
Elucidating DNA Damage-Dependent Immune System Activation

 and
and
Abstract

1. Introduction
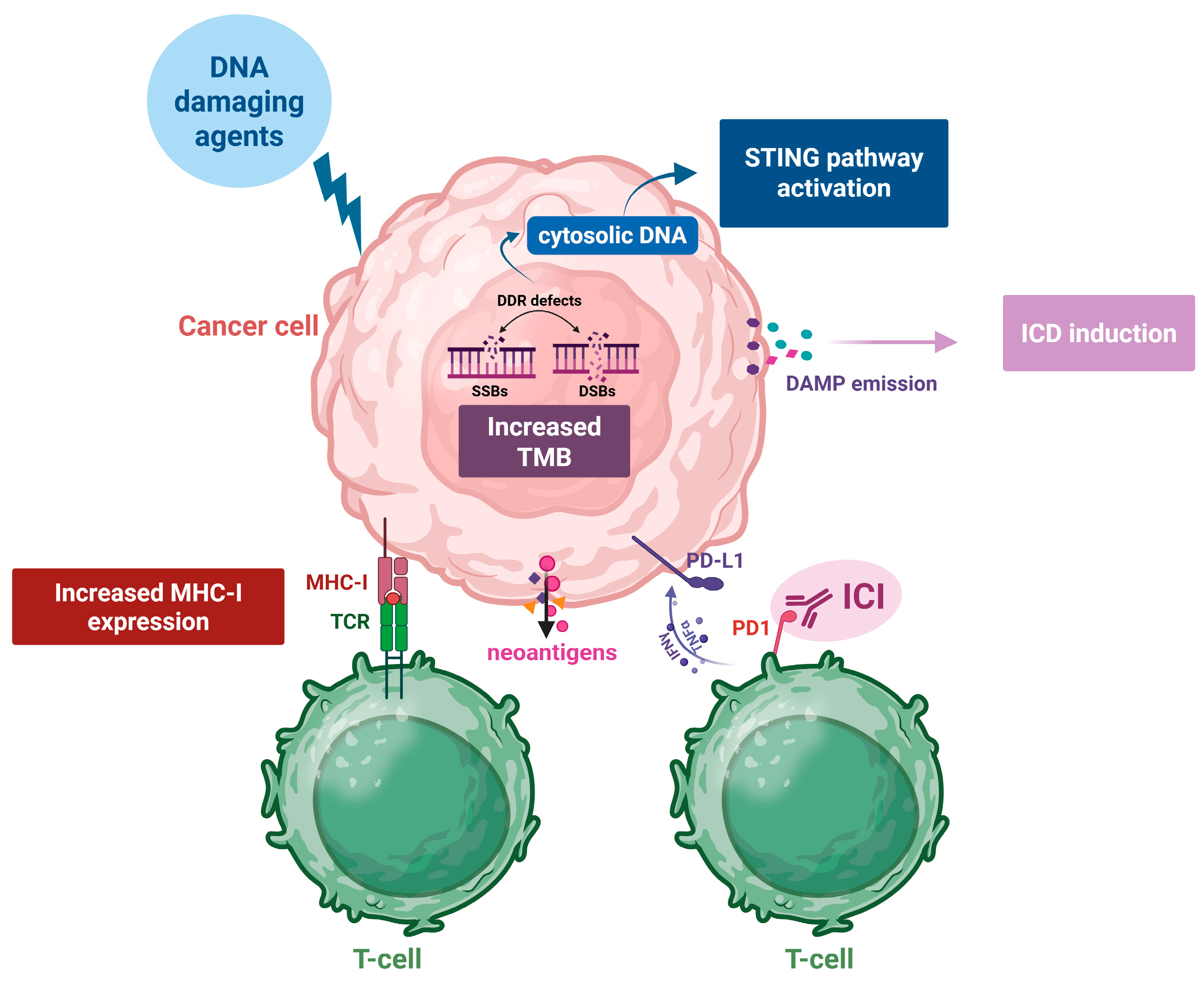
2. The Effects of DNA-Damaging Agents on the Immune System
2.1. Increased Tumor Mutational Burden (TMB) and the Synthesis of Neoantigens
2.2. Induction of Immunogenic Cell Death
2.3. Activation of the Stimulator of Interferon Genes (STING) Pathway
2.4. The Increased Expression of MHC Class I Proteins and the Subsequent Antigen Presentation
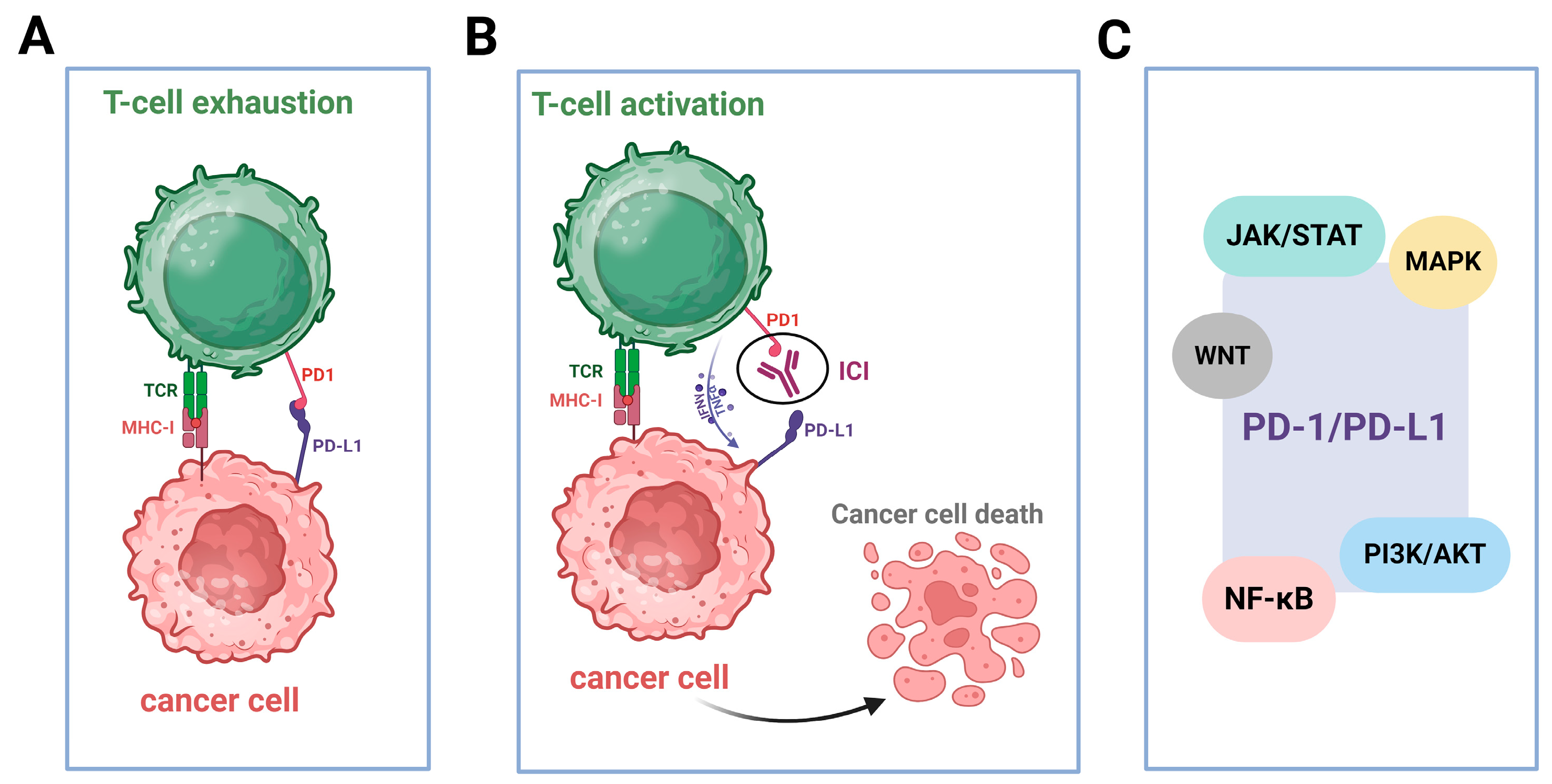
2.5. Upregulated Expression of PD-L1
2.6. The Induction of a Pro-Inflammatory Milieu in the Tumor Microenvironment (TME)
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, R.; Sun, Y.; Li, C.; Xue, Y.; Ba, X. Targeting the DNA Damage Response for Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 15907. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA Damage Response and Immune Signaling Alliance: Is It Good or Bad? Nature Decides When and Where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Liao, G.; Chen, M.; Peng, R.-W.; Yan, X.; Du, J.; Huang, R.; Pan, M.; Lin, Y.; Gong, X.; et al. Advancing Cancer Therapy: New Frontiers in Targeting DNA Damage Response. Front. Pharmacol. 2024, 15, 1474337. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Thangavel, C.; Becker, R.C.; Sadayappan, S. Monoclonal Antibody-Based Immunotherapy and Its Role in the Development of Cardiac Toxicity. Cancers 2020, 13, 86. [Google Scholar] [CrossRef]
- Verma, C.; Pawar, V.; Srivastava, S.; Tyagi, A.; Kaushik, G.; Shukla, S.; Kumar, V. Cancer Vaccines in the Immunotherapy Era: Promise and Potential. Vaccines 2023, 11, 1783. [Google Scholar] [CrossRef]
- Brzostek-Racine, S.; Gordon, C.; Van Scoy, S.; Reich, N.C. The DNA Damage Response Induces IFN. J. Immunol. 2011, 187, 5336–5345. [Google Scholar] [CrossRef] [PubMed]
- Barros, E.M.; McIntosh, S.A.; Savage, K.I. The DNA Damage Induced Immune Response: Implications for Cancer Therapy. DNA Repair 2022, 120, 103409. [Google Scholar] [CrossRef]
- He, M.; Jiang, H.; Li, S.; Xue, M.; Wang, H.; Zheng, C.; Tong, J. The Crosstalk between DNA-Damage Responses and Innate Immunity. Int. Immunopharmacol. 2024, 140, 112768. [Google Scholar] [CrossRef]
- Pálmai-Pallag, T.; Bachrati, C.Z. Inflammation-Induced DNA Damage and Damage-Induced Inflammation: A Vicious Cycle. Microbes Infect. 2014, 16, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Neves-Costa, A.; Moita, L.F. Modulation of Inflammation and Disease Tolerance by DNA Damage Response Pathways. FEBS J. 2017, 284, 680–698. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.M.; Sweasy, J.B. Interplay between DNA Repair and Inflammation, and the Link to Cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef] [PubMed]
- Fontes, F.L.; Pinheiro, D.M.L.; Oliveira, A.H.S.D.; Oliveira, R.K.D.M.; Lajus, T.B.P.; Agnez-Lima, L.F. Role of DNA Repair in Host Immune Response and Inflammation. Mutat. Res. Mol. Mech. Mutagen. 2015, 763, 246–257. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Immunogenic Chemotherapy: Dose and Schedule Dependence and Combination with Immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Wang, P.; Chen, Y.; Wang, C. Beyond Tumor Mutation Burden: Tumor Neoantigen Burden as a Biomarker for Immunotherapy and Other Types of Therapy. Front. Oncol. 2021, 11, 672677. [Google Scholar] [CrossRef]
- Wang, Q.; Douglass, J.; Hwang, M.S.; Hsiue, E.H.-C.; Mog, B.J.; Zhang, M.; Papadopoulos, N.; Kinzler, K.W.; Zhou, S.; Vogelstein, B. Direct Detection and Quantification of Neoantigens. Cancer Immunol. Res. 2019, 7, 1748–1754. [Google Scholar] [CrossRef]
- Efremova, M.; Finotello, F.; Rieder, D.; Trajanoski, Z. Neoantigens Generated by Individual Mutations and Their Role in Cancer Immunity and Immunotherapy. Front. Immunol. 2017, 8, 1679. [Google Scholar] [CrossRef]
- Tokita, S.; Kanaseki, T.; Torigoe, T. Neoantigen Prioritization Based on Antigen Processing and Presentation. Front. Immunol. 2024, 15, 1487378. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour-Specific Mutant Antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Rosenberg, S.A. Exploiting the Curative Potential of Adoptive T-cell Therapy for Cancer. Immunol. Rev. 2014, 257, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Australian Pancreatic Cancer Genome Initiative; Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; et al. Identification of Unique Neoantigen Qualities in Long-Term Survivors of Pancreatic Cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Warren, R.L.; Gibb, E.A.; Martin, S.D.; Spinelli, J.J.; Nelson, B.H.; Holt, R.A. Neo-Antigens Predicted by Tumor Genome Meta-Analysis Correlate with Increased Patient Survival. Genome Res. 2014, 24, 743–750. [Google Scholar] [CrossRef]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef]
