
Enzyme (α-Glucosidase, α-Amylase, PTP1B & VEGFR-2) Inhibition and Cytotoxicity of Fluorinated Benzenesulfonic Ester Derivatives of the 5-Substituted 2-Hydroxy-3-nitroacetophenones

 , and
, and
Abstract

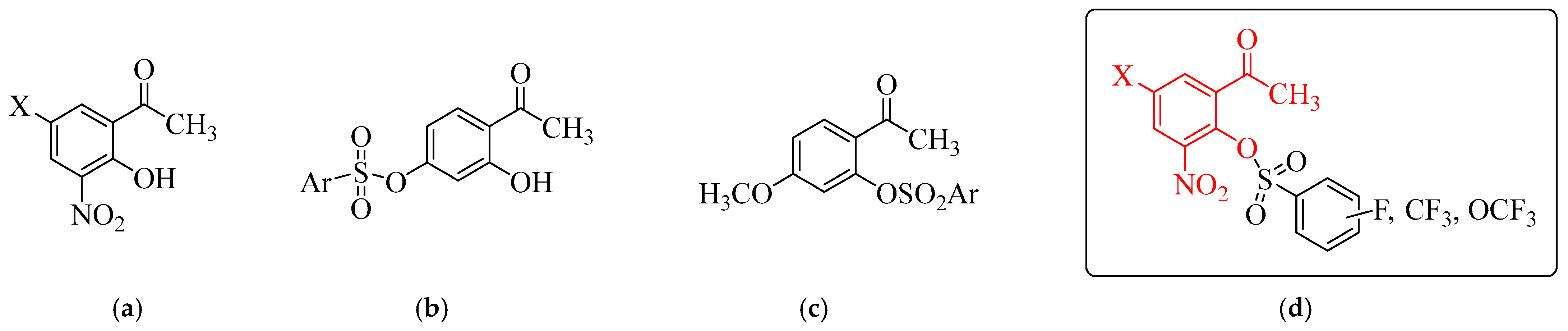
1. Introduction
2. Results and Discussion
2.1. Chemical Synthesis and Characterization
