
The Involvement of the Endocannabinoid, Glutamatergic, and GABAergic Systems in PTSD


{kind=link}
{kind=link}
{kind=link}
Abstract
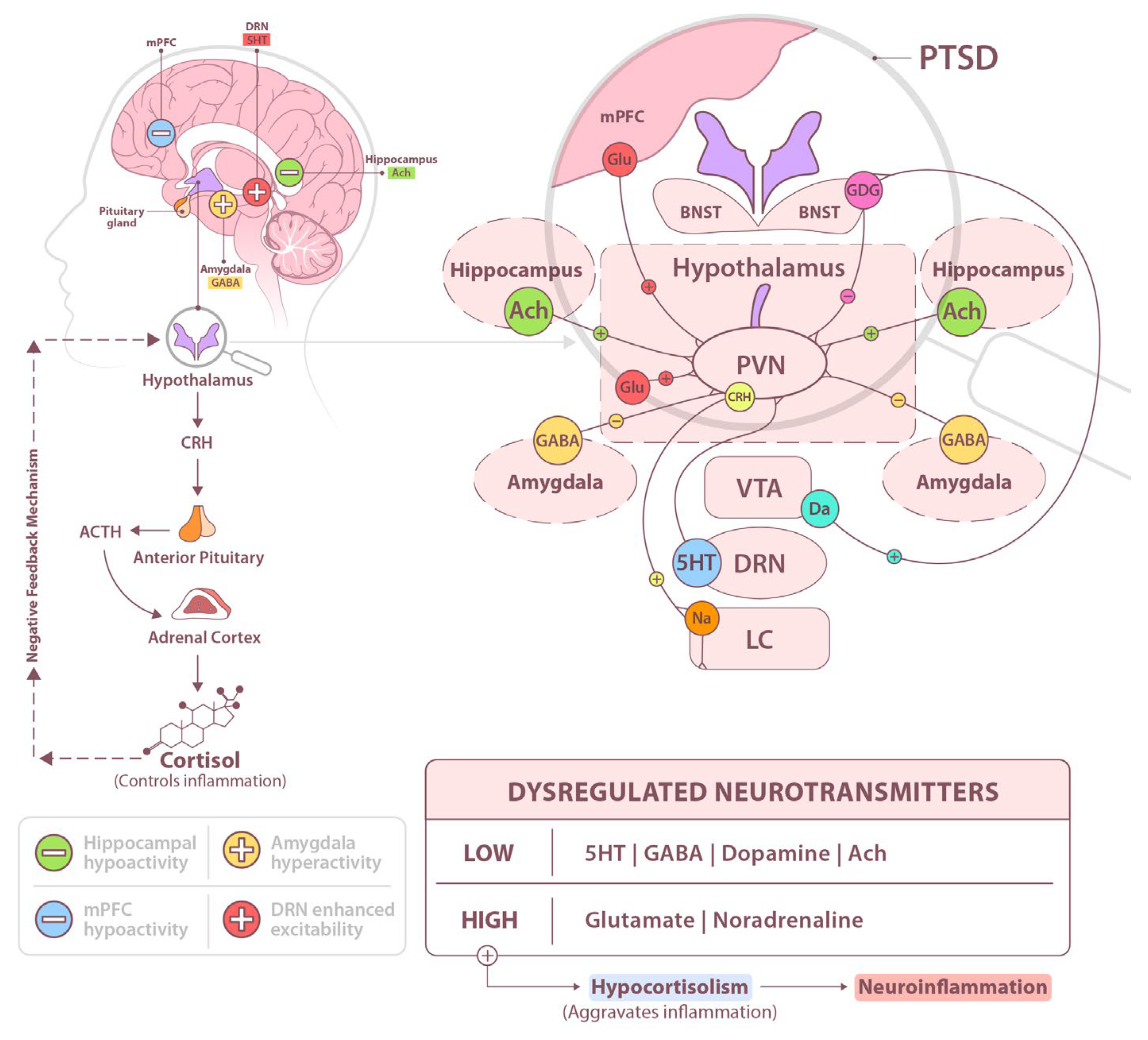
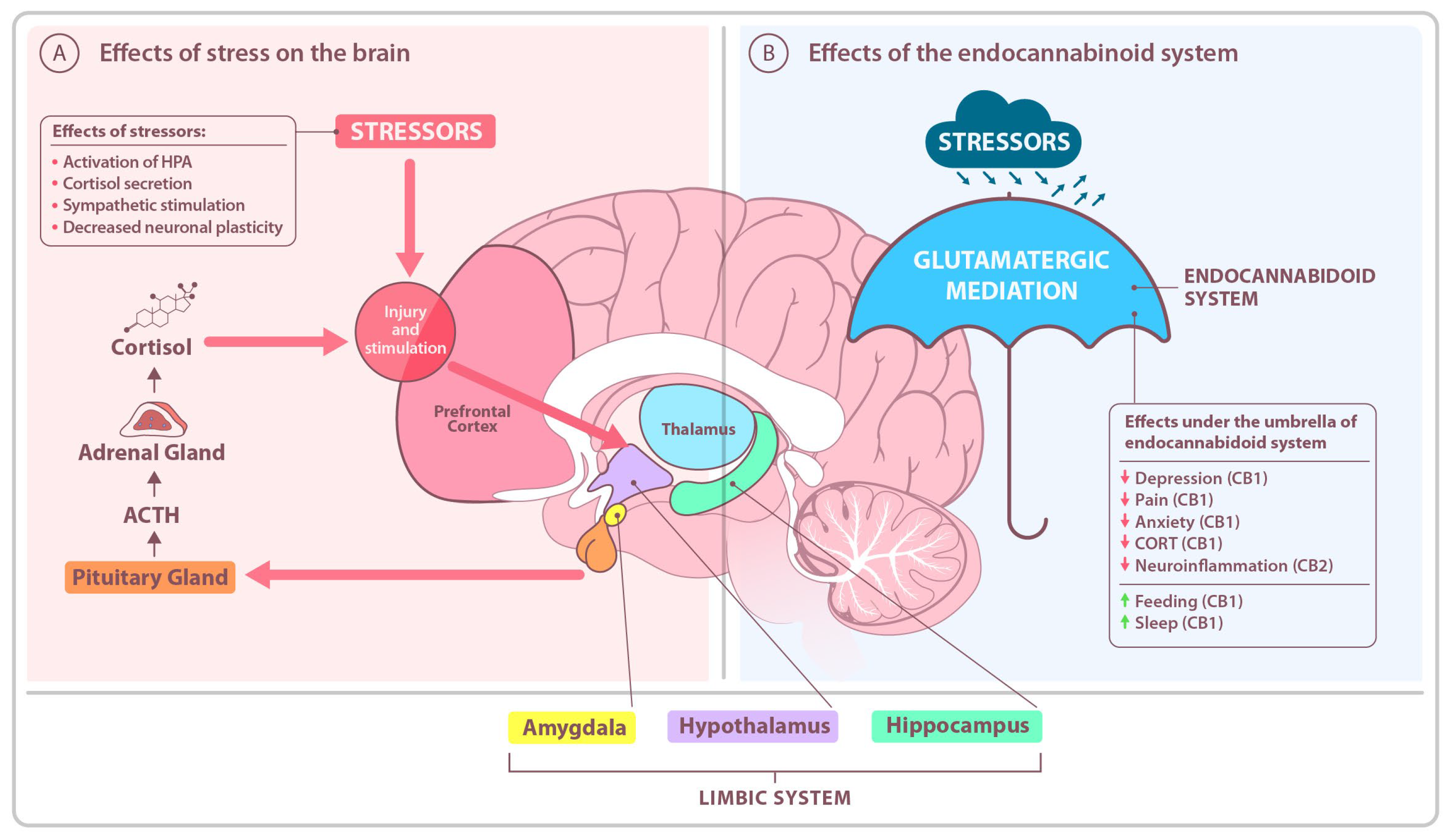
1. Introduction
2. Alterations in the Stress-Buffering Endocannabinoid System (ECS)
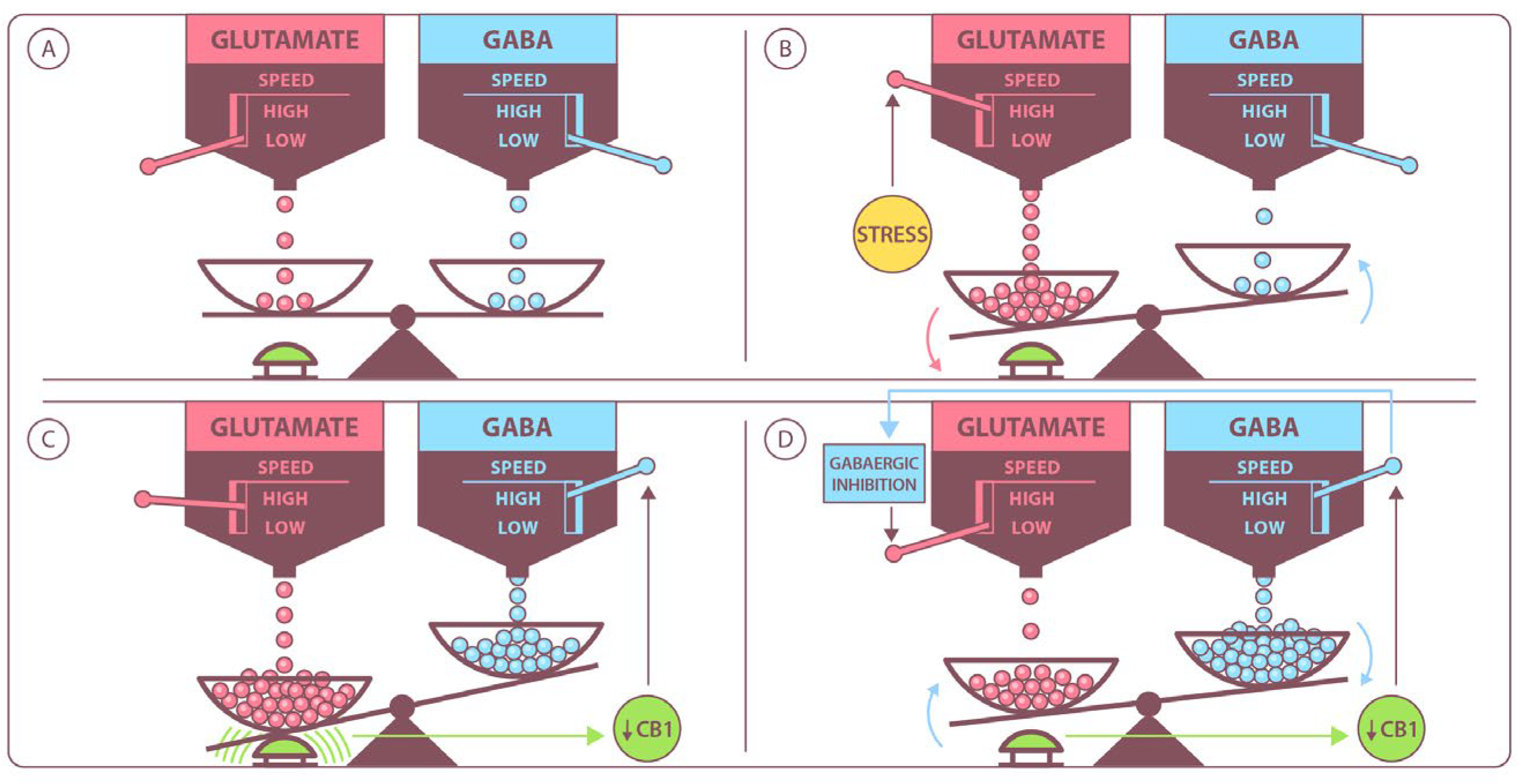
3. The Glutamatergic System
4. The GABAergic System
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PTSD | Post-Traumatic Stress Disorder |
| cAMP | cyclic adenosine monophosphate |
| F-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| mPFC | medial prefrontal cortex |
| EAATs | excitatory amino acid transporters |
| ECS | Endocannabinoid System |
| GABA | Gamma-Aminobutyric Acid |
| CB1 | Cannabinoid Receptor Type 1 |
| CB2 | Cannabinoid Receptor Type 2 |
| NMDA | N-Methyl-D-Aspartate (Receptor) |
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (Receptor) |
| HPA axis | Hypothalamic–Pituitary–Adrenal Axis |
| DSM-5 | Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition |
| SAM | Sympathetic–Adrenomedullary System |
| HPA | Hypothalamic–Pituitary–Adrenal Axis |
| CRH | Corticotropin-Releasing Hormone |
| LC | Locus Coeruleus |
| PFC | Prefrontal Cortex |
| dlPFC | Dorsolateral Prefrontal Cortex |
| NA | Noradrenaline |
| DA | Dopamine |
| PVN | Paraventricular Nucleus |
| AVP | Arginine Vasopressin |
| ACTH | Adrenocorticotropic Hormone |
| 5-HT | Serotonin (5-Hydroxytryptamine) |
| ACh | Acetylcholine |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| BNST | Bed Nucleus of the Stria Terminalis |
| VTA | Ventral Tegmental Area |
| GDG | Gaussian Delay Gaussian (Model) |
| GRs | Glucocorticoid Receptors |
| mPFC | Medial Prefrontal Cortex |
| DRN | Dorsal Raphe Nucleus |
| VGLUT3 | Vesicular Glutamate Transporter 3 |
| PAC1 receptors | Pituitary Adenylate Cyclase-Activating Polypeptide Type I Receptors |
| IL-6 | Interleukin 6 |
| IL-10 | Interleukin 10 |
| vmPFC | Ventromedial Prefrontal Cortex |
| rACC | Rostral Anterior Cingulate Cortex |
| fMRI | Functional Magnetic Resonance Imaging |
| sgACC | Subgenual Anterior Cingulate Cortex |
| CB1R | Cannabinoid Receptor Type 1 |
| CB2R | Cannabinoid Receptor Type 2 |
| CNS | Central Nervous System |
| EAATs | Excitatory Amino Acid Transporters |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate (reduced form) |
| NMDA receptor | N-Methyl-D-Aspartate Receptor |
| NLRP1 | NOD-like Receptor containing a Pyrin domain 1 |
| 1-AG | 1-Arachidonoylglycerol |
| 2-AG | 2-Arachidonoylglycerol |
| AMPAR | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid Receptor |
| DAO | D-Amino Acid Oxidase |
| DAOA | D-Amino Acid Oxidase Activator |
| HINT-1 | Histidine Triad Nucleotide-Binding Protein 1 |
| MAPK | Mitogen-Activated Protein Kinase |
| nNOS | Neuronal Nitric Oxide Synthase |
| PCP | Phencyclidine |
| THC | Tetrahydrocannabinol (delta-9-THC) |
| BDNF | Brain-Derived Neurotrophic Factor |
| CREB | cAMP Response Element-Binding Protein |
| GluA1–4 | Subunits of AMPA Receptors encoded by genes GRIA1 to GRIA4 |
| LTD | Long-Term Depression |
| LTP | Long-Term Potentiation |
| PKA | Protein Kinase A |
| RNA | Ribonucleic Acid |
| DAG | Diacylglycerol |
| ERK MAP kinase | Extracellular Signal-Regulated Kinases/Mitogen-Activated Protein Kinase |
| GluK1–5 | Subunits of Kainate Receptors (KAR) |
| GLT-1 | Glutamate Transporter-1 |
| HIP | Hippocampus |
| IP3 | Inositol-1,4,5-Triphosphate |
| KAR | Kainate Receptor |
| mGluR | Metabotropic Glutamate Receptor |
| MDD | Major Depressive Disorder |
| PET | Positron Emission Tomography |
