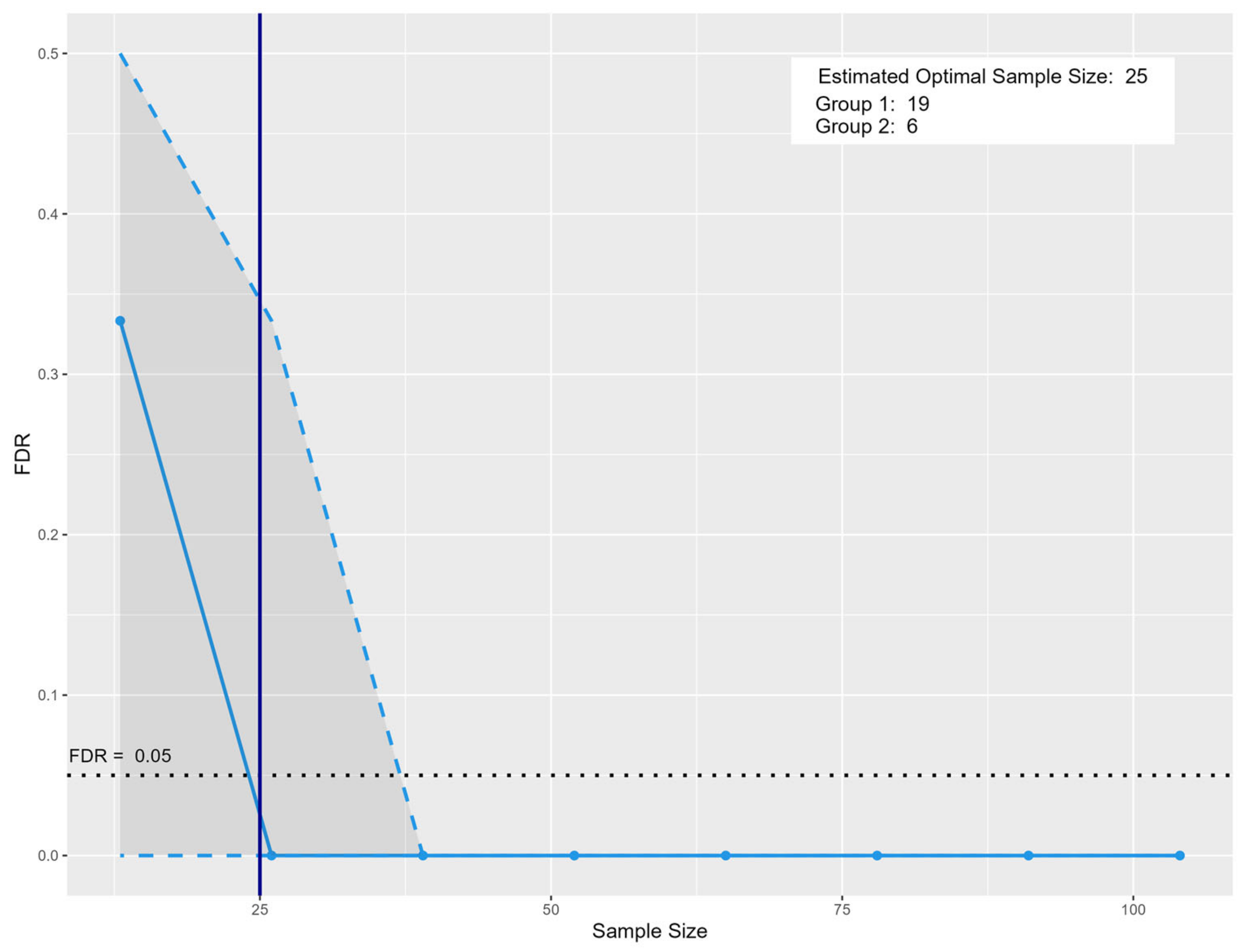
2.5. Sample Size
3. Discussion
4. Materials and Methods
4.1. Study Participants
4.2. Reagents and Chemicals
4.3. Sample Pre-Processing for Metabolomics Analysis
4.4. Analysis of UHPLC-MS/MS Using Information-Dependent Acquisition (IDA)
4.5. Development of a Metabolite Identification Database
4.6. Metabolomics Data Analysis
4.7. Data Pre-Processing and Statistical Analysis
4.8. Sample Size Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marr, K.A.; Carter, R.A.; Boeckh, M.; Martin, P.; Corey, L. Invasive aspergillosis in allogeneic stem cell transplant recipients: Changes in epidemiology and risk factors. Blood 2002, 100, 4358–4366. [Google Scholar] [CrossRef] [PubMed]
- Rayens, E.; Norris, K.A. Prevalence and Healthcare Burden of Fungal Infections in the United States, 2018. Open Forum Infect. Dis. 2022, 9, ofab593. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef] [PubMed]
- Leeflang, M.M.; Debets-Ossenkopp, Y.J.; Wang, J.; Visser, C.E.; Scholten, R.J.; Hooft, L.; Bijlmer, H.A.; Reitsma, J.B.; Zhang, M.; Bossuyt, P.M.; et al. Galactomannan detection for invasive aspergillosis in immunocompromised patients. Cochrane Database Syst. Rev. 2015, 12, CD007394. [Google Scholar] [CrossRef]
- Verweij, P.E.; Mennink-Kersten, M. Issues with galactomannan testing. Med. Mycol. 2006, 44, S179–S183. [Google Scholar] [CrossRef]
- Hadrich, I.; Makni, F.; Sellami, H.; Cheikhrouhou, F.; Sellami, A.; Bouaziz, H.; Hdiji, S.; Elloumi, M.; Ayadi, A. Invasive aspergillosis: Epidemiology and environmental study in haematology patients (Sfax, Tunisia). Mycoses 2010, 53, 443–447. [Google Scholar] [CrossRef]
- Karapinar, D. A Review of a Diagnostic Tool: Galactomannan. J. Immunol. Sci. 2018, 2, 38–42. [Google Scholar] [CrossRef]
- Alves, V.; Zamith-Miranda, D.; Frases, S.; Nosanchuk, J.D. Fungal Metabolomics: A Comprehensive Approach to Understanding Pathogenesis in Humans and Identifying Potential Therapeutics. J. Fungi 2025, 11, 93. [Google Scholar] [CrossRef]
- Fernández-García, M.; Rojo, D.; Rey-Stolle, F.; García, A.; Barbas, C. Metabolomic-Based Methods in Diagnosis and Monitoring Infection Progression; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 283–315. [Google Scholar]
- Xie, H.; Wang, X.; van der Hooft, J.J.; Medema, M.H.; Chen, Z.Y.; Yue, X.; Zhang, Q.; Li, P. Fungi population metabolomics and molecular network study reveal novel biomarkers for early detection of aflatoxigenic Aspergillus species. J. Hazard. Mater. 2022, 424, 127173. [Google Scholar] [CrossRef]
- Zhou, B.; Xiao, J.F.; Tuli, L.; Ressom, H.W. LC-MS-Based metabolomics. Mol. BioSyst. 2012, 8, 470–481. [Google Scholar] [CrossRef]
- Gupta, R.; Gaur, S. LC-MS investigated as a tool to study the metabolomic characteristics of cereal fermentation. Appl. Food Res. 2024, 4, 100365. [Google Scholar] [CrossRef]