- Matsushita, H.; Sato, Y.; Karasaki, T.; Nakagawa, T.; Kume, H.; Ogawa, S.; Homma, Y.; Kakimi, K. Neoantigen Load, Antigen Presentation Machinery, and Immune Signatures Determine Prognosis in Clear Cell Renal Cell Carcinoma. Cancer Immunol. Res. 2016, 4, 463–471. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Gurjao, C.; Tsukrov, D.; Imakaev, M.; Luquette, L.J.; Mirny, L.A. Is Tumor Mutational Burden Predictive of Response to Immunotherapy? eLife 2024, 12, RP87465. [Google Scholar]
- Puccini, A.; Poorman, K.; Salem, M.E.; Soldato, D.; Seeber, A.; Goldberg, R.M.; Shields, A.F.; Xiu, J.; Battaglin, F.; Berger, M.D.; et al. Comprehensive Genomic Profiling of Gastroenteropancreatic Neuroendocrine Neoplasms (GEP-NENs). Clin. Cancer Res. 2020, 26, 5943–5951. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, J.; Chu, Q.; Duan, J.; Zhang, J.; Bai, H.; Yang, Z.; Fang, W.; Cai, L.; Wan, R.; et al. A Novel Tumor Mutational Burden Estimation Model as a Predictive and Prognostic Biomarker in NSCLC Patients. BMC Med. 2020, 18, 232. [Google Scholar] [CrossRef] [PubMed]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Metastatic Cancers: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Yang, H.; Sun, L.; Guan, A.; Yin, H.; Liu, M.; Mao, X.; Xu, H.; Zhao, H.; Lu, X.; Sang, X.; et al. Unique TP53 Neoantigen and the Immune Microenvironment in Long-Term Survivors of Hepatocellular Carcinoma. Cancer Immunol. Immunother. 2021, 70, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.P.; Gubin, M.M.; Schreiber, R.D. The Role of Neoantigens in Naturally Occurring and Therapeutically Induced Immune Responses to Cancer. In Advances in Immunology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 130, pp. 25–74. ISBN 978-0-12-805156-6. [Google Scholar]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non–Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.-C.; Paik, S.; Kim, S. Neopepsee: Accurate Genome-Level Prediction of Neoantigens by Harnessing Sequence and Amino Acid Immunogenicity Information. Ann. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef]
- Miller, A.; Asmann, Y.; Cattaneo, L.; Braggio, E.; Keats, J.; Auclair, D.; Lonial, S.; The MMRF CoMMpass Network; Russell, S.J.; Stewart, A.K. High Somatic Mutation and Neoantigen Burden Are Correlated with Decreased Progression-Free Survival in Multiple Myeloma. Blood Cancer J. 2017, 7, e612. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.-P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation Therapy and Anti-Tumor Immunity: Exposing Immunogenic Mutations to the Immune System. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef]
- Formenti, S.C.; Demaria, S. Radiation Therapy to Convert the Tumor Into an In Situ Vaccine. Int. J. Radiat. Oncol. 2012, 84, 879–880. [Google Scholar] [CrossRef]
- Wilkins, A.; McDonald, F.; Harrington, K.; Melcher, A. Radiotherapy Enhances Responses of Lung Cancer to CTLA-4 Blockade. J. Immunother. Cancer 2019, 7, 64. [Google Scholar] [CrossRef]
- Ji, D.; Yi, H.; Zhang, D.; Zhan, T.; Li, Z.; Li, M.; Jia, J.; Qiao, M.; Xia, J.; Zhai, Z.; et al. Somatic Mutations and Immune Alternation in Rectal Cancer Following Neoadjuvant Chemoradiotherapy. Cancer Immunol. Res. 2018, 6, 1401–1416. [Google Scholar] [CrossRef]
- Mouw, K.W.; Cleary, J.M.; Reardon, B.; Pike, J.; Braunstein, L.Z.; Kim, J.; Amin-Mansour, A.; Miao, D.; Damish, A.; Chin, J.; et al. Genomic Evolution after Chemoradiotherapy in Anal Squamous Cell Carcinoma. Clin. Cancer Res. 2017, 23, 3214–3222. [Google Scholar] [CrossRef]
- Reisländer, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Hintelmann, K.; Petersen, C.; Borgmann, K. Radiotherapeutic Strategies to Overcome Resistance of Breast Cancer Brain Metastases by Considering Immunogenic Aspects of Cancer Stem Cells. Cancers 2022, 15, 211. [Google Scholar] [CrossRef]
- Li, L.; Zhang, F.; Liu, Z.; Fan, Z. Immunotherapy for Triple-Negative Breast Cancer: Combination Strategies to Improve Outcome. Cancers 2023, 15, 321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zheng, S.; Liu, Y.; Li, X.; Wu, J.; Sun, Y.; Liu, G. DNA Damage Response and PD-1/PD-L1 Pathway in Ovarian Cancer. DNA Repair. 2021, 102, 103112. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, C.; Economopoulou, P.; Gavrielatou, N.; Mavroeidi, D.; Psyrri, A.; Souliotis, V.L. UVC-Induced Oxidative Stress and DNA Damage Repair Status in Head and Neck Squamous Cell Carcinoma Patients with Different Responses to Nivolumab Therapy. Biology 2025, 14, 195. [Google Scholar] [CrossRef]