| PKC | Protein Kinase C |
| TrkB | Tropomyosin Receptor Kinase B |
| GTP-binding protein | Guanosine Triphosphate-binding protein |
| GRIK | Glutamate Receptor Ionotropic, Kainate type |
| GAD | Glutamic Acid Decarboxylase |
| GABA-T | GABA Transaminase |
| VGAT/VIAAT | Vesicular GABA Transporter/Vesicular Inhibitory Amino Acid Transporter |
| ATP | Adenosine Triphosphate |
| VGLUT1/VGLUT2 | Vesicular Glutamate Transporter 1/2 |
| EPSPs | Excitatory Postsynaptic Potentials |
| IPSPs | Inhibitory Postsynaptic Potentials |
| GABAARs | GABA_A Receptors |
| CaMKII | Calcium/Calmodulin-dependent Protein Kinase II |
| KCC2 | Potassium-Chloride Cotransporter Member 5 |
| NO | Nitric Oxide |
| cGMP | Cyclic Guanosine Monophosphate |
| GABABRs | GABA_B Receptors |
| GABAB | Gamma-Aminobutyric Acid type B receptor |
| mGluR1/mGluR5 | Metabotropic Glutamate Receptor 1/Metabotropic Glutamate Receptor 5 |
| MMP-2 | Matrix Metalloproteinase 2 |
| MMP-9 | Matrix Metalloproteinase 9 |
| DREADDs | Designer Receptors Exclusively Activated by Designer Drugs |
References
- Bakirci, A.E.; Sar, V.; Cetin, A.; Baker, L.D.; Smith, A.J. Screening for PTSD in first responders: Turkish adaptation and psychometric validation of the primary care PTSD screen for DSM-5. Psychol. Trauma 2025, ahead of print. [Google Scholar] [CrossRef]
- Brewin, C.R.; Atwoli, L.; Bisson, J.I.; Galea, S.; Koenen, K.; Lewis-Fernández, R. Post-traumatic stress disorder: Evolving conceptualization and evidence, and future research directions. World Psychiatry 2025, 24, 52–80. [Google Scholar] [CrossRef] [PubMed]
- Hoeboer, C.M.; Nava, F.; Haagen, J.F.G.; Broekman, B.F.P.; van der Gaag, R.J.; Olff, M. Epidemiology of DSM-5 PTSD and ICD-11 PTSD and complex PTSD in the Netherlands. J. Anxiety Disord. 2025, 110, 102963. [Google Scholar] [CrossRef] [PubMed]
- Wojujutari, A.K.; Idemudia, E.S.; Ugwu, L.E. The assessment of reliability generalisation of clinician-administered PTSD scale for DSM-5 (CAPS-5): A meta-analysis. Front. Psychol. 2024, 15, 1354229. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Le, S.D.T.; Bui, H.T.T.; Hoang, V.Q.; Do, C.C. Psychological Factors and Post-Traumatic Stress Disorder (PTSD) Risk in Stroke Survivors: A Cross-Sectional Study. Health Psychol. Res. 2025, 13, 129914. [Google Scholar] [CrossRef]
- Dieujuste, N.; Petri, J.M.; Mekawi, Y.; Lathan, E.C.; Carter, S.; Bradley, B.; Fani, N.; Powers, A. Investigating associations between emotion dysregulation and DSM-5 posttraumatic stress disorder (PTSD) using network analysis. J. Affect. Disord. 2025, 377, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Gottschalk, A.; Kuck, S.T.; Dyer, A.; Alpers, G.W.; Pittig, A.; Morina, N.; Ehring, T. Effectiveness in routine care: Trauma-focused treatment for PTSD. Eur. J. Psychotraumatol. 2025, 16, 2452680. [Google Scholar] [CrossRef]
- Ito, M.; Katayanagi, A.; Miyamae, M.; Inomata, T.; Takagishi, Y.; Kikuchi, A.; Makino, M.; Matsuda, Y.; Yamaguchi, K.; Nakayama, C.; et al. Cognitive Processing Therapy for Posttraumatic Stress Disorder in Japan: A Randomized Clinical Trial. JAMA Netw. Open. 2025, 8, e2458059. [Google Scholar] [CrossRef]
- Hansen, M.; Robinson, M.; Armour, C. Investigating risk factors of dissociative and complex posttraumatic stress disorder across diagnostic systems and potential implications: Latent class analyses. Psychol. Trauma 2025, ahead of print. [Google Scholar] [CrossRef]
- Rønning, L.; Zelkowitz, R.L.; Piccirillo, M.L.; Liu, J.; Thomas, J.L.; Guler, J.; Kyei, J.J.; Hoeboer, C.M.; Karchoud, J.F.; Olff, M.; et al. Gender differences in early posttraumatic stress disorder symptoms: A network analysis. Eur. J. Psychotraumatol. 2025, 16, 2448385. [Google Scholar] [CrossRef]