- Lin, C.; Tian, Q.; Guo, S.; Xie, D.; Cai, Y.; Wang, Z.; Chu, H.; Qiu, S.; Tang, S.; Zhang, A. Metabolomics for Clinical Biomarker Discovery and Therapeutic Target Identification. Molecules 2024, 29, 2198. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Shu, J.; Li, M.; Mudappathi, R.; Jin, Y.; Lewis, F.; Boon, A.; Qin, X.; Liu, L.; Gu, H. Artificial intelligence in metabolomics: A current review. TrAC Trends Anal. Chem. 2024, 178, 117852. [Google Scholar] [CrossRef]
- Hervás, C.; Martínez, A.C.; Silva, M.; Serrano, J.M. Improving the quantification of highly overlapping chromatographic peaks by using product unit neural networks modeled by an evolutionary algorithm. J. Chem. Inf. Model. 2005, 45, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Kensert, A.; Bosten, E.; Collaerts, G.; Efthymiadis, K.; Van Broeck, P.; Desmet, G.; Cabooter, D. Convolutional neural network for automated peak detection in Reversed-Phase liquid chromatography. J. Chromatogr. A 2022, 1672, 463005. [Google Scholar] [CrossRef]
- Shen, X.; Gong, X.; Cai, Y.; Guo, Y.; Tu, J.; Li, H.; Zhang, T.; Wang, J.; Xue, F.; Zhu, Z.-J. Normalization and integration of Large-Scale metabolomics data using support vector regression. Metabolomics 2016, 12, 89. [Google Scholar] [CrossRef]
- Barberis, E.; Khoso, S.; Sica, A.; Falasca, M.; Gennari, A.; Dondero, F.; Afantitis, A.; Manfredi, M. Precision Medicine Approaches with Metabolomics and Artificial Intelligence. Int. J. Mol. Sci. 2022, 23, 11269. [Google Scholar] [CrossRef]
- Quazi, S. Artificial intelligence and machine learning in precision and genomic medicine. Med. Oncol. 2022, 39, 120. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, R.; Payra, S.; Singh, S.K. Artificial intelligence and machine learning in pharmacological research: Bridging the gap between data and drug discovery. Cureus 2023, 15, e44359. [Google Scholar] [CrossRef]
- Alshannaq, A.; Yu, J.-H. Occurrence, Toxicity, and Analysis of Major Mycotoxins in Food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef]
- Kumar, P.; Kausar, M.A.; Singh, A.B.; Singh, R. Biological contaminants in the indoor air environment and their impacts on human health. Air Qual. Atmos. Health 2021, 14, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Fischbach, M.A. Small molecules from the human microbiota. Science 2015, 349, 1254766. [Google Scholar] [CrossRef] [PubMed]
- Rushing, B.R.; Selim, M.I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem. Toxicol. 2019, 124, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Nishimoto, Y.; Nozaki, H. Asperparaline A, a new paralytic alkaloid from Aspergillus japonicus JV-23. Tetrahedron Lett. 1997, 38, 5655–5658. [Google Scholar] [CrossRef]
