
CNS Resident Innate Immune Cells: Guardians of CNS Homeostasis


{kind=link}
{kind=link}
Abstract
1. Introduction
2. MS: Pathophysiology and Tools of Investigation
2.1. MS Pathophysiology
2.2. Preclinical Models of MS
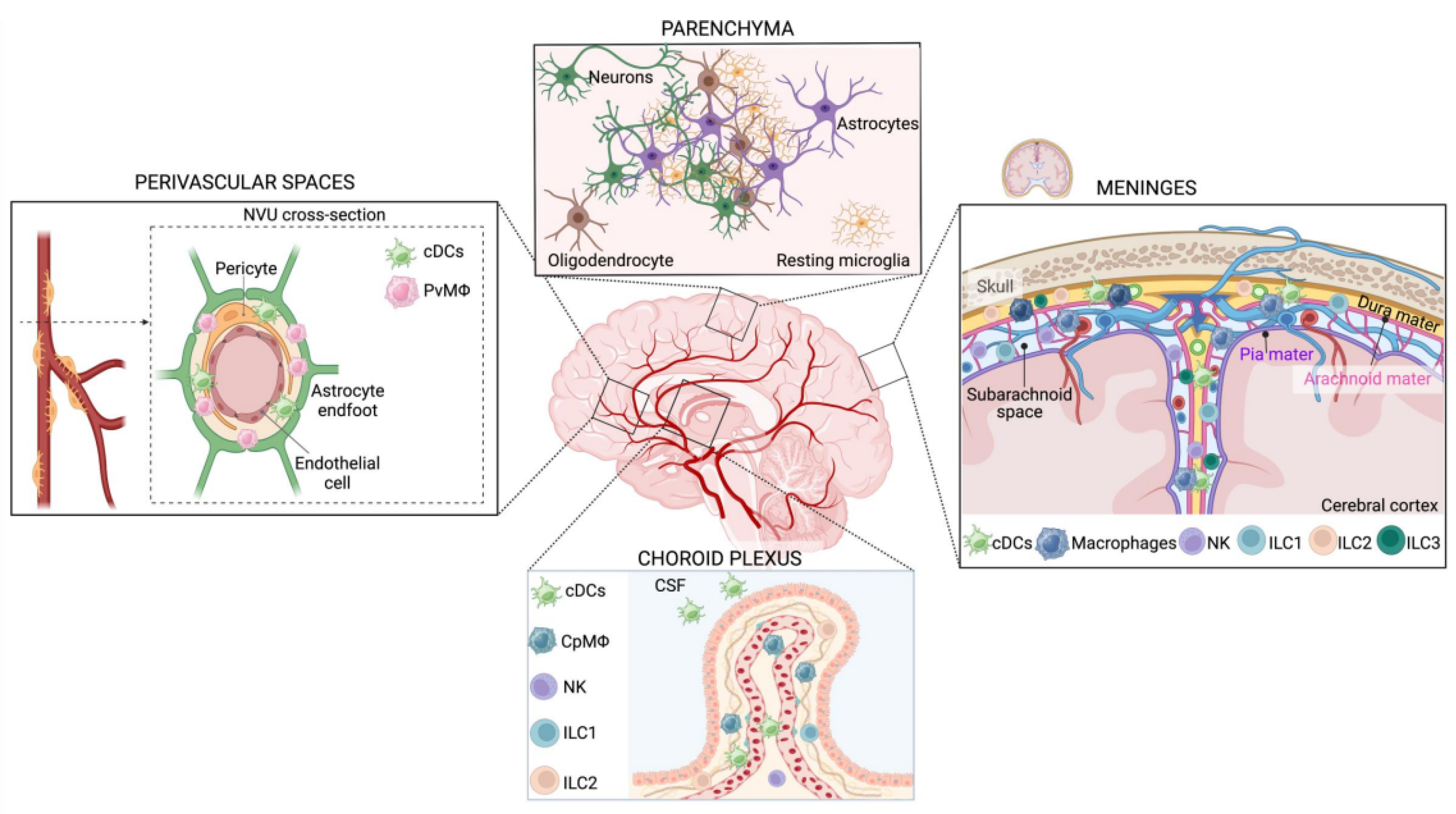
3. Innate Immunity of the CNS: Classification and Ontogeny
3.1. CNS Innate Immune Cell Classification
3.2. Microglia
3.3. Border-Associated Macrophages (BAMs)
3.4. Dendritic Cells
3.5. Innate Lymphoid Cells
3.6. Ontogeny of the CNS Resident Myeloid Compartment
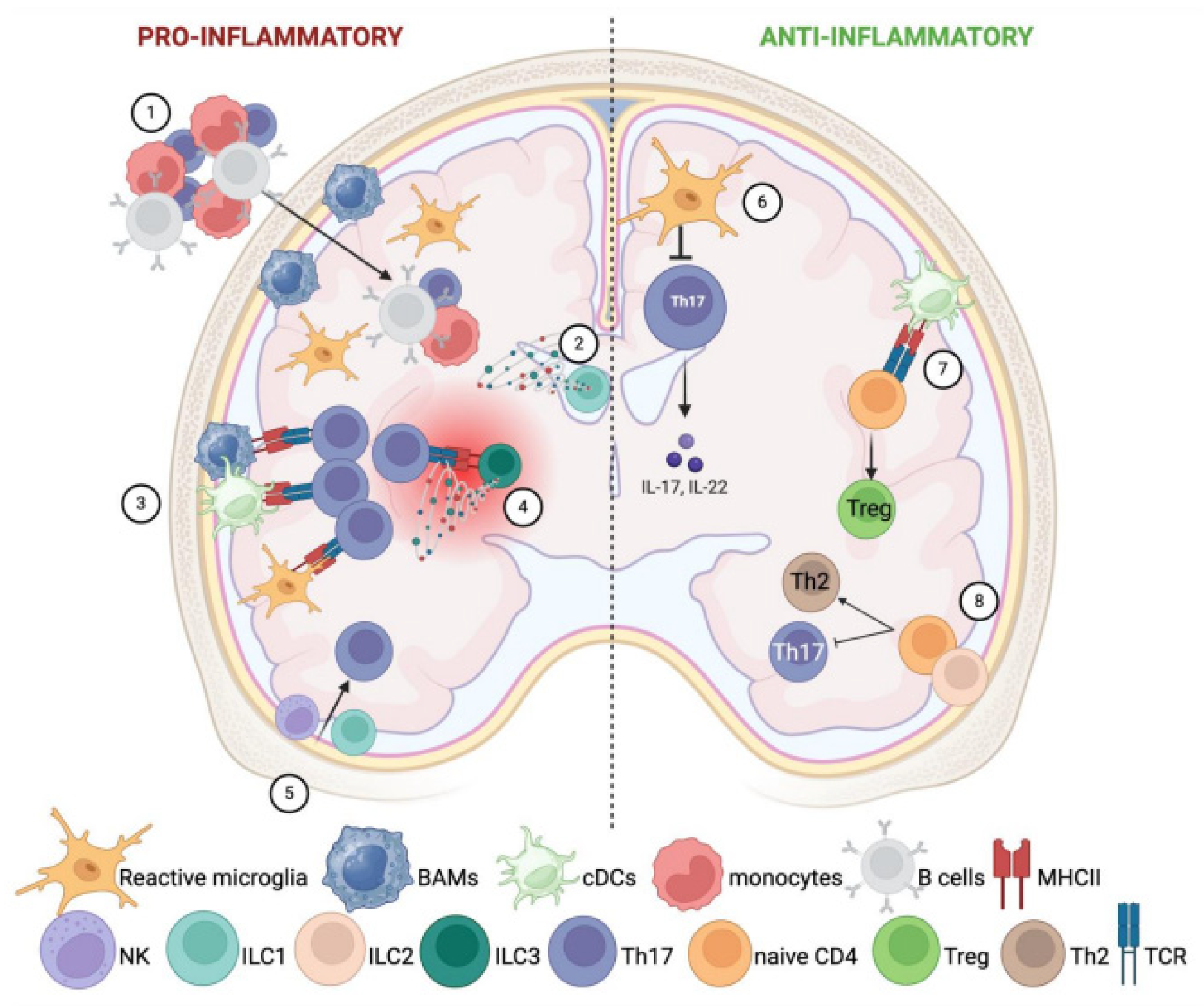
4. CNS Innate Immunity Contribution to Neuroinflammation
5. CNS Innate Immunity Contribution to Neurodegeneration
5.1. Dendritic Cells
5.2. ILCs
6. Conclusions and Therapeutic Perspective
7. Highlights
- -
- The CNS immune landscape is plastic and multifaceted. Apart from brain parenchyma, it is crucial to consider also immune cell populations residing at the borders: meninges, perivascular space, and choroid plexus.