- Massinga, L.J.; Greene, M.C.; Duarte, C.S.; Mandlate, F.; Santos, P.F.; Gouveia, L.; Oquendo, M.A.; Mello, M.F.; Wainberg, M.L. Screening for posttraumatic stress disorder (PTSD) in Mozambique: Validation of the Primary Care Posttraumatic Stress Disorder Screen for Diagnostic and Statistical Manual fifth edition (PC-PTSD-5). Psychol. Trauma 2025, 17, 88–96. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, W.; Freed, S.; DePierro, J.; Nelson, B.; Seemann, C.; McKernan, S.; Wilson, T.; Pole, N. Didn’t Even Blink: Dissociation, Complex Trauma Exposure and Decreased Startle Reactivity. J. Trauma Dissociation 2024, 17, 1–20. [Google Scholar] [CrossRef]
- King, A.R.; Kuhn, S.K.; Brezinski, S.; Jowkar, M.; Smith, K. Self-reported health profiles of trauma victims with and without psychiatric histories. Health Psychol. Rep. 2024, 12, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Di Tella, M.; Romeo, A. Posttraumatic stress symptoms and rumination: The moderator effect of time. Psychol. Health Med. 2025, 30, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.K.; Vaishnaw, A.; Shirbhate, E.; Kore, R.; Singh, V.; Veerasamy, R.; Rajak, H. Cortisol as a Target for Treating Mental Disorders: A Promising Avenue for Therapy. Mini Rev. Med. Chem. 2024, 24, 588–600. [Google Scholar] [CrossRef]
- Lehner, M.; Skórzewska, A.; Wisłowska-Stanek, A. Sex-Related Predisposition to Post-Traumatic Stress Disorder Development-The Role of Neuropeptides. Int. J. Environ. Res. Public Health 2021, 19, 314. [Google Scholar] [CrossRef]
- Zhang, E.T.; Saglimbeni, G.S.; Feng, J.; Li, Y.; Bruchas, M.R. Dentate gyrus norepinephrine ramping facilitates aversive contextual processing. Nat. Commun. 2025, 16, 454. [Google Scholar] [CrossRef]
- Chudoba, R.; Dabrowska, J. Distinct populations of corticotropin-releasing factor (CRF) neurons mediate divergent yet complementary defensive behaviors in response to a threat. Neuropharmacology 2023, 228, 109461. [Google Scholar] [CrossRef]
- Chacko, T.P.; Toole, J.T.; Morris, M.C.; Page, J.; Forsten, R.D.; Barrett, J.P.; Reinhard, M.J.; Brewster, R.C.; Costanzo, M.E.; Broderick, G. A regulatory pathway model of neuropsychological disruption in Havana syndrome. Front. Psychiatry 2023, 14, 1180929. [Google Scholar] [CrossRef]
- Glaeser-Khan, S.; Savalia, N.K.; Cressy, J.; Feng, J.; Li, Y.; Kwan, A.C.; Kaye, A.P. Spatiotemporal Organization of Prefrontal Norepinephrine Influences Neuronal Activity. eNeuro 2024, 11, ENEURO.0252-23.2024. [Google Scholar] [CrossRef]
- Calderon-Williams, D.R.; de Souza, R.R.; Tseng, C.T.; Abdi, H.; Sandoval-Flores, A.; Ploski, J.E.; Thorn, C.A.; McIntyre, C.K. Optogenetic inhibition of the locus coeruleus blocks vagus nerve stimulation-induced enhancement of extinction of conditioned fear in rats. Learn. Mem. 2024, 31, a053958. [Google Scholar] [CrossRef]
- Mavrych, V.; Riyas, F.; Bolgova, O. The Role of Basolateral Amygdala and Medial Prefrontal Cortex in Fear: A Systematic Review. Cureus 2025, 17, e78198. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Zhu, S.; Tang, E.; Xue, C.; Li, K.; Yu, H.; Zhong, T.; Li, T.; Chen, H.; Deng, W. Neural correlates of emotional processing in trauma-related narratives. Psychol. Med. 2025, 55, e33. [Google Scholar] [CrossRef]
- Furriel, B.C.R.S.; Furriel, G.P.; Cunha Xavier Pinto, M.; Lemos, R.P. Computational modeling of fear and stress responses: Validation using consolidated fear and stress protocols. Front. Syst. Neurosci. 2024, 18, 1454336. [Google Scholar] [CrossRef] [PubMed]