- Banks, R.M.; Blanchflower, S.E.; Everett, J.R.; Manger, B.R.; Reading, C. Novel Anthelmintic Metabolites from an Aspergillus Species; the Aspergillimides. J. Antibiot. 1997, 50, 840–846. [Google Scholar] [CrossRef]
- Mukhopadhyay, T.; Roy, K.; Coutinho, L.; Rupp, R.H.; Ganguli, B.N.; Fehlhaber, H.W. Fumifungin, a new antifungal antibiotic from Aspergillus fumigatus Fresenius 1863. J. Antibiot. 1987, 40, 1050–1052. [Google Scholar] [CrossRef]
- Tamiya, H.; Ochiai, E.; Kikuchi, K.; Yahiro, M.; Toyotome, T.; Watanabe, A.; Yaguchi, T.; Kamei, K. Secondary metabolite profiles and antifungal drug susceptibility of Aspergillus fumigatus and closely related species, Aspergillus lentulus, Aspergillus udagawae, and Aspergillus viridinutans. J. Infect. Chemother. 2015, 21, 385–391. [Google Scholar] [CrossRef]
- D’Enfert, C.; Diaquin, M.; Delit, A.; Wuscher, N.; Debeaupuis, J.P.; Huerre, M.; Latge, J.P. Attenuated virulence of Uridine-Uracil auxotrophs of Aspergillus fumigatus. Infect. Immun. 1996, 64, 4401–4405. [Google Scholar] [CrossRef]
- Mangrum, M.M.; Vogel, A.K.; Wagner, A.S.; King, A.E.; Miao, J.; Zhou, Y.; Phillips, E.K.; Peters, B.M.; Reynolds, T.B. Disruption to de novo uridine biosynthesis alters β-1,3-glucan masking in Candida albicans. mSphere 2024, 9, e0028724. [Google Scholar] [CrossRef]
- Ye, L.; Jiang, Y.; Zhang, M. Crosstalk between glucose metabolism, lactate production and immune response modulation. Cytokine Growth Factor Rev. 2022, 68, 81–92. [Google Scholar] [CrossRef]
- Qian, X.; Yang, Z.; Mao, E.; Chen, E. Regulation of fatty acid synthesis in immune cells. Scand. J. Immunol. 2018, 88, e12713. [Google Scholar] [CrossRef] [PubMed]
- Sameh, M.; Khalaf, H.M.; Anwar, A.M.; Osama, A.; Ahmed, E.A.; Mahgoub, S.; Ezzeldin, S.; Tanios, A.; Alfishawy, M.; Said, A.F.; et al. Integrated multiomics analysis to infer COVID-19 biological insights. Sci. Rep. 2023, 13, 1802. [Google Scholar] [CrossRef]
- Ahmed, E.A.; El-Derany, M.O.; Anwar, A.M.; Saied, E.M.; Magdeldin, S. Metabolomics and Lipidomics Screening Reveal Reprogrammed Signaling Pathways toward Cancer Development in Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2022, 24, 210. [Google Scholar] [CrossRef]
- Decaestecker, T.N.; Vande Casteele, S.R.; Wallemacq, P.E.; Van Peteghem, C.H.; Defore, D.L.; Van Bocxlaer, J.F. Information-Dependent Acquisition-Mediated LC-MS/MS screening procedure with semiquantitative potential. Anal. Chem. 2004, 76, 6365–6373. [Google Scholar] [CrossRef]
- Ramzy, A.; Abdelmoneim, T.K.; Arafat, M.; Mokhtar, M.; Bakkar, A.; Mokhtar, A.; Anwar, W.; Magdeldin, S.; Enany, S. Metabolomic analysis reveals key changes in amino acid metabolism in colorectal cancer patients. Amino Acids 2025, 57, 22. [Google Scholar] [CrossRef]