- -
- The resident CNS innate immune cells are involved in multiple aspects of CNS homeostasis and pathology. Here, we focus on neuroinflammation and neurodegeneration, two interdependent facets of multiple sclerosis, as well as other neurological disorders.
- -
- The same cellular subset can have either a beneficial or detrimental role in the frame of MS, according to the disease phase.
- -
- When analyzing the effect driven by DMTs, it is pivotal to consider both adaptive immunity and innate immunity since they mutually influence each other.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef] [PubMed]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Giovannoni, G.; Salvetti, M. Epstein-Barr virus as a cause of multiple sclerosis: Opportunities for prevention and therapy. Lancet Neurol. 2023, 22, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Baumann, R.; Gao, X.; Mendoza, M.; Singh, S.; Sand, I.K.; Xia, Z.; Cox, L.M.; Chitnis, T.; Yoon, H.; et al. Gut microbiome of multiple sclerosis patients and paired household healthy controls reveal associations with disease risk and course. Cell 2022, 185, 3467–3486.e16. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L.; White, R.; Köchert, K.; Simon, K.C.; Polman, C.H.; Freedman, M.S.; Hartung, H.-P.; Miller, D.H.; Montalbán, X.; et al. Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA Neurol. 2014, 71, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2016, 13, 25–36. [Google Scholar] [CrossRef]
- Lublin, F.D. New Multiple Sclerosis Phenotypic Classification. Eur. Neurol. 2014, 72, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Klineova, S.; Lublin, F.D. Clinical Course of Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028928. [Google Scholar] [CrossRef]
- Hauser, S.L.; Cree, B.A. Treatment of Multiple Sclerosis: A Review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef]
- Pozzilli, C.; Pugliatti, M.; Vermersch, P.; Grigoriadis, N.; Alkhawajah, M.; Airas, L.; Oreja-Guevara, C. Diagnosis and treatment of progressive multiple sclerosis: A position paper. Eur. J. Neurol. 2022, 30, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [PubMed]
- Babaloo, Z.; Aliparasti, M.R.; Babaiea, F.; Almasi, S.; Baradaran, B.; Farhoudi, M. The role of Th17 cells in patients with relapsing-remitting multiple sclerosis: Interleukin-17A and interleukin-17F serum levels. Immunol. Lett. 2015, 164, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Kovacs, G.G.; Kutzelnigg, A.; Lassmann, H.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Moccia, M.; Coetzee, T.; A Cohen, J.; Correale, J.; Graves, J.; Marrie, R.A.; Montalban, X.; Yong, V.W.; Thompson, A.J.; et al. Multiple sclerosis progression: Time for a new mechanism-driven framework. Lancet Neurol. 2023, 22, 78–88. [Google Scholar] [CrossRef]
- Mayhew, C.N.; Singhania, R. A review of protocols for brain organoids and applications for disease modeling. STAR Protoc. 2023, 4, 101860. [Google Scholar] [CrossRef] [PubMed]
- Marangon, D.; Caporale, N.; Boccazzi, M.; Abbracchio, M.P.; Testa, G.; Lecca, D. Novel in vitro Experimental Approaches to Study Myelination and Remyelination in the Central Nervous System. Front. Cell. Neurosci. 2021, 15, 748849. [Google Scholar] [CrossRef] [PubMed]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Dixit, A.; Savage, H.S.; Greer, J.M. An appraisal of emerging therapeutic targets for multiple sclerosis derived from current preclinical models. Expert Opin. Ther. Targets 2023, 27, 553–574. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, S.; Bettelli, E. Experimental Autoimmune Encephalomyelitis (EAE) as Animal Models of Multiple Sclerosis (MS). Cold Spring Harb. Perspect. Med. 2018, 8, a028977. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M. How to Use the Cuprizone Model to Study De- and Remyelination. Int. J. Mol. Sci. 2024, 25, 1445. [Google Scholar] [CrossRef] [PubMed]
- Hart, B.A. Why does multiple sclerosis only affect human primates? Mult. Scler. J. 2016, 22, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Rivers, T.M.; Sprunt, D.H.; Berry, G.P. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J. Exp. Med. 1933, 58, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Goverman, J.M. Passive induction of experimental allergic encephalomyelitis. Nat. Protoc. 2006, 1, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Zamvil, S.S.; Steinman, L. The T Lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol. 1990, 8, 579–621. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Pelech, S.; Zhang, H.; Bond, J.; Spach, K.; Noubade, R.; Blankenhorn, E.P.; Teuscher, C. Pertussis toxin induces angiogenesis in brain microvascular endothelial cells. J. Neurosci. Res. 2008, 86, 2624–2640. [Google Scholar] [CrossRef]
- Raphael, I.; Mahesula, S.; Purkar, A.; Black, D.; Catala, A.; Gelfond, J.A.L.; Forsthuber, T.G.; Haskins, W.E. Microwave & Magnetic (M2) proteomics reveals CNS-specific protein expression waves that precede clinical symptoms of experimental autoimmune encephalomyelitis. Sci. Rep. 2014, 4, srep06210. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef]
- Leo, H.; Kipp, M. Remyelination in Multiple Sclerosis: Findings in the Cuprizone Model. Int. J. Mol. Sci. 2022, 23, 16093. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Mann, T.; Joost, S.; Behrangi, N.; Frank, M.; Kipp, M. The Cuprizone Model: Dos and Do Nots. Cells 2020, 9, 843. [Google Scholar] [CrossRef]