- Aupperle, R.; Berg, H.; Armstrong, J. Fears Worth Testing Out: A Systematic Review of the Neural Mechanisms of Treatment Outcome for Anxiety-Related Disorders. Curr. Top Behav. Neurosci. 2024; ahead of print. [Google Scholar] [CrossRef]
- Daskalakis, N.P.; Iatrou, A.; Chatzinakos, C.; Jajoo, A.; Snijders, C.; Wylie, D.; DiPietro, C.P.; Tsatsani, I.; Chen, C.Y.; Pernia, C.D.; et al. Systems biology dissection of PTSD and MDD across brain regions, cell types, and blood. Science 2024, 384, eadh3707. [Google Scholar] [CrossRef]
- Theodoratou, M.; Kougioumtzis, G.A.; Yotsidi, V.; Sofologi, M.; Katsarou, D.; Megari, K. Neuropsychological Consequences of Massive Trauma: Implications and Clinical Interventions. Medicina 2023, 59, 2128. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Huang, G.D.; Xue, Y.X.; Yang, M.; Jia, X.J. The neural circuits and molecular mechanisms underlying fear dysregulation in posttraumatic stress disorder. Front. Neurosci. 2023, 17, 1281401. [Google Scholar] [CrossRef]
- Helpman, L.; Marin, M.F.; Papini, S.; Zhu, X.; Sullivan, G.M.; Schneier, F.; Neria, M.; Shvil, E.; Malaga Aragon, M.J.; Markowitz, J.C.; et al. Neural changes in extinction recall following prolonged exposure treatment for PTSD: A longitudinal fMRI study. Neuroimage Clin. 2016, 12, 715–723. [Google Scholar] [CrossRef]
- Fullana, M.A.; Albajes-Eizagirre, A.; Soriano-Mas, C.; Vervliet, B.; Cardoner, N.; Benet, O.; Radua, J.; Harrison, B.J. Fear extinction in the human brain: A meta-analysis of fMRI studies in healthy participants. Neurosci. Biobehav. Rev. 2018, 88, 16–25. [Google Scholar] [CrossRef]
- Fullana, M.A.; Albajes-Eizagirre, A.; Soriano-Mas, C.; Vervliet, B.; Cardoner, N.; Benet, O.; Radua, J.; Harrison, B.J. Amygdala where art thou? Neurosci. Biobehav. Rev. 2019, 102, 430–431. [Google Scholar] [CrossRef] [PubMed]
- Rahn, E.J.; Guzman-Karlsson, M.C.; David Sweatt, J. Cellular, molecular, and epigenetic mechanisms in non-associative conditioning: Implications for pain and memory. Neurobiol. Learn. Mem. 2013, 105, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Shin, L.M.; Orr, S.P.; Carson, M.A.; Rauch, S.L.; Macklin, M.L.; Lasko, N.B.; Peters, P.M.; Metzger, L.J.; Dougherty, D.D.; Cannistraro, P.A.; et al. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch. Gen. Psychiatry 2024, 61, 168–176. [Google Scholar] [CrossRef]
- Rajasekera, T.A.; Joseph, A.; Pan, H.; Dreyfuss, J.M.; Fida, D.; Wilson, J.; Behee, M.; Fichorova, R.N.; Cinar, R.; Spagnolo, P.A. Sex Differences in Endocannabinoid and Inflammatory Markers Associated with Posttraumatic Stress Disorder. medRxiv 2025. [Google Scholar] [CrossRef]
- Mazurka, R.; Harkness, K.L.; Hassel, S.; Stensson, N.; Nogovitsyn, N.; Poppenk, J.; Foster, J.A.; Squires, S.D.; Rowe, J.; Milev, R.V.; et al. Endocannabinoid concentrations in major depression: Effects of childhood maltreatment and relation to hippocampal volume. Transl. Psychiatry 2024, 14, 431. [Google Scholar] [CrossRef]
- Hill, M.N.; Eiland, L.; Lee, T.T.Y.; Hillard, C.J.; McEwen, B.S. Early life stress alters the developmental trajectory of corticolimbic endocannabinoid signaling in male rats. Neuropharmacology 2019, 146, 154–162. [Google Scholar] [CrossRef]
- Zabik, N.L.; Iadipaolo, A.; Peters, C.A.; Baglot, S.L.; Hill, M.N.; Rabinak, C.A. Dose-dependent effect of acute THC on extinction memory recall and fear renewal: A randomized, double-blind, placebo-controlled study. Psychopharmacology, 2024; ahead of print. [Google Scholar] [CrossRef]