- Papanikolaou, C.; Economopoulou, P.; Spathis, A.; Kotsantis, I.; Gavrielatou, N.; Anastasiou, M.; Moutafi, M.; Kyriazoglou, A.; Foukas, G.-R.P.; Lelegiannis, I.M.; et al. Association of DNA Damage Response Signals and Oxidative Stress Status with Nivolumab Efficacy in Patients with Head and Neck Squamous Cell Carcinoma. Br. J. Cancer 2025. [Google Scholar] [CrossRef]
- Bever, K.M.; Le, D.T. DNA Repair Defects and Implications for Immunotherapy. J. Clin. Investig. 2018, 128, 4236–4242. [Google Scholar] [CrossRef]
- Criscuolo, D.; Morra, F.; Giannella, R.; Visconti, R.; Cerrato, A.; Celetti, A. New Combinatorial Strategies to Improve the PARP Inhibitors Efficacy in the Urothelial Bladder Cancer Treatment. J. Exp. Clin. Cancer Res. 2019, 38, 91. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Tani, K. Multimodal Immunogenic Cancer Cell Death as a Consequence of Anticancer Cytotoxic Treatments. Cell Death Differ. 2014, 21, 39–49. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Birmpilis, A.I.; Paschalis, A.; Mourkakis, A.; Christodoulou, P.; Kostopoulos, I.V.; Antimissari, E.; Terzoudi, G.; Georgakilas, A.G.; Armpilia, C.; Papageorgis, P.; et al. Immunogenic Cell Death, DAMPs and Prothymosin α as a Putative Anticancer Immune Response Biomarker. Cells 2022, 11, 1415. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Gomes-de-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial Strategies for the Induction of Immunogenic Cell Death. Front. Immunol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Zhai, J.; Gu, X.; Liu, Y.; Hu, Y.; Jiang, Y.; Zhang, Z. Chemotherapeutic and Targeted Drugs-Induced Immunogenic Cell Death in Cancer Models and Antitumor Therapy: An Update Review. Front. Pharmacol. 2023, 14, 1152934. [Google Scholar] [CrossRef]
- Naito, S.; Kajiwara, T.; Karasawa, H.; Ono, T.; Saito, T.; Funayama, R.; Nakayama, K.; Ohnuma, S.; Unno, M. Calreticulin Exposure Induced by Anticancer Drugs Is Associated with the P53 Signaling Pathway in Colorectal Cancer Cells. Biochem. Biophys. Res. Commun. 2024, 733, 150665. [Google Scholar] [CrossRef]
- Combès, E.; Andrade, A.F.; Tosi, D.; Michaud, H.-A.; Coquel, F.; Garambois, V.; Desigaud, D.; Jarlier, M.; Coquelle, A.; Pasero, P.; et al. Inhibition of Ataxia-Telangiectasia Mutated and RAD3-Related (ATR) Overcomes Oxaliplatin Resistance and Promotes Antitumor Immunity in Colorectal Cancer. Cancer Res. 2019, 79, 2933–2946. [Google Scholar] [CrossRef]
- Chen, F.; Zhao, W.; Du, C.; Chen, Z.; Du, J.; Zhou, M. Bleomycin Induces Senescence and Repression of DNA Repair via Downregulation of Rad51. Mol. Med. 2024, 30, 54. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, H.; Bruchard, M.; Berger, H.; Derangère, V.; Odoul, L.; Euvrard, R.; Ladoire, S.; Chalmin, F.; Végran, F.; Rébé, C.; et al. Bleomycin Exerts Ambivalent Antitumor Immune Effect by Triggering Both Immunogenic Cell Death and Proliferation of Regulatory T Cells. PLoS ONE 2013, 8, e65181. [Google Scholar] [CrossRef]
- Li, C.; Sun, H.; Wei, W.; Liu, Q.; Wang, Y.; Zhang, Y.; Lian, F.; Liu, F.; Li, C.; Ying, K.; et al. Mitoxantrone Triggers Immunogenic Prostate Cancer Cell Death via P53-Dependent PERK Expression. Cell Oncol. 2020, 43, 1099–1116. [Google Scholar] [CrossRef]
- Qin, J.; Kunda, N.M.; Qiao, G.; Tulla, K.; Prabhakar, B.S.; Maker, A.V. Vaccination With Mitoxantrone-Treated Primary Colon Cancer Cells Enhances Tumor-Infiltrating Lymphocytes and Clinical Responses in Colorectal Liver Metastases. J. Surg. Res. 2019, 233, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Hu, J.; Zhang, H.; Xu, F.; He, W.; Wang, X.; Li, M.; Lu, W.; Zeng, G.; et al. cGAS/STING Axis Mediates a Topoisomerase II Inhibitor–Induced Tumor Immunogenicity. J. Clin. Investig. 2019, 129, 4850–4862. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-Induced “BRCAness” Sensitizes Multiple Myeloma Cells to PARP Inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib Induces Anti–Multiple Myeloma Immune Response Mediated by cGAS/STING Pathway Activation. Blood Cancer Discov. 2021, 2, 468–483. [Google Scholar] [CrossRef]
- Hui, Y.-J.; Yu, T.-T.; Li, L.-G.; Peng, X.-C.; Di, M.-J.; Liu, H.; Gu, W.-L.; Li, T.-F.; Zhao, K.-L.; Wang, W.-X. B-Myb Deficiency Boosts Bortezomib-Induced Immunogenic Cell Death in Colorectal Cancer. Sci. Rep. 2024, 14, 7733. [Google Scholar] [CrossRef]
- Zhang, X.; Wen, J.; Zhang, G.; Fan, W.; Tan, J.; Liu, H.; Li, J. Identification and Validation of Novel Immunogenic Cell Death- and DNA Damage Response-Related Molecular Patterns Correlated with Immune Status and Prognosis in Hepatocellular Carcinoma. Transl. Oncol. 2023, 27, 101600. [Google Scholar] [CrossRef]