- Fischbach, F.; Nedelcu, J.; Leopold, P.; Zhan, J.; Clarner, T.; Nellessen, L.; Beißel, C.; van Heuvel, Y.; Goswami, A.; Weis, J.; et al. Cuprizone-induced graded oligodendrocyte vulnerability is regulated by the transcription factor DNA damage-inducible transcript 3. Glia 2019, 67, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, L.A.; Calatayud, C.A.; Uña, A.L.B.; Millet, V.; Pasquini, J.M.; Soto, E.F. The Neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem. Res. 2007, 32, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, A.; Walker, T.; Avila, R.; Huang, H.; Caporoso, J.; Manandhar, E.; Leeper, T.C.; Modarelli, D.A.; Medicetty, S.; Shriver, L.P. Cuprizone Intoxication Induces Cell Intrinsic Alterations in Oligodendrocyte Metabolism Independent of Copper Chelation. Biochemistry 2017, 56, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.L.; Teo, W.; Hernandez, Y.; Brideau, C.; Cummins, K.; Kuipers, H.F.; Stys, P.K. Cuprizone-induced Demyelination in Mouse Brain is not due to Depletion of Copper. ASN Neuro 2022, 14, 17590914221126367. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhou, Y.; Cai, Z.; Terekhova, M.; Swain, A.; Andhey, P.S.; Guimaraes, R.M.; Antonova, A.U.; Qiu, T.; Sviben, S.; et al. Transcriptomic atlas and interaction networks of brain cells in mouse CNS demyelination and remyelination. Cell Rep. 2023, 42, 112293. [Google Scholar] [CrossRef]
- Zirngibl, M.; Assinck, P.; Sizov, A.; Caprariello, A.V.; Plemel, J.R. Oligodendrocyte death and myelin loss in the cuprizone model: An updated overview of the intrinsic and extrinsic causes of cuprizone demyelination. Mol. Neurodegener. 2022, 17, 34. [Google Scholar] [CrossRef]
- Salvador, F.; Deramoudt, L.; Leprêtre, F.; Figeac, M.; Guerrier, T.; Boucher, J.; Bas, M.; Journiac, N.; Peters, A.; Mars, L.T.; et al. A Spontaneous Model of Experimental Autoimmune Encephalomyelitis Provides Evidence of MOG-Specific B Cell Recruitment and Clonal Expansion. Front. Immunol. 2022, 13, 755900. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein–specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Pöllinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bösl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System–Associated Macrophages—From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Mildenberger, W.; A Stifter, S.; Greter, M. Diversity and function of brain-associated macrophages. Curr. Opin. Immunol. 2022, 76, 102181. [Google Scholar] [CrossRef] [PubMed]
- Neniskyte, U.; Gross, C.T. Errant gardeners: Glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat. Rev. Neurosci. 2017, 18, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, J.; Huang, M.; Chen, Z.; Liu, J.; Zhang, Q.; Zhang, C.; Xiang, Y.; Zen, K.; Li, L. Loss of microglial SIRPα promotes synaptic pruning in preclinical models of neurodegeneration. Nat. Commun. 2021, 12, 2030. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Myhre, C.L.; Thygesen, C.; Villadsen, B.; Vollerup, J.; Ilkjær, L.; Krohn, K.T.; Grebing, M.; Zhao, S.; Khan, A.M.; Dissing-Olesen, L.; et al. Microglia Express Insulin-Like Growth Factor-1 in the Hippocampus of Aged APPswe/PS1ΔE9 Transgenic Mice. Front. Cell. Neurosci. 2019, 13, 308. [Google Scholar] [CrossRef]
- De Lucia, C.; Rinchon, A.; Olmos-Alonso, A.; Riecken, K.; Fehse, B.; Boche, D.; Perry, V.H.; Gomez-Nicola, D. Microglia regulate hippocampal neurogenesis during chronic neurodegeneration. Brain Behav. Immun. 2016, 55, 179–190. [Google Scholar] [CrossRef]
- Oosterhof, N.; Kuil, L.E.; van der Linde, H.C.; Burm, S.M.; Berdowski, W.; van Ijcken, W.F.; van Swieten, J.C.; Hol, E.M.; Verheijen, M.H.; van Ham, T.J. Colony-Stimulating Factor 1 Receptor (CSF1R) Regulates Microglia Density and Distribution, but Not Microglia Differentiation In Vivo. Cell Rep. 2018, 24, 1203–1217.e6. [Google Scholar] [CrossRef]
- Stratoulias, V.; Ruiz, R.; Kanatani, S.; Osman, A.M.; Keane, L.; Armengol, J.A.; Rodríguez-Moreno, A.; Murgoci, A.-N.; García-Domínguez, I.; Alonso-Bellido, I.; et al. ARG1-expressing microglia show a distinct molecular signature and modulate postnatal development and function of the mouse brain. Nat. Neurosci. 2023, 26, 1008–1020. [Google Scholar] [CrossRef]
- Harley, S.B.R.; Willis, E.F.; Shaikh, S.N.; Blackmore, D.G.; Sah, P.; Ruitenberg, M.J.; Bartlett, P.F.; Vukovic, J. Selective ablation of BDNF from microglia reveals novel roles in Self-renewal and hippocampal neurogenesis. J. Neurosci. 2021, 41, 4172–4186. [Google Scholar] [CrossRef]
- Zhang, J.; Rong, P.; Zhang, L.; He, H.; Zhou, T.; Fan, Y.; Mo, L.; Zhao, Q.; Han, Y.; Li, S.; et al. IL4-driven microglia modulate stress resilience through BDNF-dependent neurogenesis. Sci. Adv. 2021, 7, eabb9888. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodríguez-Iglesias, N.; Márquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J. Neurosci. 2020, 40, 1453–1482. [Google Scholar] [CrossRef]
- Tan, Y.-L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Muntslag, T.A.; Martin-Estebané, M.; Barry-Carroll, L.; Chapman, M.A.; Adorjan, I.; Tyler, T.; Turnbull, B.; Rose-Zerilli, M.J.; Nicoll, J.A.; et al. The spatiotemporal dynamics of microglia across the human lifespan. Dev. Cell 2022, 57, 2127–2139.e6. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef]
- Rusin, D.; Becirovic, L.V.; Lyszczarz, G.; Krueger, M.; Benmamar-Badel, A.; Mathiesen, C.V.; Schiöth, E.S.; Lambertsen, K.L.; Wlodarczyk, A. Microglia-Derived Insulin-like Growth Factor 1 Is Critical for Neurodevelopment. Cells 2024, 13, 184. [Google Scholar] [CrossRef]
- Tagliatti, E.; Desiato, G.; Mancinelli, S.; Bizzotto, M.; Gagliani, M.C.; Faggiani, E.; Hernández-Soto, R.; Cugurra, A.; Poliseno, P.; Miotto, M.; et al. Trem2 expression in microglia is required to maintain normal neuronal bioenergetics during development. Immunity 2023, 57, 86–105.e9. [Google Scholar] [CrossRef] [PubMed]