- Gil-de-la-Fuente, A.; Mamani-Huanca, M.; Stroe, M.C.; Saugar, S.; Garcia-Alvarez, A.; Brakhage, A.A.; Barbas, C.; Otero, A. Aspergillus Metabolome Database for Mass Spectrometry Metabolomics. J. Fungi 2021, 7, 387. [Google Scholar] [CrossRef]
- El-Elimat, T.; Figueroa, M.; Ehrmann, B.M.; Cech, N.B.; Pearce, C.J.; Oberlies, N.H. High-Resolution MS, MS/MS, and UV database of fungal secondary metabolites as a dereplication protocol for bioactive natural products. J. Nat. Prod. 2013, 76, 1709–1716. [Google Scholar] [CrossRef]
- Vivek-Ananth, R.P.; Sahoo, A.K.; Kumaravel, K.; Mohanraj, K.; Samal, A. MeFSAT: A curated natural product database specific to secondary metabolites of medicinal fungi. RSC Adv. 2021, 11, 2596–2607. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-Independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Chan, B.K.C. Data Analysis Using R Programming; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 47–122. [Google Scholar]
- Gotsmy, M.; Brunmair, J.; Büschl, C.; Gerner, C.; Zanghellini, J. Probabilistic quotient’s work and pharmacokinetics’ contribution: Countering size effect in metabolic time series measurements. BMC Bioinform. 2022, 23, 379. [Google Scholar] [CrossRef]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing Value Imputation Approach for Mass Spectrometry-Based Metabolomics Data. Sci. Rep. 2018, 8, 663. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R.; Broadhurst, D.; Smilde, A.K.; Kristal, B.S.; Baker, J.D.; Beger, R.; Bessant, C.; Connor, S.; Capuani, G.; Craig, A.; et al. Proposed minimum reporting standards for data analysis in metabolomics. Metabolomics 2007, 3, 231–241. [Google Scholar] [CrossRef]
- Mishra, P.; Pandey, C.M.; Singh, U.; Gupta, A.; Sahu, C.; Keshri, A. Descriptive statistics and normality tests for statistical data. Ann. Card. Anaesth. 2019, 22, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Dexter, F. Wilcoxon-Mann-Whitney Test Used for Data That Are Not Normally Distributed. Anesth. Analg. 2013, 117, 537–538. [Google Scholar] [CrossRef]
- Nyamundanda, G.; Gormley, I.C.; Fan, Y.; Gallagher, W.M.; Brennan, L. MetSizeR: Selecting the optimal sample size for metabolomic studies using an analysis based approach. BMC Bioinform. 2013, 14, 338. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Non-Fungal Pediatric Cancer (NPC) | Aspergillus-Infected Cancer (AIC) | |
|---|---|---|
| Age (Mean, ±SD) | 7 ± 3.3 | 11.6 ± 6.6 |
| Males (N) | 4 | NA |
| Females (N) | 6 | 3 |
| Type of cancer (N) | 3 ALL 2 WT 3 AML 1 NB 1 Lymphoma | 2 ALL 1 WT |
| Metabolite Name | log2 (FC) | p. Adjusted | Status |
|---|---|---|---|
| Citric acid * | −2.44 | 0.022 | Relatively quantified in both cohorts |
| Aspergillimide * | 1.82 | 0.022 | Relatively quantified in both cohorts |
| Aflatoxin B1 * | 1.71 | 0.022 | Relatively quantified in both cohorts |