- Gao, M.; He, Y.; Tang, H.; Chen, X.; Liu, S.; Tao, Y. cGAS/STING: Novel Perspectives of the Classic Pathway. Mol. Biomed. 2020, 1, 7. [Google Scholar] [CrossRef]
- Durante, M.; Formenti, S.C. Radiation-Induced Chromosomal Aberrations and Immunotherapy: Micronuclei, Cytosolic DNA, and Interferon-Production Pathway. Front. Oncol. 2018, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis Checkpoints as New Targets for Cancer Immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 2020, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Alem, F.; Olanrewaju, A.A.; Omole, S.; Hobbs, H.E.; Ahsan, N.; Matulis, G.; Brantner, C.A.; Zhou, W.; Petricoin, E.F.; Liotta, L.A.; et al. Exosomes Originating from Infection with the Cytoplasmic Single-Stranded RNA Virus Rift Valley Fever Virus (RVFV) Protect Recipient Cells by Inducing RIG-I Mediated IFN-B Response That Leads to Activation of Autophagy. Cell Biosci. 2021, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Dopkins, N.; Nixon, D.F. Two-Step Recognition of HIV-1 DNA in the Cytosol. Trends Microbiol. 2023, 31, 430–431. [Google Scholar] [CrossRef]
- Patrick, K.L.; Bell, S.L.; Watson, R.O. For Better or Worse: Cytosolic DNA Sensing during Intracellular Bacterial Infection Induces Potent Innate Immune Responses. J. Mol. Biol. 2016, 428, 3372–3386. [Google Scholar] [CrossRef]
- Shi, W.; Zhou, Q.; Lu, L.; Zhang, Y.; Zhang, H.; Pu, Y.; Yin, L. Copper Induced Cytosolic Escape of Mitochondrial DNA and Activation of cGAS-STING-NLRP3 Pathway-Dependent Pyroptosis in C8-D1A Cells. Ecotoxicol. Environ. Saf. 2024, 285, 117085. [Google Scholar] [CrossRef]
- Lv, Q.-M.; Lei, H.-M.; Wang, S.-Y.; Zhang, K.-R.; Tang, Y.-B.; Shen, Y.; Lu, L.-M.; Chen, H.-Z.; Zhu, L. Cancer Cell-Autonomous cGAS-STING Response Confers Drug Resistance. Cell Chem. Biol. 2023, 30, 591–605.e4. [Google Scholar] [CrossRef]
- Storozynsky, Q.; Hitt, M.M. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int. J. Mol. Sci. 2020, 21, 8877. [Google Scholar] [CrossRef]
- Técher, H.; Kemiha, S.; Aobuli, X.; Kolinjivadi, A.M. Oncogenic RAS in Cancers from the DNA Replication Stress and Senescence Perspective. Cancers 2024, 16, 3993. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- Suptela, A.J.; Radwan, Y.; Richardson, C.; Yan, S.; Afonin, K.A.; Marriott, I. cGAS Mediates the Inflammatory Responses of Human Microglial Cells to Genotoxic DNA Damage. Inflammation 2024, 47, 822–836. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Liu, F. The cGAS-cGAMP-STING Pathway: A Molecular Link Between Immunity and Metabolism. Diabetes 2019, 68, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between Cellular Responses to DNA Damage and Cancer Immunotherapy. Genes. Dev. 2021, 35, 602–618. [Google Scholar] [CrossRef]
- Paludan, S.R.; Reinert, L.S.; Hornung, V. DNA-Stimulated Cell Death: Implications for Host Defence, Inflammatory Diseases and Cancer. Nat. Rev. Immunol. 2019, 19, 141–153. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Shevtsov, M.; Sato, H.; Multhoff, G.; Shibata, A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front. Oncol. 2019, 9, 156. [Google Scholar] [CrossRef]
- Van Limbergen, E.J.; De Ruysscher, D.K.; Olivo Pimentel, V.; Marcus, D.; Berbee, M.; Hoeben, A.; Rekers, N.; Theys, J.; Yaromina, A.; Dubois, L.J.; et al. Combining Radiotherapy with Immunotherapy: The Past, the Present and the Future. Br. J. Radiol. 2017, 90, 20170157. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Konstantinopoulos, P.A. Combined PARP and Immune Checkpoint Inhibition in Ovarian Cancer. Trends Cancer 2019, 5, 524–528. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Peng, G.; Yap, T.A. A Tale of Two Checkpoints: ATR Inhibition and PD-(L)1 Blockade. Annu. Rev. Med. 2022, 73, 231–250. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP Inhibition Enhances Tumor Cell–Intrinsic Immunity in ERCC1-Deficient Non–Small Cell Lung Cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, P.M.; Kok, Y.P.; Wierenga, E.; Bakker, B.; Foijer, F.; Spierings, D.C.J.; Van Vugt, M.A.T.M. Premature Mitotic Entry Induced by ATR Inhibition Potentiates Olaparib Inhibition-mediated Genomic Instability, Inflammatory Signaling, and Cytotoxicity in BRCA2-deficient Cancer Cells. Mol. Oncol. 2019, 13, 2422–2440. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- De Oliveira Mann, C.C.; Kranzusch, P.J. cGAS Conducts Micronuclei DNA Surveillance. Trends Cell Biol. 2017, 27, 697–698. [Google Scholar] [CrossRef]
- Ahn, J.; Xia, T.; Konno, H.; Konno, K.; Ruiz, P.; Barber, G.N. Inflammation-Driven Carcinogenesis Is Mediated through STING. Nat. Commun. 2014, 5, 5166. [Google Scholar] [CrossRef]