- Kiialainen, A.; Hovanes, K.; Paloneva, J.; Kopra, O.; Peltonen, L. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol. Dis. 2005, 18, 314–322. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Microglia in steady state. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Itriago, A.; Radford, R.A.W.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Front. Immunol. 2022, 13, 997786. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Yang, S.; Qin, C.; Hu, Z.-W.; Zhou, L.-Q.; Yu, H.-H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.-J.; Tian, D.-S. Microglia reprogram metabolic profiles for phenotype and function changes in central nervous system. Neurobiol. Dis. 2021, 152, 105290. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Savinova, O.V.; Hoffmann, A.; Ghosh, G. The Nfkb1 and Nfkb2 Proteins p105 and p100 Function as the Core of High-Molecular-Weight Heterogeneous Complexes. Mol. Cell 2009, 34, 591–602. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-κB Function in the Nervous System. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef] [PubMed]
- Kyrargyri, V.; Vega-Flores, G.; Gruart, A.; Delgado-García, J.M.; Probert, L. Differential contributions of microglial and neuronal IKKβ to synaptic plasticity and associative learning in alert behaving mice. Glia 2015, 63, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Yue, Y.; Stone, S. Role of nuclear factor κB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural Regen. Res. 2018, 13, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Avloniti, M.; Evangelidou, M.; Gomini, M.; Loupis, T.; Emmanouil, M.; Mitropoulou, A.; Tselios, T.; Lassmann, H.; Gruart, A.; Delgado-García, J.M.; et al. IKKβ deletion from CNS macrophages increases neuronal excitability and accelerates the onset of EAE, while from peripheral macrophages reduces disease severity. J. Neuroinflamm. 2024, 21, 34. [Google Scholar] [CrossRef]
- Amann, L.; Masuda, T.; Prinz, M. Mechanisms of myeloid cell entry to the healthy and diseased central nervous system. Nat. Immunol. 2023, 24, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner , F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Lively, S.; Schlichter, L.C. Microglia responses to pro-inflammatory stimuli (LPS, IFNγ+TNFα) and reprogramming by resolving cytokines (IL-4, IL-10). Front. Cell Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Derk, J.; Jones, H.E.; Como, C.; Pawlikowski, B.; Siegenthaler, J.A. Living on the Edge of the CNS: Meninges Cell Diversity in Health and Disease. Front. Cell. Neurosci. 2021, 15, 703944. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zheng, H.; Shi, C.; Li, X.; Zhang, S.; Guo, G.; Yu, W.; Zhang, S.; Hu, Z.; Yang, J.; et al. Meningeal immunity and neurological diseases: New approaches, new insights. J. Neuroinflamm. 2023, 20, 125. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Drieu, A.; Mamuladze, T.; de Lima, K.A.; Dykstra, T.; Wall, M.; Papadopoulos, Z.; Kanamori, M.; Salvador, A.F.; Baker, W.; et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 2021, 184, 1000–1016.e27. [Google Scholar] [CrossRef] [PubMed]
- Rebejac, J.; Eme-Scolan, E.; Paroutaud, L.A.; Kharbouche, S.; Teleman, M.; Spinelli, L.; Gallo, E.; Roussel-Queval, A.; Zarubica, A.; Sansoni, A.; et al. Meningeal macrophages protect against viral neuroinfection. Immunity 2022, 55, 2103–2117.e10. [Google Scholar] [CrossRef] [PubMed]
- Lun, M.P.; Monuki, E.S.; Lehtinen, M.K. Development and functions of the choroid plexus–cerebrospinal fluid system. Nat. Rev. Neurosci. 2015, 16, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Xu, H.; Lehtinen, M.K. Macrophages on the margin: Choroid plexus immune responses. Trends Neurosci. 2021, 44, 864–875. [Google Scholar] [CrossRef]
- Dani, N.; Herbst, R.H.; McCabe, C.; Green, G.S.; Kaiser, K.; Head, J.P.; Cui, J.; Shipley, F.B.; Jang, A.; Dionne, D.; et al. A cellular and spatial map of the choroid plexus across brain ventricles and ages. Cell 2021, 184, 3056–3074.e21. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Gokhan, S.; Dai, X.-M.; Wei, S.; Enikolopov, G.; Lin, H.; Mehler, M.F.; Stanley, E.R. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev. Biol. 2012, 367, 100–113. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Robert, S.M.; Reeves, B.C.; Kiziltug, E.; Duy, P.Q.; Karimy, J.K.; Mansuri, M.S.; Marlier, A.; Allington, G.; Greenberg, A.B.; DeSpenza, T.; et al. The choroid plexus links innate immunity to CSF dysregulation in hydrocephalus. Cell 2023, 186, 764–785.e21. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Karam, M.; Janbon, H.; Malkinson, G.; Brunet, I. Heterogeneity and developmental dynamics of LYVE-1 perivascular macrophages distribution in the mouse brain. J. Cereb. Blood Flow Metab. 2022, 42, 1797–1812. [Google Scholar] [CrossRef] [PubMed]
- Császár, E.; Lénárt, N.; Cserép, C.; Környei, Z.; Fekete, R.; Pósfai, B.; Balázsfi, D.; Hangya, B.; Schwarcz, A.D.; Szabadits, E.; et al. Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. J. Exp. Med. 2022, 219, e20211071. [Google Scholar] [CrossRef] [PubMed]
- Polfliet, M.M.J.; Zwijnenburg, P.J.G.; van Furth, A.M.; van der Poll, T.; Döpp, E.A.; de Lavalette, C.R.; van Kesteren-Hendrikx, E.M.L.; van Rooijen, N.; Dijkstra, C.D.; van den Berg, T.K. Meningeal and Perivascular Macrophages of the Central Nervous System Play a Protective Role During Bacterial Meningitis. J. Immunol. 2001, 167, 4644–4650. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Sugiyama, Y.; Lane, D.; Garcia-Bonilla, L.; Chang, H.; Santisteban, M.M.; Racchumi, G.; Murphy, M.; Van Rooijen, N.; Anrather, J.; et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J. Clin. Investig. 2016, 126, 4674–4689. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.; Garwood, C.; Ray, D. A size selective vascular barrier in the rat area postrema formed by perivascular macrophages and the extracellular matrix. Neuroscience 2007, 150, 498–509. [Google Scholar] [CrossRef]
- Zhang, W.; Dai, M.; Fridberger, A.; Hassan, A.; DeGagne, J.; Neng, L.; Zhang, F.; He, W.; Ren, T.; Trune, D.; et al. Perivascular-resident macrophage-like melanocytes in the inner ear are essential for the integrity of the intrastrial fluid–blood barrier. Proc. Natl. Acad. Sci. USA 2012, 109, 10388–10393. [Google Scholar] [CrossRef]