| Fumifungin * | 1.35 | 0.022 | Relatively quantified in both cohorts |
| Uridine * | 1.24 | 0.022 | Relatively quantified in both cohorts |
| Penitrem A * | −1.13 | 0.022 | Relatively quantified in both cohorts |
| Aflatoxin B2 | 0.97 | 0.022 | Relatively quantified in both cohorts |
| Kotanin | −0.91 | 0.022 | Relatively quantified in both cohorts |
| Phenylacetic acid | −0.89 | 0.022 | Relatively quantified in both cohorts |
| Glyoxylic acid | −0.82 | 0.022 | Relatively quantified in both cohorts |
| Ochratoxin A | −0.71 | 0.022 | Relatively quantified in both cohorts |
| Xanthomegnin | −0.39 | 0.022 | Relatively quantified in both cohorts |
| Violaceol I | 0.17 | 0.022 | Relatively quantified in both cohorts |
| Penicillic acid * | −1.58 | 0.038 | Relatively quantified in both cohorts |
| Neoxaline * | −1.56 | 0.038 | Relatively quantified in both cohorts |
| Succinic acid | 2.16 | 0.064 | Relatively quantified in both cohorts |
| D-Tryptophan | −1.05 | 0.064 | Relatively quantified in both cohorts |
| Rubrofusarin | 0.55 | 0.064 | Relatively quantified in both cohorts |
| 4-Hydroxybenzaldehyde | −3.24 | 0.091 | Relatively quantified in both cohorts |
| Indole | −1.10 | 0.091 | Relatively quantified in both cohorts |
| Caproic acid | −0.42 | 0.091 | Relatively quantified in both cohorts |
| Kojic acid | −0.27 | 0.091 | Relatively quantified in both cohorts |
| Itaconic acid | −1.07 | 0.137 | Relatively quantified in both cohorts |
| Citrinin | −0.85 | 0.191 | Relatively quantified in both cohorts |
| Malformin | −4.28 | 0.244 | Relatively quantified in both cohorts |
| fumagillin | −0.90 | 0.244 | Relatively quantified in both cohorts |
| Silybin B | 0.17 | 0.244 | Relatively quantified in both cohorts |
| Mycophenolic acid | −2.13 | 0.306 | Relatively quantified in both cohorts |
| Gentisyl alcohol | 0.25 | 0.306 | Relatively quantified in both cohorts |
| Emodin | −0.94 | 0.379 | Relatively quantified in both cohorts |
| Fumiquinazoline F | 0.62 | 0.379 | Relatively quantified in both cohorts |
| Sulochrin | −2.06 | 0.460 | Relatively quantified in both cohorts |
| Warfarin | −0.11 | 0.460 | Relatively quantified in both cohorts |
| Lovastatin acid | 1.05 | 0.653 | Relatively quantified in both cohorts |
| Fumigaclavine A | 0.96 | 0.653 | Relatively quantified in both cohorts |
| Viridicatin | 0.20 | 0.653 | Relatively quantified in both cohorts |
| Cyclopeptine | −0.63 | 0.899 | Relatively quantified in both cohorts |
| Fumigaclavine C | −0.36 | 0.960 | Relatively quantified in both cohorts |
| Aflatoxin G | −0.17 | 0.960 | Relatively quantified in both cohorts |
| Imperatorin | −0.07 | 0.960 | Relatively quantified in both cohorts |
| Dehydrocyclopeptine | 0.01 | 1 | Relatively quantified in both cohorts |
| Taichunamide H | NA | NA | Uniquely quantified in NPC cohort |