- Zheng, Z.; Jia, S.; Shao, C.; Shi, Y. Irradiation Induces Cancer Lung Metastasis through Activation of the cGAS–STING–CCL5 Pathway in Mesenchymal Stromal Cells. Cell Death Dis. 2020, 11, 326. [Google Scholar] [CrossRef]
- Li, S.; Mirlekar, B.; Johnson, B.M.; Brickey, W.J.; Wrobel, J.A.; Yang, N.; Song, D.; Entwistle, S.; Tan, X.; Deng, M.; et al. STING-Induced Regulatory B Cells Compromise NK Function in Cancer Immunity. Nature 2022, 610, 373–380. [Google Scholar] [CrossRef]
- Liang, D.; Xiao-Feng, H.; Guan-Jun, D.; Er-Ling, H.; Sheng, C.; Ting-Ting, W.; Qin-Gang, H.; Yan-Hong, N.; Ya-Yi, H. Activated STING Enhances Tregs Infiltration in the HPV-Related Carcinogenesis of Tongue Squamous Cells via the c-Jun/CCL22 Signal. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 2494–2503. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef]
- Tang, C.-H.A.; Zundell, J.A.; Ranatunga, S.; Lin, C.; Nefedova, Y.; Del Valle, J.R.; Hu, C.-C.A. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016, 76, 2137–2152. [Google Scholar] [CrossRef]
- Curran, E.; Chen, X.; Corrales, L.; Kline, D.E.; Dubensky, T.W.; Duttagupta, P.; Kortylewski, M.; Kline, J. STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia. Cell Rep. 2016, 15, 2357–2366. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Shin, E.-C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC Class I Antigen Processing and Presenting Machinery: Organization, Function, and Defects in Tumor Cells. JNCI J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced β2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef]
- Yoo, S.H.; Keam, B.; Ock, C.-Y.; Kim, S.; Han, B.; Kim, J.-W.; Lee, K.-W.; Jeon, Y.K.; Jung, K.C.; Chung, E.-J.; et al. Prognostic Value of the Association between MHC Class I Downregulation and PD-L1 Upregulation in Head and Neck Squamous Cell Carcinoma Patients. Sci. Rep. 2019, 9, 7680. [Google Scholar] [CrossRef]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC Proteins Confer Differential Sensitivity to CTLA-4 and PD-1 Blockade in Untreated Metastatic Melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef]
- Friedman, L.A.; Bullock, T.N.; Sloan, E.A.; Ring, K.L.; Mills, A.M. MHC Class I Loss in Endometrial Carcinoma: A Potential Resistance Mechanism to Immune Checkpoint Inhibition. Mod. Pathol. 2021, 34, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef]
- Del Campo, A.B.; Carretero, J.; Muñoz, J.A.; Zinchenko, S.; Ruiz-Cabello, F.; González-Aseguinolaza, G.; Garrido, F.; Aptsiauri, N. Adenovirus Expressing Β2-Microglobulin Recovers HLA Class I Expression and Antitumor Immunity by Increasing T-Cell Recognition. Cancer Gene Ther. 2014, 21, 317–332. [Google Scholar] [CrossRef]
- Zhou, Y.-F.; Xiao, Y.; Jin, X.; Di, G.-H.; Jiang, Y.-Z.; Shao, Z.-M. Integrated Analysis Reveals Prognostic Value of HLA-I LOH in Triple-Negative Breast Cancer. J. Immunother. Cancer 2021, 9, e003371. [Google Scholar] [CrossRef]
- Larson, A.C.; Knoche, S.M.; Brumfield, G.L.; Doty, K.R.; Gephart, B.D.; Moore-Saufley, P.R.; Solheim, J.C. Gemcitabine Modulates HLA-I Regulation to Improve Tumor Antigen Presentation by Pancreatic Cancer Cells. Int. J. Mol. Sci. 2024, 25, 3211. [Google Scholar] [CrossRef]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.-C.; Liu, L.F. Chemotherapeutics and Radiation Stimulate MHC Class I Expression through Elevated Interferon-Beta Signaling in Breast Cancer Cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Trung, V.; Bedi, D.; Shaddox, S.; Gunturu, D.; Yates, C.; Datta, P.; Samuel, T. Interference with Pathways Activated by Topoisomerase Inhibition Alters the Surface Expression of PD-L1 and MHC I in Colon Cancer Cells. Oncol. Lett. 2022, 25, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bastian, I.N.; Long, M.D.; Dow, M.; Li, W.; Liu, T.; Ngu, R.K.; Antonucci, L.; Huang, J.Y.; Phung, Q.T.; et al. Activation of NF-κB and P300/CBP Potentiates Cancer Chemoimmunotherapy through Induction of MHC-I Antigen Presentation. Proc. Natl. Acad. Sci. USA 2021, 118, e2025840118. [Google Scholar] [CrossRef]
- Walle, T.; Martinez Monge, R.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation Effects on Antitumor Immune Responses: Current Perspectives and Challenges. Ther. Adv. Med. Oncol. 2018, 10, 1758834017742575. [Google Scholar] [CrossRef]
- Zebertavage, L.K.; Alice, A.; Crittenden, M.R.; Gough, M.J. Transcriptional Upregulation of NLRC5 by Radiation Drives STING- and Interferon-Independent MHC-I Expression on Cancer Cells and T Cell Cytotoxicity. Sci. Rep. 2020, 10, 7376. [Google Scholar] [CrossRef]