- Qin, J.; Lovelace, M.D.; Mitchell, A.J.; de Koning-Ward, T.; Grau, G.E.; Pai, S. Perivascular macrophages create an intravascular niche for CD8+ T cell localisation prior to the onset of fatal experimental cerebral malaria. Clin. Transl. Immunol. 2021, 10, e1273. [Google Scholar] [CrossRef]
- Lee, C.H.; Kim, H.J.; Lee, Y.-S.; Kang, G.M.; Lim, H.S.; Lee, S.-H.; Song, D.K.; Kwon, O.; Hwang, I.; Son, M.; et al. Hypothalamic Macrophage Inducible Nitric Oxide Synthase Mediates Obesity-Associated Hypothalamic Inflammation. Cell Rep. 2018, 25, 934–946.e5. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Solas, M.; Backes, H.; Chaurasia, B.; Kleinridders, A.; Theurich, S.; Mauer, J.; Steculorum, S.M.; Hampel, B.; Goldau, J.; et al. Myeloid-Cell-Derived VEGF Maintains Brain Glucose Uptake and Limits Cognitive Impairment in Obesity. Cell 2016, 165, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Mendes, N.F.; Velloso, L.A. Perivascular macrophages in high-fat diet-induced hypothalamic inflammation. J. Neuroinflamm. 2022, 19, 136. [Google Scholar] [CrossRef] [PubMed]
- Gerganova, G.; Riddell, A.; Miller, A.A. CNS border-associated macrophages in the homeostatic and ischaemic brain. Pharmacol. Ther. 2022, 240, 108220. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Uekawa, K.; Garcia-Bonilla, L.; Koizumi, K.; Murphy, M.; Pistik, R.; Younkin, L.; Younkin, S.; Zhou, P.; Carlson, G.; et al. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ. Res. 2017, 121, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Iyonaga, T.; Shinohara, K.; Mastuura, T.; Hirooka, Y.; Tsutsui, H. Brain perivascular macrophages contribute to the development of hypertension in stroke-prone spontaneously hypertensive rats via sympathetic activation. Hypertens. Res. 2019, 43, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; da Costa, M.P.; e Sousa, C.R. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.T.; Brian, B.F.; Senevirathne, S.E.; Freedman, T.S. Regulation of myeloid-cell activation. Curr. Opin. Immunol. 2021, 73, 34–42. [Google Scholar] [CrossRef]
- Hornero, R.A.; Idoyaga, J. Plasmacytoid dendritic cells: A dendritic cell in disguise. Mol. Immunol. 2023, 159, 38–45. [Google Scholar] [CrossRef]
- Pashenkov, M.; Huang, Y.-M.; Kostulas, V.; Haglund, M.; Söderström, M.; Link, H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain 2001, 124, 480–492. [Google Scholar] [CrossRef]
- Longhini, A.L.F.; von Glehn, F.; Brandão, C.O.; de Paula, R.F.; Pradella, F.; Moraes, A.S.; Farias, A.S.; Oliveira, E.C.; Quispe-Cabanillas, J.G.; Abreu, C.H.; et al. Plasmacytoid dendritic cells are increased in cerebrospinal fluid of untreated patients during multiple sclerosis relapse. J. Neuroinflamm. 2011, 8, 2. [Google Scholar] [CrossRef]
- Esaulova, E.; Cantoni, C.; Shchukina, I.; Zaitsev, K.; Bucelli, R.C.; Wu, G.F.; Artyomov, M.N.; Cross, A.H.; Edelson, B.T. Single-cell RNA-seq analysis of human CSF microglia and myeloid cells in neuroinflammation. Neurol.-Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef]
- Mundt, S.; Greter, M.; Becher, B. The CNS mononuclear phagocyte system in health and disease. Neuron 2022, 110, 3497–3512. [Google Scholar] [CrossRef]
- Ludewig, P.; Gallizioli, M.; Urra, X.; Behr, S.; Brait, V.H.; Gelderblom, M.; Magnus, T.; Planas, A.M. Dendritic cells in brain diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 352–367. [Google Scholar] [CrossRef]
- Croese, T.; Castellani, G.; Schwartz, M. Immune cell compartmentalization for brain surveillance and protection. Nat. Immunol. 2021, 22, 1083–1092. [Google Scholar] [CrossRef]
- Wang, S.; van de Pavert, S.A. Innate Lymphoid Cells in the Central Nervous System. Front. Immunol. 2022, 13, 837250. [Google Scholar] [CrossRef]
- Romero-Suárez, S.; Serrato, A.D.R.; Bueno, R.J.; Brunotte-Strecker, D.; Stehle, C.; Figueiredo, C.A.; Hertwig, L.; Dunay, I.R.; Romagnani, C.; Infante-Duarte, C. The Central Nervous System Contains ILC1s That Differ From NK Cells in the Response to Inflammation. Front. Immunol. 2019, 10, 2337. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Herbert, D.R.; Douglas, B.; Zullo, K. Group 2 Innate Lymphoid Cells (ILC2): Type 2 Immunity and Helminth Immunity. Int. J. Mol. Sci. 2019, 20, 2276. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Wolterink, R.G.J.K.; Godinho-Silva, C.; Domingues, R.G.; Ribeiro, H.; da Silva, J.A.; Mahú, I.; Domingos, A.I.; Veiga-Fernandes, H. Neuro-mesenchymal units control ILC2 and obesity via a brain–adipose circuit. Nature 2021, 597, 410–414. [Google Scholar] [CrossRef]
- Fung, I.T.H.; Sankar, P.; Zhang, Y.; Robison, L.S.; Zhao, X.; D’souza, S.S.; Salinero, A.E.; Wang, Y.; Qian, J.; Kuentzel, M.L.; et al. Activation of group 2 innate lymphoid cells alleviates aging-associated cognitive decline. J. Exp. Med. 2020, 217, e20190915. [Google Scholar] [CrossRef]
- Golomb, S.M.; Guldner, I.H.; Zhao, A.; Wang, Q.; Palakurthi, B.; Aleksandrovic, E.A.; Lopez, J.A.; Lee, S.W.; Yang, K.; Zhang, S. Multi-modal Single-Cell Analysis Reveals Brain Immune Landscape Plasticity during Aging and Gut Microbiota Dysbiosis. Cell Rep. 2020, 33, 108438. [Google Scholar] [CrossRef]
- van de Pavert, S.A.; Vivier, E. Differentiation and function of group 3 innate lymphoid cells, from embryo to adult. Int. Immunol. 2016, 28, 35–42. [Google Scholar] [CrossRef]
- Hatfield, J.K.; Brown, M.A. Group 3 innate lymphoid cells accumulate and exhibit disease-induced activation in the meninges in EAE. Cell. Immunol. 2015, 297, 69–79. [Google Scholar] [CrossRef]
- Prinz, M.; Erny, D.; Hagemeyer, N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 2017, 18, 385–392. [Google Scholar] [CrossRef]
- Cugurra, A.; Mamuladze, T.; Rustenhoven, J.; Dykstra, T.; Beroshvili, G.; Greenberg, Z.J.; Baker, W.; Papadopoulos, Z.; Drieu, A.; Blackburn, S.; et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science 2021, 373, eabf7844. [Google Scholar] [CrossRef]