2.2. Biological Activity Studies with SAR
2.2.1. Inhibition of α-Glucosidase
2.2.2. Inhibition of α-Amylase
2.2.3. Inhibition of PTP1B
2.2.4. Inhibition of VEGFR-2
2.2.5. Cytotoxicity of Compounds 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x
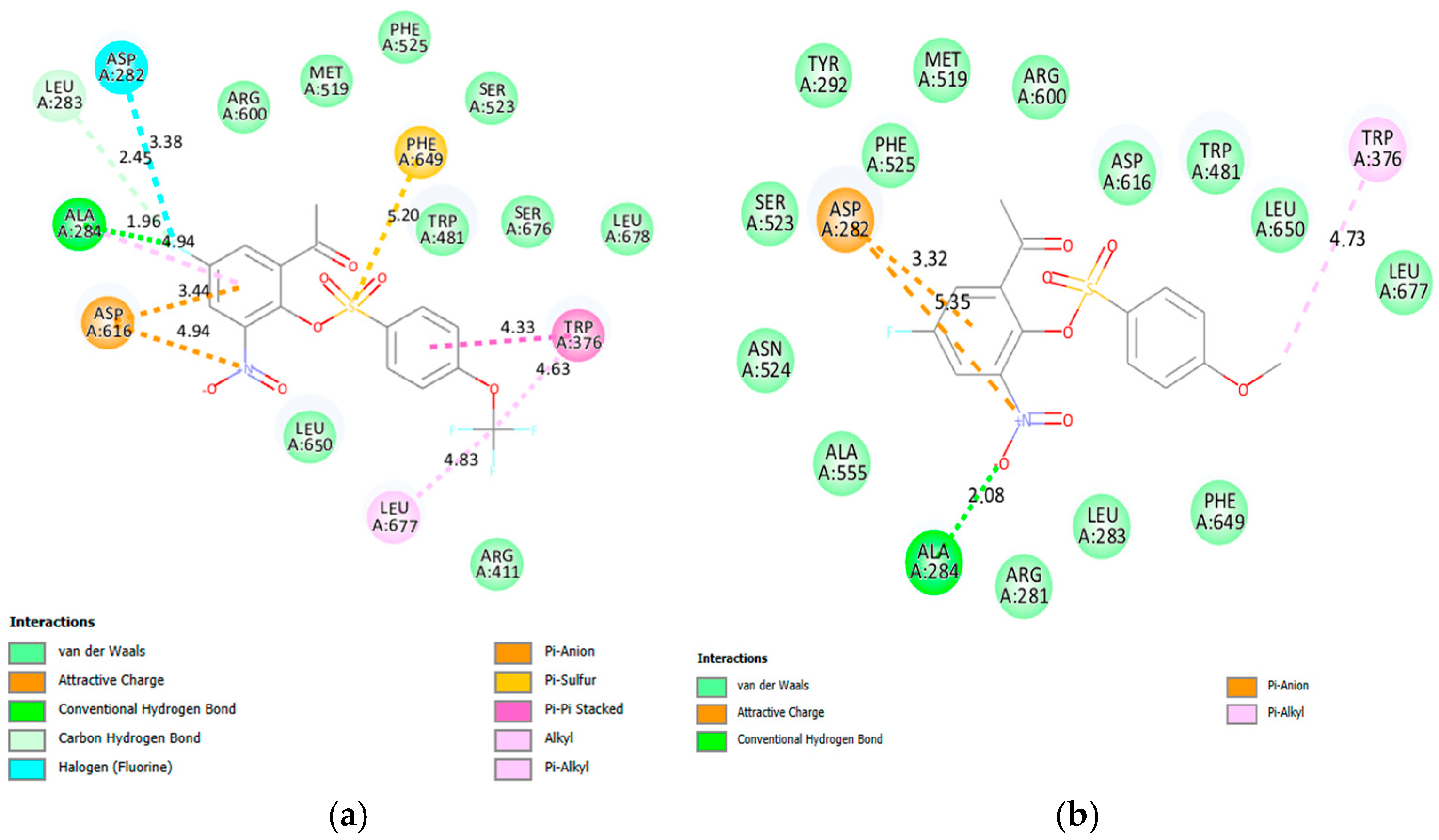
2.3. Molecular Docking Studies on 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x