- Deng, S.; Wang, J.; Hu, Y.; Sun, Y.; Yang, X.; Zhang, B.; Deng, Y.; Wei, W.; Zhang, Z.; Wen, L.; et al. Irradiated Tumour Cell-Derived Microparticles Upregulate MHC-I Expression in Cancer Cells via DNA Double-Strand Break Repair Pathway. Cancer Lett. 2024, 592, 216898. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, B.; Tu, J.; Liu, C.; Wang, Y.; Chen, J.; Huang, Y.; Liu, B.; Yuan, X. ATM Inhibition Enhance Immunotherapy by Activating STING Signaling and Augmenting MHC Class I. Cell Death Dis. 2024, 15, 519. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Yadollahi, P.; Jeon, Y.-K.; Ng, W.L.; Choi, I. Current Understanding of Cancer-Intrinsic PD-L1: Regulation of Expression and Its Protumoral Activity. BMB Rep. 2021, 54, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Du, W. PD-1/PD-L1 Signaling Pathway and Tumor Immune Escape. J. Biosci. Med. 2023, 11, 9–16. [Google Scholar] [CrossRef]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of Programmed Death 1 Ligands by Murine T Cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef]
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 Expression during Normal and Autoimmune Responses. Eur. J. Immunol. 2003, 33, 2706–2716. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T Cell Exhaustion in Infection and Cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef]
- Guan, H.; Wan, Y.; Lan, J.; Wang, Q.; Wang, Z.; Li, Y.; Zheng, J.; Zhang, X.; Wang, Z.; Shen, Y.; et al. PD-L1 Is a Critical Mediator of Regulatory B Cells and T Cells in Invasive Breast Cancer. Sci. Rep. 2016, 6, 35651. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front. Pharmacol. 2021, 12, 731798. [Google Scholar] [CrossRef] [PubMed]
- Zarour, H.M. Reversing T-Cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Co-Inhibitory Molecules of the B7–CD28 Family in the Control of T-Cell Immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, L. Inhibitory B7-Family Molecules in the Tumour Microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of Inflammatory Response with B7-H1 Expression in Human Melanocytic Lesions Supports an Adaptive Resistance Mechanism of Immune Escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Immunosuppressive Cells in Tumor Immune Escape and Metastasis. J. Mol. Med. 2016, 94, 509–522. [Google Scholar] [CrossRef]
- Wang, D.; Lin, J.; Yang, X.; Long, J.; Bai, Y.; Yang, X.; Mao, Y.; Sang, X.; Seery, S.; Zhao, H. Combination Regimens with PD-1/PD-L1 Immune Checkpoint Inhibitors for Gastrointestinal Malignancies. J. Hematol. Oncol. 2019, 12, 42. [Google Scholar] [CrossRef]
- Sharma, V.R.; Gupta, G.K.; Sharma, A.K.; Batra, N.; Sharma, D.K.; Joshi, A.; Sharma, A.K. PI3K/Akt/mTOR Intracellular Pathway and Breast Cancer: Factors, Mechanism and Regulation. Curr. Pharm. Des. 2017, 23, 1633–1638. [Google Scholar] [CrossRef]
- Peng, Q.; Deng, Z.; Pan, H.; Gu, L.; Liu, O.; Tang, Z. Mitogen-Activated Protein Kinase Signaling Pathway in Oral Cancer (Review). Oncol. Lett. 2017, 15, 1379–1388. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Jeong, M.; Bazer, F.W.; Song, G. Curcumin Suppresses Proliferation and Migration and Induces Apoptosis on Human Placental Choriocarcinoma Cells via ERK1/2 and SAPK/JNK MAPK Signaling Pathways. Biol. Reprod. 2016, 95, 83. [Google Scholar] [CrossRef]
- Wang, N.-H.; Lei, Z.; Yang, H.-N.; Tang, Z.; Yang, M.-Q.; Wang, Y.; Sui, J.-D.; Wu, Y.-Z. Radiation-Induced PD-L1 Expression in Tumor and Its Microenvironment Facilitates Cancer-Immune Escape: A Narrative Review. Ann. Transl. Med. 2022, 10, 1406. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Honjo, T. Combination Therapy Strategies for Improving PD-1 Blockade Efficacy: A New Era in Cancer Immunotherapy. J. Intern. Med. 2018, 283, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using Immunotherapy to Boost the Abscopal Effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-Based Mechanisms of Cytotoxic Chemotherapy: Implications for the Design of Novel and Rationale-Based Combined Treatments against Cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef]
- Tsai, T.-F.; Lin, J.-F.; Lin, Y.-C.; Chou, K.-Y.; Chen, H.-E.; Ho, C.-Y.; Chen, P.-C.; Hwang, T.I.-S. Cisplatin Contributes to Programmed Death-Ligand 1 Expression in Bladder Cancer through ERK1/2-AP-1 Signaling Pathway. Biosci. Rep. 2019, 39, BSR20190362. [Google Scholar] [CrossRef] [PubMed]