- Szente, L.; Balla, G.Y.; Varga, Z.K.; Toth, B.; Biro, L.; Balogh, Z.; Hill, M.N.; Toth, M.; Mikics, E.; Aliczki, M. Endocannabinoid and neuroplasticity-related changes as susceptibility factors in a rat model of posttraumatic stress disorder. Neurobiol. Stress 2024, 32, 100662. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.G.; Papagianni, E.P.; Stevenson, C.W.; Stubbendorff, C. In it together? The case for endocannabinoid-noradrenergic interactions in fear extinction. Eur. J. Neurosci. 2022, 55, 952–970. [Google Scholar] [CrossRef]
- Hodebourg, R.; Meyerink, M.E.; Crow, A.D.; Reichel, C.M.; Kalivas, P.W.; Garcia-Keller, C. Cannabinoid use is enhanced by stress and changes conditioned stress responses. Neuropsychopharmacology 2022, 47, 1037–1045. [Google Scholar] [CrossRef]
- Portugalov, A.; Peled, G.; Zorin, S.; Akirav, I. Cannabidiol Modulates Neuroinflammatory Markers in a PTSD Model Conducted on Female Rats. Biomolecules 2024, 14, 1384. [Google Scholar] [CrossRef] [PubMed]
- Lisboa, S.F.; Stern, C.A.J.; Gazarini, L.; Bertoglio, L.J. Cannabidiol effects on fear processing and implications for PTSD: Evidence from rodent and human studies. Int. Rev. Neurobiol. 2024, 177, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Lutz, G.; Marsicano, G.; Maldonado, R.; Hillard, C.J. The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci. 2015, 16, 705–718. [Google Scholar] [CrossRef]
- Claire, A.; Vasefi, M. Cannabidiol and the corticoraphe circuit in post-traumatic stress disorder. IBRO Neurosci. Rep. 2021, 11, 88–102. [Google Scholar] [CrossRef]
- Korem, N.; Hillmer, A.T.; D’Souza, D.C.; Bassir Nia, A.; Levy, I.; Pietrzak, R.H.; Harpaz-Rotem, I. Amygdala Cannabinoid 1 Receptor, Pain Response, and Emotional Numbing in Trauma-Exposed Individuals. JAMA Netw. Open. 2024, 7, e2432387. [Google Scholar] [CrossRef]
- Battaglia, S.; Fazio, C.D.; Borgomaneri, S.; Avenanti, A. Cortisol Imbalance and Fear Learning in PTSD: Therapeutic Approaches to Control Abnormal Fear Responses. Curr. Neuropharmacol. 2025, 23, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Qin, Y.; Wu, N.; Han, X.; Li, J. Single-Nucleus Transcriptome Profiling from the Hippocampus of a PTSD Mouse Model and CBD-Treated Cohorts. Genes 2024, 15, 519. [Google Scholar] [CrossRef]
- Soler-Cedeño, O.; Torres-Rodríguez, O.; Bernard, F.; Maldonado, L.; Hernández, A.; Porter, J.T. Plasticity of NMDA Receptors at Ventral Hippocampal Synapses in the Infralimbic Cortex Regulates Cued Fear. eNeuro 2019, 6, ENEURO.0354-18.2019. [Google Scholar] [CrossRef]
- Ghosh, S.; Mohammed, Z.; Singh, I. Bruton’s tyrosine kinase drives neuroinflammation and anxiogenic behavior in mouse models of stress. J. Neuroinflamm. 2021, 18, 289. [Google Scholar] [CrossRef]
- Gunduz-Cinar, O.; Castillo, L.I.; Xia, M.; Van Leer, E.; Brockway, E.T.; Pollack, G.A.; Yasmin, F.; Bukalo, O.; Limoges, A.; Oreizi-Esfahani, S.; et al. A cortico-amygdala neural substrate for endocannabinoid modulation of fear extinction. Neuron 2023, 111, 3053–3067.e10. [Google Scholar] [CrossRef]
- Pineles, S.L.; Nillni, Y.I.; Pinna, G.; Webb, A.; Arditte Hall, K.A.; Fonda, J.R.; Irvine, J.; King, M.W.; Hauger, R.L.; Resick, P.A.; et al. Associations between PTSD-Related extinction retention deficits in women and plasma steroids that modulate brain GABAA and NMDA receptor activity. Neurobiol. Stress 2020, 13, 100225. [Google Scholar] [CrossRef] [PubMed]
- Sheth, C.; Prescot, A.P.; Legarreta, M.; Renshaw, P.F.; McGlade, E.; Yurgelun-Todd, D. Reduced gamma-amino butyric acid (GABA) and glutamine in the anterior cingulate cortex (ACC) of veterans exposed to trauma. J. Affect. Disord. 2019, 248, 166–174. [Google Scholar] [CrossRef]