- Bennett, F.C.; Bennett, M.L.; Yaqoob, F.; Mulinyawe, S.B.; Grant, G.A.; Gephart, M.H.; Plowey, E.D.; Barres, B.A. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 2018, 98, 1170–1183.e8. [Google Scholar] [CrossRef]
- Chen, B.; Zhu, L.; Yang, S.; Su, W. Unraveling the Heterogeneity and Ontogeny of Dendritic Cells Using Single-Cell RNA Sequencing. Front. Immunol. 2021, 12, 711329. [Google Scholar] [CrossRef]
- Lin, D.S.; Tian, L.; Tomei, S.; Amann-Zalcenstein, D.; Baldwin, T.M.; Weber, T.S.; Schreuder, J.; Stonehouse, O.J.; Rautela, J.; Huntington, N.D.; et al. Single-cell analyses reveal the clonal and molecular aetiology of Flt3L-induced emergency dendritic cell development. Nature 2021, 23, 219–231. [Google Scholar] [CrossRef]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef]
- Attfield, K.E.; Jensen, L.T.; Kaufmann, M.; Friese, M.A.; Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022, 22, 734–750. [Google Scholar] [CrossRef]
- Wasser, B.; Luchtman, D.; Löffel, J.; Robohm, K.; Birkner, K.; Stroh, A.; Vogelaar, C.F.; Zipp, F.; Bittner, S. CNS-localized myeloid cells capture living invading T cells during neuroinflammation. J. Exp. Med. 2020, 217, e20190812. [Google Scholar] [CrossRef]
- Nissen, J.C.; Thompson, K.K.; West, B.L.; Tsirka, S.E. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp. Neurol. 2018, 307, 24–36. [Google Scholar] [CrossRef]
- Montilla, A.; Zabala, A.; Er-Lukowiak, M.; Rissiek, B.; Magnus, T.; Rodriguez-Iglesias, N.; Sierra, A.; Matute, C.; Domercq, M. Microglia and meningeal macrophages depletion delays the onset of experimental autoimmune encephalomyelitis. Cell Death Dis. 2023, 14, 16. [Google Scholar] [CrossRef]
- Mato, M.; Ookawara, S.; Sakamoto, A.; Aikawa, E.; Ogawa, T.; Mitsuhashi, U.; Masuzawa, T.; Suzuki, H.; Honda, M.; Yazaki, Y.; et al. Involvement of specific macrophage-lineage cells surrounding arterioles in barrier and scavenger function in brain cortex. Proc. Natl. Acad. Sci. USA 1996, 93, 3269–3274. [Google Scholar] [CrossRef]
- Pedragosa, J.; Salas-Perdomo, A.; Gallizioli, M.; Cugota, R.; Miró-Mur, F.; Briansó, F.; Justicia, C.; Pérez-Asensio, F.; Marquez-Kisinousky, L.; Urra, X.; et al. CNS-border associated macrophages respond to acute ischemic stroke attracting granulocytes and promoting vascular leakage. Acta Neuropathol. Commun. 2018, 6, 76. [Google Scholar] [CrossRef]
- Dong, Y.; Yong, V.W. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat. Rev. Neurol. 2019, 15, 704–717. [Google Scholar] [CrossRef]
- Mundt, S.; Mrdjen, D.; Utz, S.G.; Greter, M.; Schreiner, B.; Becher, B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol. 2019, 4, eaau8380. [Google Scholar] [CrossRef]
- Giles, D.A.; Duncker, P.C.; Wilkinson, N.M.; Washnock-Schmid, J.M.; Segal, B.M. CNS-resident classical DCs play a critical role in CNS autoimmune disease. J. Clin. Investig. 2018, 128, 5322–5334. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef]
- Keller, C.W.; Sina, C.; Kotur, M.B.; Ramelli, G.; Mundt, S.; Quast, I.; Ligeon, L.-A.; Weber, P.; Becher, B.; Münz, C.; et al. ATG-dependent phagocytosis in dendritic cells drives myelin-specific CD4 + T cell pathogenicity during CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E11228–E11237. [Google Scholar] [CrossRef]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In Vivo Requirement for Atg5 in Antigen Presentation by Dendritic Cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef]
- Keller, C.W.; Kotur, M.B.; Mundt, S.; Dokalis, N.; Ligeon, L.-A.; Shah, A.M.; Prinz, M.; Becher, B.; Münz, C.; Lünemann, J.D. CYBB/NOX2 in conventional DCs controls T cell encephalitogenicity during neuroinflammation. Autophagy 2020, 17, 1244–1258. [Google Scholar] [CrossRef]
- Hatterer, E.; Touret, M.; Belin, M.-F.; Honnorat, J.; Nataf, S. Cerebrospinal Fluid Dendritic Cells Infiltrate the Brain Parenchyma and Target the Cervical Lymph Nodes under Neuroinflammatory Conditions. PLoS ONE 2008, 3, e3321. [Google Scholar] [CrossRef] [PubMed]
- King, I.L.; Dickendesher, T.L.; Segal, B.M. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009, 113, 3190–3197. [Google Scholar] [CrossRef]
- Miller, S.D.; McMahon, E.J.; Schreiner, B.; Bailey, S.L. Antigen Presentation in the CNS by Myeloid Dendritic Cells Drives Progression of Relapsing Experimental Autoimmune Encephalomyelitis. Ann. N. Y. Acad. Sci. 2007, 1103, 179–191. [Google Scholar] [CrossRef]
- Bailey, S.L.; Schreiner, B.; McMahon, E.J.; Miller, S.D. CNS myeloid DCs presenting endogenous myelin peptides ’preferentially’ polarize CD4+ TH-17 cells in relapsing EAE. Nat. Immunol. 2007, 8, 172–180. [Google Scholar] [CrossRef]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef]
- Gallizioli, M.; Miró-Mur, F.; Otxoa-De-Amezaga, A.; Cugota, R.; Salas-Perdomo, A.; Justicia, C.; Brait, V.H.; Ruiz-Jaén, F.; Arbaizar-Rovirosa, M.; Pedragosa, J.; et al. Dendritic Cells and Microglia Have Non-redundant Functions in the Inflamed Brain with Protective Effects of Type 1 cDCs. Cell Rep. 2020, 33, 108291. [Google Scholar] [CrossRef] [PubMed]
- Kwong, B.; Rua, R.; Gao, Y.; Flickinger, J., Jr.; Wang, Y.; Kruhlak, M.J.; Zhu, J.; Vivier, E.; McGavern, D.B.; Lazarevic, V. T-bet-dependent NKp46 + innate lymphoid cells regulate the onset of T H 17-induced neuroinflammation. Nat. Immunol. 2017, 18, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liu, R.; Piao, W.; Zhou, Q.; Vollmer, T.L.; Campagnolo, D.I.; Xiang, R.; La Cava, A.; Van Kaer, L.; Shi, F.-D. Central nervous system (CNS)–resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J. Exp. Med. 2010, 207, 1907–1921. [Google Scholar] [CrossRef]
- Kunis, G.; Baruch, K.; Rosenzweig, N.; Kertser, A.; Miller, O.; Berkutzki, T.; Schwartz, M. IFN-γ-dependent activation of the brain’s choroid plexus for CNS immune surveillance and repair. Brain 2013, 136, 3427–3440. [Google Scholar] [CrossRef]