| 6,8-dihydroxy-3-methylisocoumarin | NA | NA | Uniquely quantified in NPC cohort |
| Aflatoxin G2 | NA | NA | Uniquely quantified in NPC cohort |
| Orcinol | NA | NA | Uniquely quantified in NPC cohort |
| Oxaline | NA | NA | Uniquely quantified in NPC cohort |
| Silybin B | NA | NA | Uniquely quantified in NPC cohort |
| Viridicatumtoxin | NA | NA | Uniquely quantified in AIC cohort |
| Dihydroaflatoxin G1 | NA | NA | Uniquely quantified in AIC cohort |
| Asterric acid | NA | NA | Uniquely quantified in AIC cohort |
| Chrysophanol | NA | NA | Uniquely quantified in AIC cohort |
| Cyclopenol | NA | NA | Uniquely quantified in AIC cohort |
| Physcion | NA | NA | Uniquely quantified in AIC cohort |
| Sterigmatocystin | NA | NA | Uniquely quantified in AIC cohort |
| Trypacidin | NA | NA | Uniquely quantified in AIC cohort |
| Metabolite Name | log2 (FC) | p. Ajusted | Regulation |
|---|---|---|---|
| Citric acid | −2.438 | 0.022 | Downregulated |
| Penicillic acid | −1.585 | 0.038 | Downregulated |
| Neoxaline | −1.555 | 0.038 | Downregulated |
| Penitrem A | −1.133 | 0.022 | Downregulated |
| Uridine | 1.237 | 0.022 | Upregulated |
| Fumifungin | 1.351 | 0.022 | Upregulated |
| Aflatoxin B1 | 1.708 | 0.022 | Upregulated |
| Aspergillimide | 1.816 | 0.022 | Upregulated |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelmoneim, T.K.; Ramzy, A.; Zaki, M.A.; Karam, A.; Hesham, A.; Osama, A.; Sabar, N.; Mokhtar, M.; Youssef, N.A.; Ahmed, E.A.; et al. A Pilot Metabolomic Study for Diagnosing Aspergillus Infection in Immunocompromised Pediatric Cancer Patients. Int. J. Mol. Sci. 2025, 26, 5926. https://doi.org/10.3390/ijms26135926
Abdelmoneim TK, Ramzy A, Zaki MA, Karam A, Hesham A, Osama A, Sabar N, Mokhtar M, Youssef NA, Ahmed EA, et al. A Pilot Metabolomic Study for Diagnosing Aspergillus Infection in Immunocompromised Pediatric Cancer Patients. International Journal of Molecular Sciences. 2025; 26(13):5926. https://doi.org/10.3390/ijms26135926
Chicago/Turabian StyleAbdelmoneim, Taghreed Khaled, Asmaa Ramzy, Mostafa Ahmed Zaki, Ahmed Karam, Ahmed Hesham, Aya Osama, Nabila Sabar, Maha Mokhtar, Nada A. Youssef, Eman A. Ahmed, and et al. 2025. "A Pilot Metabolomic Study for Diagnosing Aspergillus Infection in Immunocompromised Pediatric Cancer Patients" International Journal of Molecular Sciences 26, no. 13: 5926. https://doi.org/10.3390/ijms26135926
APA StyleAbdelmoneim, T. K., Ramzy, A., Zaki, M. A., Karam, A., Hesham, A., Osama, A., Sabar, N., Mokhtar, M., Youssef, N. A., Ahmed, E. A., Shalaby, L., Salama, A., Kamel, A., Elenany, M., & Magdeldin, S. (2025). A Pilot Metabolomic Study for Diagnosing Aspergillus Infection in Immunocompromised Pediatric Cancer Patients. International Journal of Molecular Sciences, 26(13), 5926. https://doi.org/10.3390/ijms26135926