- Simões, P.F.; Silva, A.P.; Pereira, F.C.; Marques, E.; Grade, S.; Milhazes, N.; Borges, F.; Ribeiro, C.F.; Macedo, T.R. Methamphetamine induces alterations on hippocampal NMDA and AMPA receptor subunit levels and impairs spatial working memory. Neuroscience 2007, 150, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Haney, M.; Hillard, C.J.; Karhson, D.S.; Vecchiarelli, H.A. The endocannabinoid system as a putative target for the development of novel drugs for the treatment of psychiatric illnesses. Psychol. Med. 2023, 53, 7006–7024. [Google Scholar] [CrossRef]
- Zhigulin, A.S.; Barygin, O.I. Mechanisms of NMDA receptor inhibition by vortioxetine—Comparison with fluoxetine. Eur. J. Pharmacol. 2025, 998, 177460. [Google Scholar] [CrossRef]
- Veeraiah, P.; Noronha, J.M.; Maitra, S.; Bagga, P.; Khandelwal, N.; Chakravarty, S.; Kumar, A.; Patel, A.B. Dysfunctional glutamatergic and γ-aminobutyric acidergic activities in prefrontal cortex of mice in social defeat model of depression. Biol. Psychiatry 2014, 76, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Wolosker, H.; Balu, D.T. D-Serine as the gatekeeper of NMDA receptor activity: Implications for the pharmacologic management of anxiety disorders. Transl. Psychiatry 2020, 10, 184. [Google Scholar] [CrossRef]
- Matias, M.E.; Radulski, D.R.; Rodrigues da Silva, T.; Raymundi, A.M.; Stern, C.A.J.; Zampronio, A.R. Involvement of cannabinoid receptors and neuroinflammation in early sepsis: Implications for posttraumatic stress disorder. Int. Immunopharmacol. 2023, 123, 110745. [Google Scholar] [CrossRef]
- Reitich-Stolero, T.; Halperin, D.; Morris, G.; Goldstein, L.; Bergman, L.; Fahoum, F.; Strauss, I.; Paz, R. Aversive generalization in human amygdala neurons. Curr. Biol. 2025, 35, 1137–1144.e3. [Google Scholar] [CrossRef]
- Li, Q.; Liu, S.; Zhu, X.; Mi, W.; Maoying, Q.; Wang, J.; Yu, J.; Wang, Y. Hippocampal PKR/NLRP1 Inflammasome Pathway Is Required for the Depression-Like Behaviors in Rats with Neuropathic Pain. Neuroscience 2019, 412, 16–28. [Google Scholar] [CrossRef]
- Crombie, K.M.; Leitzelar, B.N.; Brellenthin, A.G.; Hillard, C.J.; Koltyn, K.F. Loss of exercise- and stress-induced increases in circulating 2-arachidonoylglycerol concentrations in adults with chronic PTSD. Biol. Psychol. 2019, 145, 1–7. [Google Scholar] [CrossRef]
- Clancy, K.J.; Chen, X.; Song, X.; Song, T.; Zhou, S.; Akman, E.; Ostrand, C.; Ren, B.; Du, F.; Rosso, I.M. Multimodal associations between posterior hippocampus glutamate metabolism, visual cortex connectivity, and intrusive trauma reexperiencing symptoms. medRxiv 2025. [Google Scholar] [CrossRef]
- Lamberti, M.; van Putten, M.J.A.M.; Marzen, S.; le Feber, J. The role of NMDA receptors in memory and prediction in cultured neural networks. J. Neural. Eng. 2025, 22, 016053. [Google Scholar] [CrossRef]
- Knox, D.; Perrine, S.A.; George, S.A.; Galloway, M.P.; Liberzon, I. Single prolonged stress decreases glutamate, glutamine, and creatine concentrations in the rat medial prefrontal cortex. Neurosci. Lett. 2010, 480, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Ouardouz, M.; Sastry, B.R. Mechanisms underlying LTP of inhibitory synaptic transmission in the deep cerebellar nuclei. J. Neurophysiol. 2000, 84, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Qi, Y.; Hou, D.N.; Ji, Y.Y.; Zheng, C.Y.; Li, C.Y.; Yung, W.H.; Lu, B.; Huang, Y. BDNF val66met Polymorphism Impairs Hippocampal Long-Term Depression by Down-Regulation of 5-HT3 Receptors. Front. Cell Neurosci. 2017, 11, 306. [Google Scholar] [CrossRef]