- Russi, A.E.; Ebel, M.E.; Yang, Y.; Brown, M.A. Male-specific IL-33 expression regulates sex-dimorphic EAE susceptibility. Proc. Natl. Acad. Sci. USA 2018, 115, E1520–E1529. [Google Scholar] [CrossRef]
- Grigg, J.B.; Shanmugavadivu, A.; Regen, T.; Parkhurst, C.N.; Ahmed, A.; Joseph, A.M.; Mazzucco, M.; Gronke, K.; Diefenbach, A.; Eberl, G.; et al. Antigen-presenting innate lymphoid cells orchestrate neuroinflammation. Nature 2021, 600, 707–712. [Google Scholar] [CrossRef]
- Zhan, J.; Kipp, M.; Han, W.; Kaddatz, H. Ectopic lymphoid follicles in progressive multiple sclerosis: From patients to animal models. Immunology 2021, 164, 450–466. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef]
- Choi, S.R.; Howell, O.W.; Carassiti, D.; Magliozzi, R.; Gveric, D.; Muraro, P.A.; Nicholas, R.; Roncaroli, F.; Reynolds, R. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012, 135, 2925–2937. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Multiple sclerosis: Role of meningeal lymphoid aggregates in progression independent of relapse activity. Trends Immunol. 2023, 44, 266–275. [Google Scholar] [CrossRef]
- Hsieh, J.; Aimone, J.B.; Kaspar, B.K.; Kuwabara, T.; Nakashima, K.; Gage, F.H. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J. Cell Biol. 2004, 164, 111–122. [Google Scholar] [CrossRef]
- Kamma, E.; Lasisi, W.; Libner, C.; Ng, H.S.; Plemel, J.R. Central nervous system macrophages in progressive multiple sclerosis: Relationship to neurodegeneration and therapeutics. J. Neuroinflamm. 2022, 19, 45. [Google Scholar] [CrossRef]
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef]
- Locatelli, G.; Theodorou, D.; Kendirli, A.; Jordão, M.J.C.; Staszewski, O.; Phulphagar, K.; Cantuti-Castelvetri, L.; Dagkalis, A.; Bessis, A.; Simons, M.; et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat. Neurosci. 2018, 21, 1196–1208. [Google Scholar] [CrossRef]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11, 584303. [Google Scholar] [CrossRef]
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Schlaphoff, L.; Depp, C.; Düking, T.; Winchenbach, J.; Neuber, J.; Ewers, D.; et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 2021, 24, 47–60. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.-T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef]
- Gouna, G.; Klose, C.; Bosch-Queralt, M.; Liu, L.; Gokce, O.; Schifferer, M.; Cantuti-Castelvetri, L.; Simons, M. TREM2-dependent lipid droplet biogenesis in phagocytes is required for remyelination. J. Exp. Med. 2021, 218, e20210227. [Google Scholar] [CrossRef]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef]
- Pham, H.; Ramp, A.A.; Klonis, N.; Ng, S.W.; Klopstein, A.; Ayers, M.M.; Orian, J.M. The astrocytic response in early experimental autoimmune encephalomyelitis occurs across both the grey and white matter compartments. J. Neuroimmunol. 2009, 208, 30–39. [Google Scholar] [CrossRef]
- Heß, K.; Starost, L.; Kieran, N.W.; Thomas, C.; Vincenten, M.C.J.; Antel, J.; Martino, G.; Huitinga, I.; Healy, L.; Kuhlmann, T. Lesion stage-dependent causes for impaired remyelination in MS. Acta Neuropathol. 2020, 140, 359–375. [Google Scholar] [CrossRef]
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Höftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative damage in multiple sclerosis lesions. Brain 2011, 134, 1914–1924. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Bando, Y. Roads to Formation of Normal Myelin Structure and Pathological Myelin Structure. Adv. Exp. Med. Biol. 2019, 1190, 257–264. [Google Scholar]
- Wagner, C.A.; Roqué, P.J.; Goverman, J.M. Pathogenic T cell cytokines in multiple sclerosis. J. Exp. Med. 2019, 217, e20210227. [Google Scholar] [CrossRef]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef]
- Häusler, D.; Akgün, K.; Stork, L.; Lassmann, H.; Ziemssen, T.; Brück, W.; Metz, I. CNS inflammation after natalizumab therapy for multiple sclerosis: A retrospective histopathological and CSF cohort study. Brain Pathol. 2021, 31, e12969. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.S.-H.; Ho, Y.-S.; Chang, R.C.-C. The role of meningeal populations of type II innate lymphoid cells in modulating neuroinflammation in neurodegenerative diseases. Exp. Mol. Med. 2021, 53, 1251–1267. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, C.; Yao, S.-Y.; Sriram, S. TLR3 Agonist Poly-IC Induces IL-33 and Promotes Myelin Repair. PLoS ONE 2016, 11, e0152163. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Zhang, Y.; Zuloaga, K.; Yang, Q. The role of innate lymphocytes in regulating brain and cognitive function. Neurobiol. Dis. 2023, 179, 106061. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Smirnov, I.; Wiltbank, A.T.; Overall, C.C.; Kipnis, J. Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J. Exp. Med. 2017, 214, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Wang, J.; Yang, B.; Weng, Q.; He, Q. Targeting microglia and macrophages: A potential treatment strategy for multiple sclerosis. Front. Pharmacol. 2019, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Djedović, N.; Stanisavljevic, S.; Jevtić, B.; Momčilović, M.; Lavrnja, I.; Miljković, D. Anti-encephalitogenic effects of ethyl pyruvate are reflected in the central nervous system and the gut. Biomed. Pharmacother. 2017, 96, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Krämer, J.; Bar-Or, A.; Turner, T.J.; Wiendl, H. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat. Rev. Neurol. 2023, 19, 289–304. [Google Scholar] [CrossRef]
- Vermersch, P.; Arnold, D.L.; Wolinsky, J.; Havrdova, E.K.; Kinkolykh, A.; Hyvert, Y.; Tomic, D.; Montalban, X. MRI and Clinical Outcomes of Evobrutinib, a Bruton’s Tyrosine Kinase Inhibitor, in Relapsing Multiple Sclerosis Over 2.5 Years of the Open-Label Extension to a Phase 2 Trial. Mult. Scler. Relat. Disord. 2023, 71, 104360. [Google Scholar] [CrossRef]