- Permata, T.B.M.; Hagiwara, Y.; Sato, H.; Yasuhara, T.; Oike, T.; Gondhowiardjo, S.; Held, K.D.; Nakano, T.; Shibata, A. Base Excision Repair Regulates PD-L1 Expression in Cancer Cells. Oncogene 2019, 38, 4452–4466. [Google Scholar] [CrossRef]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and Pemetrexed with or without Pembrolizumab for Advanced, Non-Squamous Non-Small-Cell Lung Cancer: A Randomised, Phase 2 Cohort of the Open-Label KEYNOTE-021 Study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Mouw, K.W.; Konstantinopoulos, P.A. From Checkpoint to Checkpoint: DNA Damage ATR/Chk1 Checkpoint Signalling Elicits PD-L1 Immune Checkpoint Activation. Br. J. Cancer 2018, 118, 933–935. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Li, W.; Su, Q.; Huang, Z.; Zhang, X.; Chen, H.; Mo, C.; Huang, B.; Ou, W.; et al. ATM/NEMO Signaling Modulates the Expression of PD-L1 Following Docetaxel Chemotherapy in Prostate Cancer. J. Immunother. Cancer 2021, 9, e001758. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Shi, Y.; Lees-Miller, S.P.; Tainer, J.A. Function and Molecular Mechanism of the DNA Damage Response in Immunity and Cancer Immunotherapy. Front. Immunol. 2021, 12, 797880. [Google Scholar] [CrossRef]
- Pham, M.M.; Ngoi, N.Y.L.; Peng, G.; Tan, D.S.P.; Yap, T.A. Development of Poly(ADP-Ribose) Polymerase Inhibitor and Immunotherapy Combinations: Progress, Pitfalls, and Promises. Trends Cancer 2021, 7, 958–970. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Sun, L.-L.; Yang, R.-Y.; Li, C.-W.; Chen, M.-K.; Shao, B.; Hsu, J.-M.; Chan, L.-C.; Yang, Y.; Hsu, J.L.; Lai, Y.-J.; et al. Inhibition of ATR Downregulates PD-L1 and Sensitizes Tumor Cells to T Cell-Mediated Killing. Am. J. Cancer Res. 2018, 8, 1307–1316. [Google Scholar]
- Mavroeidi, D.; Georganta, A.; Panagiotou, E.; Syrigos, K.; Souliotis, V.L. Targeting ATR Pathway in Solid Tumors: Evidence of Improving Therapeutic Outcomes. Int. J. Mol. Sci. 2024, 25, 2767. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef]
- Bonadio, R.C.; Estevez-Diz, M.D.P. Perspectives on PARP Inhibitor Combinations for Ovarian Cancer. Front. Oncol. 2021, 11, 754524. [Google Scholar] [CrossRef]
- Van Wilpe, S.; Tolmeijer, S.H.; Koornstra, R.H.T.; De Vries, I.J.M.; Gerritsen, W.R.; Ligtenberg, M.; Mehra, N. Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers 2021, 13, 2249. [Google Scholar] [CrossRef] [PubMed]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; Lalonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR Kinase Inhibitor AZD6738 Potentiates CD8+ T Cell–Dependent Antitumor Activity Following Radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef]
- Moutafi, M.; Economopoulou, P.; Rimm, D.; Psyrri, A. PARP Inhibitors in Head and Neck Cancer: Molecular Mechanisms, Preclinical and Clinical Data. Oral Oncol. 2021, 117, 105292. [Google Scholar] [CrossRef] [PubMed]
- Moutafi, M.; Koliou, G.-A.; Papaxoinis, G.; Economopoulou, P.; Kotsantis, I.; Gkotzamanidou, M.; Anastasiou, M.; Pectasides, D.; Kyrodimos, E.; Delides, A.; et al. Phase II Window Study of Olaparib Alone or with Cisplatin or Durvalumab in Operable Head and Neck Cancer. Cancer Res. Commun. 2023, 3, 1514–1523. [Google Scholar] [CrossRef]
- Oliva, M.; Llop, S.; Vidales, Z.; Arrazubi, V.; Baste, N.; Brana, I.; Cirauqui, B.; Cacicedo, J.; Giralt, J.; Marruecos, J.; et al. TTCC-2022-01: Niraparib and Dostarlimab in Locally-Advanced Head and Neck Squamous Cell Carcinoma Treated with (Chemo) Radiotherapy (RADIAN). J. Clin. Oncol. 2024, 42, TPS6125. [Google Scholar] [CrossRef]
- Harano, K.; Nakao, T.; Nishio, S.; Katsuta, T.; Tasaki, K.; Takehara, K.; Yokoyama, T.; Furuya, H.; Hongo, K.; Asano, M.; et al. Neoadjuvant Combination Treatment of Olaparib and Pembrolizumab for Patients with HRD-Positive Advanced Ovarian Cancer. J. Clin. Oncol. 2024, 42, 5545. [Google Scholar] [CrossRef]
- Mayer, E.; Leon-Ferre, R. TBCRC 056: A Phase II Study of Neoadjuvant Niraparib with Dostarlimab for Patients with BRCA- or PALB2-Mutated Breast Cancer: Results from the ER+/HER2- Cohort. In Proceedings of the San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 10–13 December 2024. [Google Scholar]
- Harrington, K.J.; Burtness, B.; Greil, R.; Soulières, D.; Tahara, M.; De Castro, G.; Psyrri, A.; Brana, I.; Basté, N.; Neupane, P.; et al. Pembrolizumab With or Without Chemotherapy in Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma: Updated Results of the Phase III KEYNOTE-048 Study. J. Clin. Oncol. 2023, 41, 790–802. [Google Scholar] [CrossRef]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Garassino, M.C.; Gadgeel, S.; Speranza, G.; Felip, E.; Esteban, E.; Dómine, M.; Hochmair, M.J.; Powell, S.F.; Bischoff, H.G.; Peled, N.; et al. Pembrolizumab Plus Pemetrexed and Platinum in Nonsquamous Non–Small-Cell Lung Cancer: 5-Year Outcomes From the Phase 3 KEYNOTE-189 Study. J. Clin. Oncol. 2023, 41, 1992–1998. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-Line Nivolumab plus Ipilimumab Combined with Two Cycles of Chemotherapy in Patients with Non-Small-Cell Lung Cancer (CheckMate 9LA): An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Doki, Y.; Ajani, J.A.; Kato, K.; Xu, J.; Wyrwicz, L.; Motoyama, S.; Ogata, T.; Kawakami, H.; Hsu, C.-H.; Adenis, A.; et al. Nivolumab Combination Therapy in Advanced Esophageal Squamous-Cell Carcinoma. N. Engl. J. Med. 2022, 386, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Aivaliotis, I.L.; Pateras, I.S.; Papaioannou, M.; Glytsou, C.; Kontzoglou, K.; Johnson, E.O.; Zoumpourlis, V. How Do Cytokines Trigger Genomic Instability? J. Biomed. Biotechnol. 2012, 2012, 536761. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef]
- Yi, M.; Li, T.; Niu, M.; Zhang, H.; Wu, Y.; Wu, K.; Dai, Z. Targeting Cytokine and Chemokine Signaling Pathways for Cancer Therapy. Sig Transduct. Target. Ther. 2024, 9, 176. [Google Scholar] [CrossRef]
- An, L.; Li, M.; Jia, Q. Mechanisms of Radiotherapy Resistance and Radiosensitization Strategies for Esophageal Squamous Cell Carcinoma. Mol. Cancer 2023, 22, 140. [Google Scholar] [CrossRef]
- Zhang, H. CAF-Secreted CXCL1 Conferred Radioresistance by Regulating DNA Damage Response in a ROS-Dependent Manner in Esophageal Squamous Cell Carcinoma. Cell Death Dis. 2017, 8, e2790. [Google Scholar] [CrossRef]
- Guo, Y.; Shen, R.; Wang, F.; Wang, Y.; Xia, P.; Wu, R.; Liu, X.; Ye, W.; Tian, Y.; Wang, D. Carbon Ion Irradiation Induces DNA Damage in Melanoma and Optimizes the Tumor Microenvironment Based on the cGAS–STING Pathway. J. Cancer Res. Clin. Oncol. 2023, 149, 6315–6328. [Google Scholar] [CrossRef]
- Meidenbauer, J.; Wachter, M.; Schulz, S.R.; Mostafa, N.; Zülch, L.; Frey, B.; Fietkau, R.; Gaipl, U.S.; Jost, T. Inhibition of ATM or ATR in Combination with Hypo-Fractionated Radiotherapy Leads to a Different Immunophenotype on Transcript and Protein Level in HNSCC. Front. Oncol. 2024, 14, 1460150. [Google Scholar] [CrossRef]
- Chen, W.-T.; Ebelt, N.D.; Stracker, T.H.; Xhemalce, B.; Van Den Berg, C.L.; Miller, K.M. ATM Regulation of IL-8 Links Oxidative Stress to Cancer Cell Migration and Invasion. eLife 2015, 4, e07270. [Google Scholar] [CrossRef]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-Induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-L.; Tang, C.; Zhang, M.-Y.; Huang, W.-L.; Xu, Y.; Sun, H.-Y.; Yang, F.; Song, L.-L.; Wang, H.; Mu, L.-L.; et al. Blocking ATM-Dependent NF-κB Pathway Overcomes Niche Protection and Improves Chemotherapy Response in Acute Lymphoblastic Leukemia. Leukemia 2019, 33, 2365–2378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-N.; Yang, K.-D.; Chen, C.; He, Z.-C.; Wang, Q.-H.; Feng, H.; Lv, S.-Q.; Wang, Y.; Mao, M.; Liu, Q.; et al. Pericytes Augment Glioblastoma Cell Resistance to Temozolomide through CCL5-CCR5 Paracrine Signaling. Cell Res. 2021, 31, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.E.; Maallin, S.; Haston, S.; Martinez-Barbera, J.P.; Hodivala-Dilke, K.M.; Pedrosa, A. Effects of Senescence on the Tumour Microenvironment and Response to Therapy. FEBS J. 2024, 291, 2306–2319. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sources | Ref. | |
|---|---|---|
| External factors | virus | [76] |
| retrovirus | [77] | |
| bacteria | [78] | |
| oxidative stress | [79] | |
| chemotherapy | [80] | |
| radiation | [81] | |
| Internal factors | damaged nuclear DNA | [71] |
| mitochondrial DNA | [71] | |
| mitotic defects | [71] | |
| DNA from micronuclei | [71] | |
| hyperactivation of oncogene signaling | [82] | |
| low chromosome instability | [83] | |
| cell debris | [84] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deligianni, E.; Papanikolaou, C.; Terpos, E.; Souliotis, V.L. Elucidating DNA Damage-Dependent Immune System Activation. Int. J. Mol. Sci. 2025, 26, 5849. https://doi.org/10.3390/ijms26125849
Deligianni E, Papanikolaou C, Terpos E, Souliotis VL. Elucidating DNA Damage-Dependent Immune System Activation. International Journal of Molecular Sciences. 2025; 26(12):5849. https://doi.org/10.3390/ijms26125849
Chicago/Turabian StyleDeligianni, Elisavet, Christina Papanikolaou, Evangelos Terpos, and Vassilis L. Souliotis. 2025. "Elucidating DNA Damage-Dependent Immune System Activation" International Journal of Molecular Sciences 26, no. 12: 5849. https://doi.org/10.3390/ijms26125849
APA StyleDeligianni, E., Papanikolaou, C., Terpos, E., & Souliotis, V. L. (2025). Elucidating DNA Damage-Dependent Immune System Activation. International Journal of Molecular Sciences, 26(12), 5849. https://doi.org/10.3390/ijms26125849