- Smiley, C.E.; McGonigal, J.T.; Valvano, T.; Newsom, R.J.; Otero, N.; Gass, J.T. The infralimbic cortex and mGlu5 mediate the effects of chronic intermittent ethanol exposure on fear learning and memory. Psychopharmacology 2020, 237, 3417–3433. [Google Scholar] [CrossRef]
- Perez-Garcia, G.; De Gasperi, R.; Gama Sosa, M.A.; Perez, G.M.; Otero-Pagan, A.; Tschiffely, A.; McCarron, R.M.; Ahlers, S.T.; Elder, G.A.; Gandy, S. PTSD-Related Behavioral Traits in a Rat Model of Blast-Induced mTBI Are Reversed by the mGluR2/3 Receptor Antagonist BCI-838. eNeuro 2018, 5, ENEURO.0357-17.2018. [Google Scholar] [CrossRef]
- Iqbal, J.; Huang, G.D.; Shen, D.; Xue, Y.X.; Yang, M.; Jia, X.J. Single prolonged stress induces behavior and transcriptomic changes in the medial prefrontal cortex to increase susceptibility to anxiety-like behavior in rats. Front. Psychiatry 2024, 15, 1472194. [Google Scholar] [CrossRef]
- Fendt, M.; Bürki, H.; Imobersteg, S.; van der Putten, H.; McAllister, K.; Leslie, J.C.; Shaw, D.; Hölscher, C. The effect of mGlu8 deficiency in animal models of psychiatric diseases. Genes Brain Behav. 2010, 9, 33–44. [Google Scholar] [CrossRef]
- Huang, J.; Xu, F.; Yang, L.; Tuolihong, L.; Wang, X.; Du, Z.; Zhang, Y.; Yin, X.; Li, Y.; Lu, K.; et al. Involvement of the GABAergic system in PTSD and its therapeutic significance. Front. Mol. Neurosci. 2023, 16, 1052288. [Google Scholar] [CrossRef] [PubMed]
- Maples-Keller, J.L.; Watkins, L.; Hellman, N.; Phillips, N.L.; Rothbaum, B.O. Treatment Approaches for Posttraumatic Stress Disorder Derived From Basic Research on Fear Extinction. Biol. Psychiatry 2025, 97, 382–391. [Google Scholar] [CrossRef]
- Kheirkhah, M.; Duncan, W.C., Jr.; Yuan, Q.; Wang, P.R.; Jamalabadi, H.; Leistritz, L.; Walter, M.; Goldman, D.; Zarate, C.A., Jr.; Hejazi, N.S. REM density predicts rapid antidepressant response to ketamine in individuals with treatment-resistant depression. Neuropsychopharmacology 2025, 50, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.T.K.; Marques, L.S.; Brambila, C.A.; da Cruz Weber Fulco, B.; Nogueira, C.W.; Zeni, G. Social-Single Prolonged Stress affects contextual fear conditioning in male and female Wistar rats: Molecular insights in the amygdala. Prog. Neuropsychopharmacol. Biol. Psychiatry 2024, 133, 111021. [Google Scholar] [CrossRef] [PubMed]
- Polepalli, J.S.; Gooch, H.; Sah, P. Diversity of interneurons in the lateral and basal amygdala. NPJ Sci. Learn. 2020, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, H.; Bink, D.I.; Daniels, E.G.; Pandit, R.; Piriou, C.; Slieker, R.; Westphal, K.G.; Olivier, B.; Oosting, R.S. Re-exposure and environmental enrichment reveal NPY-Y1 as a possible target for post-traumatic stress disorder. Neuropharmacology 2012, 63, 733–742. [Google Scholar] [CrossRef]
- Pinna, G.; Ponomareva, O.; Stalcup, G.L.; Rasmusson, A.M. Neuroactive Steroids and the Pathophysiology of PTSD: Biomarkers for Treatment Targeting. Neurosci. Biobehav. Rev. 2025, 172, 106085. [Google Scholar] [CrossRef]
- Dong, W.; Todd, A.C.; Bröer, A.; Hulme, S.R.; Bröer, S.; Billups, B. PKC-Mediated Modulation of Astrocyte SNAT3 Glutamine Transporter Function at Synapses in Situ. Int. J. Mol. Sci. 2018, 19, 924. [Google Scholar] [CrossRef]
- Tsuda, S.; Hou, J.; Thompson, F.J.; Bose, P.K. Traumatic brain injury-induced anxiety: Injury and plasticity of the central noradrenergic system. Exp. Neurol. 2025, 388, 115182. [Google Scholar] [CrossRef]