- Reich, D.S.; Arnold, D.L.; Vermersch, P.; Bar-Or, A.; Fox, R.J.; Matta, A.; Turner, T.; Wallström, E.; Zhang, X.; Mareš, M.; et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: A phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 729–738. [Google Scholar] [CrossRef]
- Zoubi, S.A.; Esam, H.; Elzawi, E. Impact of Dyslipidemia on Progression of Multiple Sclerosis. Mult. Scler. Relat. Disord. 2023, 71, 104367. [Google Scholar] [CrossRef]
- Rudick, R.; Polman, C.; Clifford, D.; Miller, D.; Steinman, L. Natalizumab. JAMA Neurol. 2013, 70, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.Z.; Cravens, P.C.; Doelger, R.; Dentel, B.; Herndon, E.; Loof, N.; Tsai, P.; Okuda, D.T.; Racke, M.K.; Stüve, O. TLR3 agonism re-establishes CNS immune competence during α4-integrin deficiency. Ann. Clin. Transl. Neurol. 2018, 5, 1543–1561. [Google Scholar] [CrossRef]
- Hassanabadi, N.S.; Broux, B.; Marinović, S.; Gotthardt, D. Innate Lymphoid Cells—Neglected Players in Multiple Sclerosis. Front. Immunol. 2022, 13, 909275. [Google Scholar] [CrossRef]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Wiendl, H.; Marcenaro, E.; de Rosbo, N.K.; Uccelli, A.; Laroni, A. Regulatory Functions of Natural Killer Cells in Multiple Sclerosis. Front. Immunol. 2016, 7, 606. [Google Scholar] [CrossRef] [PubMed]
- Skarica, M.; Eckstein, C.; Whartenby, K.A.; Calabresi, P.A. Novel mechanisms of immune modulation of natalizumab in multiple sclerosis patients. J. Neuroimmunol. 2011, 235, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Rünzi, A.; Kuhlmann, T.; Posevitz-Fejfár, A.; Schwab, N.; Schneider-Hohendorf, T.; Herich, S.; Held, K.; Konjević, M.; et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2973–E2982. [Google Scholar] [CrossRef]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Hanning, U.; Posevitz-Fejfár, A.; Korsukewitz, C.; Schwab, N.; Meuth, S.G.; Wiendl, H.; Klotz, L. Distinct pattern of lesion distribution in multiple sclerosis is associated with different circulating T-helper and helper-like innate lymphoid cell subsets. Mult. Scler. J. 2017, 23, 1025–1030. [Google Scholar] [CrossRef]
- Perry, J.S.A.; Han, S.; Xu, Q.; Herman, M.L.; Kennedy, L.B.; Csako, G.; Bielekova, B. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci. Transl. Med. 2012, 4, 145ra106. [Google Scholar] [CrossRef]
- Eken, A.; Yetkin, M.F.; Vural, A.; Okus, F.Z.; Erdem, S.; Azizoglu, Z.B.; Haliloglu, Y.; Cakir, M.; Turkoglu, E.M.; Kilic, O.; et al. Fingolimod alters tissue distribution and cytokine production of human and murine innate lymphoid cells. Front. Immunol. 2019, 10, 217. [Google Scholar] [CrossRef]
- Acar, N.P.; Tuncer, A.; Ozkazanc, D.; Ozbay, F.G.; Karaosmanoglu, B.; Goksen, S.; Sayat, G.; Taskiran, E.Z.; Esendagli, G.; Karabudak, R. An immunological and transcriptomics approach on differential modulation of NK cells in multiple sclerosis patients under interferon-β1 and fingolimod therapy. J. Neuroimmunol. 2020, 347, 577353. [Google Scholar] [CrossRef]
- Thell, K.; Hellinger, R.; Sahin, E.; Michenthaler, P.; Gold-Binder, M.; Haider, T.; Kuttke, M.; Liutkevičiūtė, Z.; Göransson, U.; Gründemann, C.; et al. Oral activity of a nature-derived cyclic peptide for the treatment of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, K.; Chen, X.; Sun, M.-A.; Kawabe, T.; Li, W.; Usher, N.; Zhu, J.; Urban, J.F., Jr.; Paul, W.E.; et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 2018, 359, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Hermann, R.; Krajcsi, P.; Fluck, M.; Seithel-Keuth, A.; Bytyqi, A.; Galazka, A.; Munafo, A. Cladribine as a Potential Object of Nucleoside Transporter-Based Drug Interactions. Clin. Pharmacokinet. 2022, 61, 167–187. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Ruck, T.; Muraro, P.A.; Bar’Or, A.; Wiendl, H. Immune reconstitution therapies: Concepts for durable remission in multiple sclerosis. Nat. Rev. Neurol. 2020, 16, 56–62. [Google Scholar] [CrossRef]
- Aglas-Leitner, F.T.; Juillard, P.; Juillard, A.; Byrne, S.N.; Hawke, S.; Grau, G.E.; Marsh-Wakefield, F. Mass cytometry reveals cladribine-induced resets among innate lymphoid cells in multiple sclerosis. Sci. Rep. 2022, 12, 20411. [Google Scholar] [CrossRef]
- Lancet, T. End of the road for daclizumab in multiple sclerosis. Lancet 2018, 391, 1000. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.; Tobenkin, A.; Brinker, A.; Kortepeter, C.M. Tumefactive multiple sclerosis in association with fingolimod initiation and discontinuation. Mult. Scler. J. 2021, 27, 903–912. [Google Scholar] [CrossRef]
- Jeung, L.; Smits, L.; Hoogervorst, E.; van Oosten, B.; Frequin, S. A tumefactive demyelinating lesion in a person with MS after five years of fingolimod. Mult. Scler. Relat. Disord. 2020, 40, 101978. [Google Scholar] [CrossRef]
- İriş, M.; Kızılkılıç, O.; Saip, S.; Uygunoğlu, U. Recurrent tumefactive demyelination under fingolimod treatment. Neurol. Sci. 2024, 45, 2377–2378. [Google Scholar] [CrossRef]
- Faissner, S.; Hoepner, R.; Lukas, C.; Chan, A.; Gold, R.; Ellrichmann, G. Tumefactive multiple sclerosis lesions in two patients after cessation of fingolimod treatment. Ther. Adv. Neurol. Disord. 2015, 8, 233–238. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muzio, L.; Perego, J. CNS Resident Innate Immune Cells: Guardians of CNS Homeostasis. Int. J. Mol. Sci. 2024, 25, 4865. https://doi.org/10.3390/ijms25094865
Muzio L, Perego J. CNS Resident Innate Immune Cells: Guardians of CNS Homeostasis. International Journal of Molecular Sciences. 2024; 25(9):4865. https://doi.org/10.3390/ijms25094865
Chicago/Turabian StyleMuzio, Luca, and Jessica Perego. 2024. "CNS Resident Innate Immune Cells: Guardians of CNS Homeostasis" International Journal of Molecular Sciences 25, no. 9: 4865. https://doi.org/10.3390/ijms25094865
APA StyleMuzio, L., & Perego, J. (2024). CNS Resident Innate Immune Cells: Guardians of CNS Homeostasis. International Journal of Molecular Sciences, 25(9), 4865. https://doi.org/10.3390/ijms25094865