- Li, H.Q.; Jiang, W.; Ling, L.; Pratelli, M.; Chen, C.; Gupta, V.; Godavarthi, S.K.; Spitzer, N.C. Generalized fear after acute stress is caused by change in neuronal cotransmitter identity. Science 2024, 383, 1252–1259. [Google Scholar] [CrossRef]
- Coelho, A.A.; Lima-Bastos, S.; Gobira, P.H.; Lisboa, S.F. Endocannabinoid signaling and epigenetics modifications in the neurobiology of stress-related disorders. Neuronal Signal. 2023, 7, NS20220034. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 2018, 19, 81–94. [Google Scholar] [CrossRef]
- Leone, G.; Casanave, H.; Postel, C.; Fraisse, F.; Vallée, T.; de La Sayette, V.; Dayan, J.; Peschanski, D.; Eustache, F.; Gagnepain, P. Plasticity of human resilience mechanisms. Sci. Adv. 2025, 11, eadq8336. [Google Scholar] [CrossRef]
- Wang, L.; Tu, P.; Bonet, L.; Aubrey, K.R.; Supplisson, S. Cytosolic transmitter concentration regulates vesicle cycling at hippocampal GABAergic terminals. Neuron 2013, 80, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Singewald, N.; Sartori, S.B.; Reif, A.; Holmes, A. Alleviating anxiety and taming trauma: Novel pharmacotherapeutics for anxiety disorders and posttraumatic stress disorder. Neuropharmacology 2023, 226, 109418. [Google Scholar] [CrossRef]
- Ueda, H.H.; Nagasawa, Y.; Sato, A.; Onda, M.; Murakoshi, H. Chronic neuronal excitation leads to dual metaplasticity in the signaling for structural long-term potentiation. Cell Rep. 2022, 38, 110153. [Google Scholar] [CrossRef]
- Suo, Z.W.; Fan, Q.Q.; Yang, X.; Hu, X.D. Ca2+/calmodulin-dependent protein kinase II in spinal dorsal horn contributes to the pain hypersensitivity induced by γ-aminobutyric acid type a receptor inhibition. J. Neurosci. Res. 2013, 91, 1473–1482. [Google Scholar] [CrossRef]
- Beesley, P.W.; Herrera-Molina, R.; Smalla, K.H.; Seidenbecher, C. The Neuroplastin adhesion molecules: Key regulators of neuronal plasticity and synaptic function. J. Neurochem. 2014, 131, 268–283. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.M.; McCafferty, C.P.; Bravo, J.A.; Singewald, N.; Holmes, A.; Cryan, J.F. Increased amygdalar metabotropic glutamate receptor 7 mRNA in a genetic mouse model of impaired fear extinction. Psychopharmacology 2019, 236, 265–272. [Google Scholar] [CrossRef]
- Chen, C.; Li, S.; Zhou, Y.; Huang, H.; Lin, J.T.; Wu, W.F.; Qiu, Y.K.; Dong, W.; Wan, J.; Liu, Q.; et al. Neuronal excitation-inhibition imbalance in the basolateral amygdala is involved in propofol-mediated enhancement of fear memory. Commun. Biol. 2024, 7, 1408. [Google Scholar] [CrossRef]
- Wu, L.; Jin, M. Clinical Significance of Serum MMP-9, S100-β and GFAP in Patients with Mental Disorders after Traumatic Brain Injury. Actas Esp. Psiquiatr. 2025, 53, 11–18. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grzesińska, A.D. The Involvement of the Endocannabinoid, Glutamatergic, and GABAergic Systems in PTSD. Int. J. Mol. Sci. 2025, 26, 5929. https://doi.org/10.3390/ijms26135929
Grzesińska AD. The Involvement of the Endocannabinoid, Glutamatergic, and GABAergic Systems in PTSD. International Journal of Molecular Sciences. 2025; 26(13):5929. https://doi.org/10.3390/ijms26135929
Chicago/Turabian StyleGrzesińska, Anna Dorota. 2025. "The Involvement of the Endocannabinoid, Glutamatergic, and GABAergic Systems in PTSD" International Journal of Molecular Sciences 26, no. 13: 5929. https://doi.org/10.3390/ijms26135929
APA StyleGrzesińska, A. D. (2025). The Involvement of the Endocannabinoid, Glutamatergic, and GABAergic Systems in PTSD. International Journal of Molecular Sciences, 26(13), 5929. https://doi.org/10.3390/ijms26135929