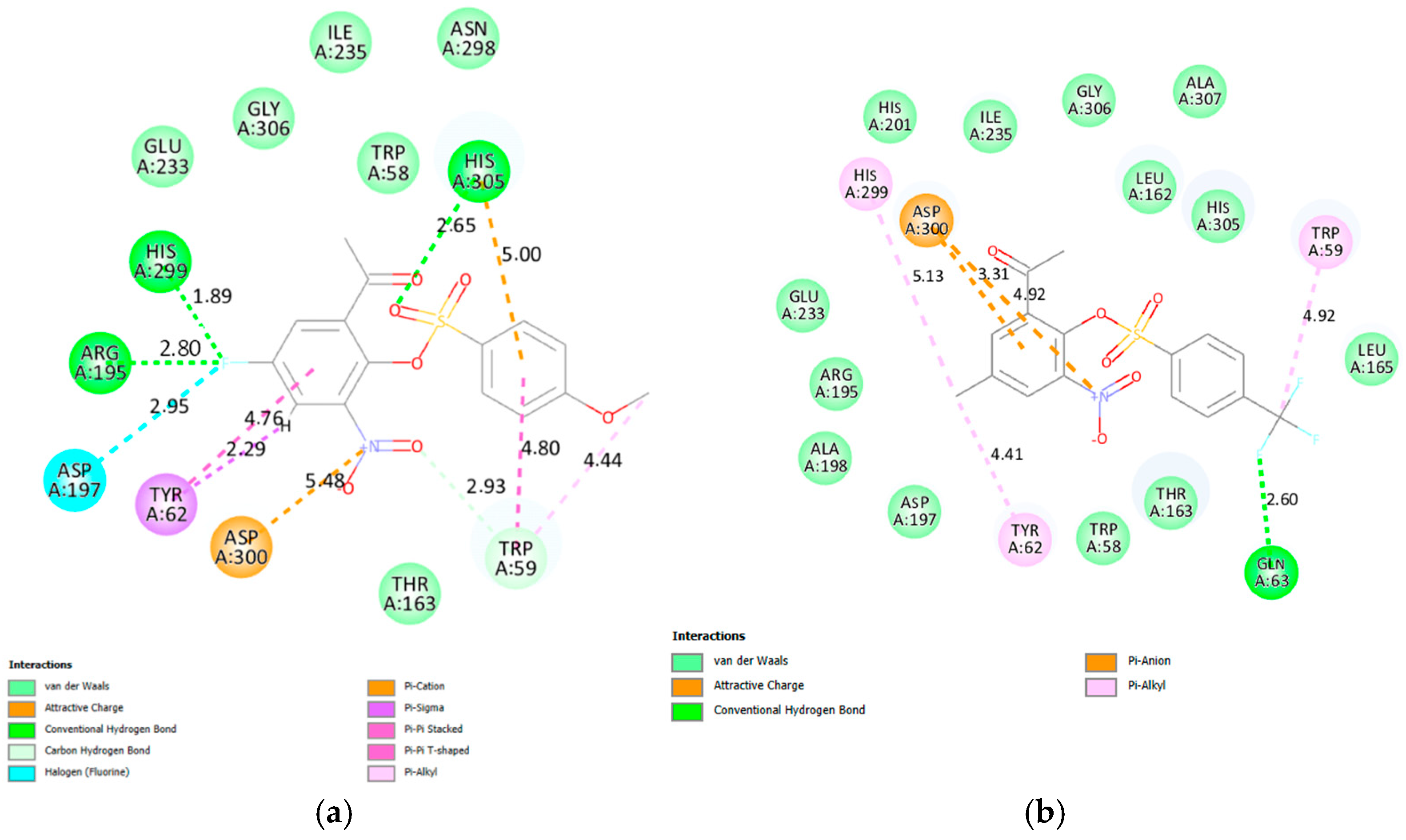
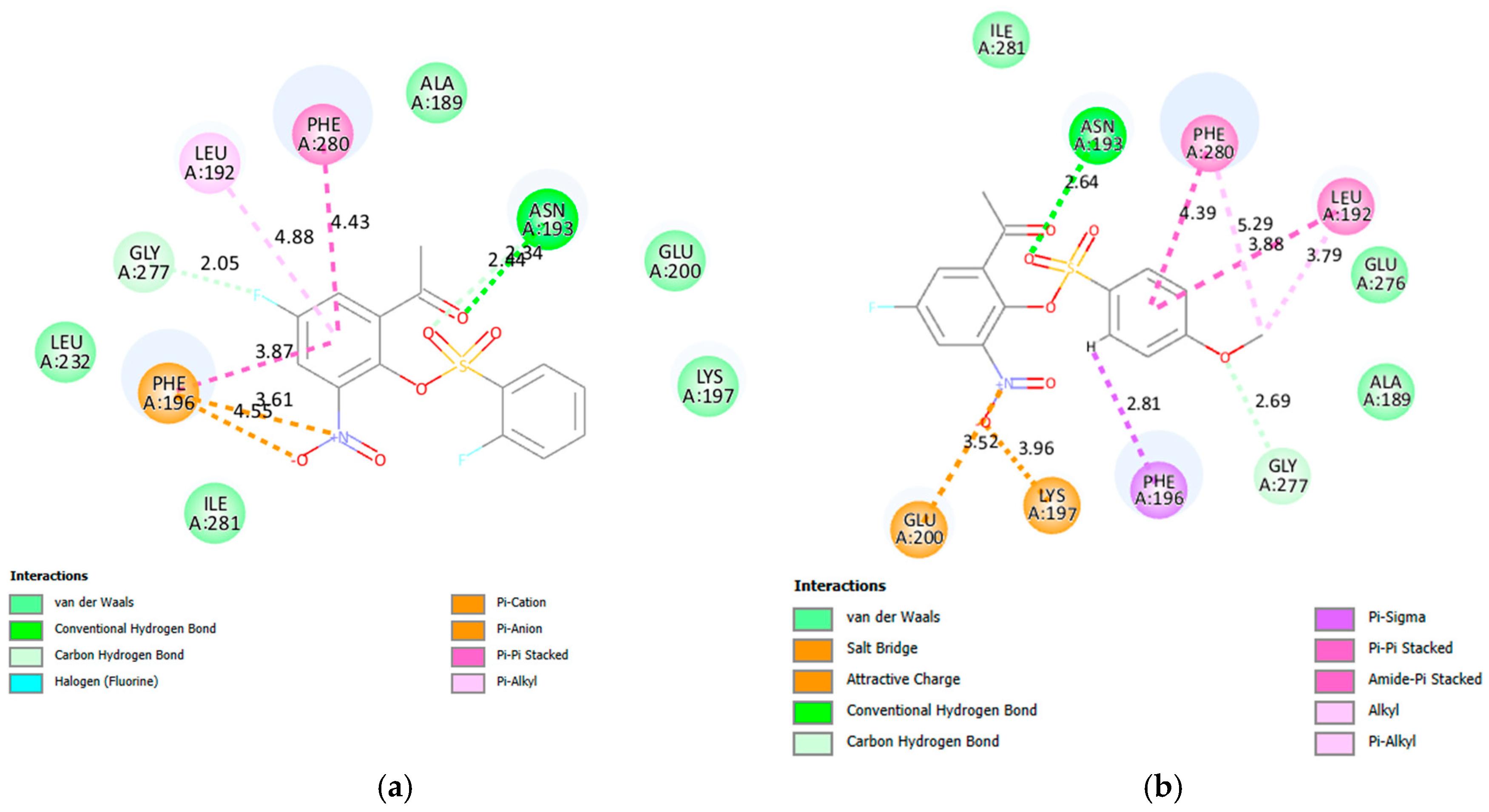
2.3.1. Molecular Docking of 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x into α-Glucosidase Active Site
2.3.2. Molecular Docking of 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x into α-Amylase Active Site
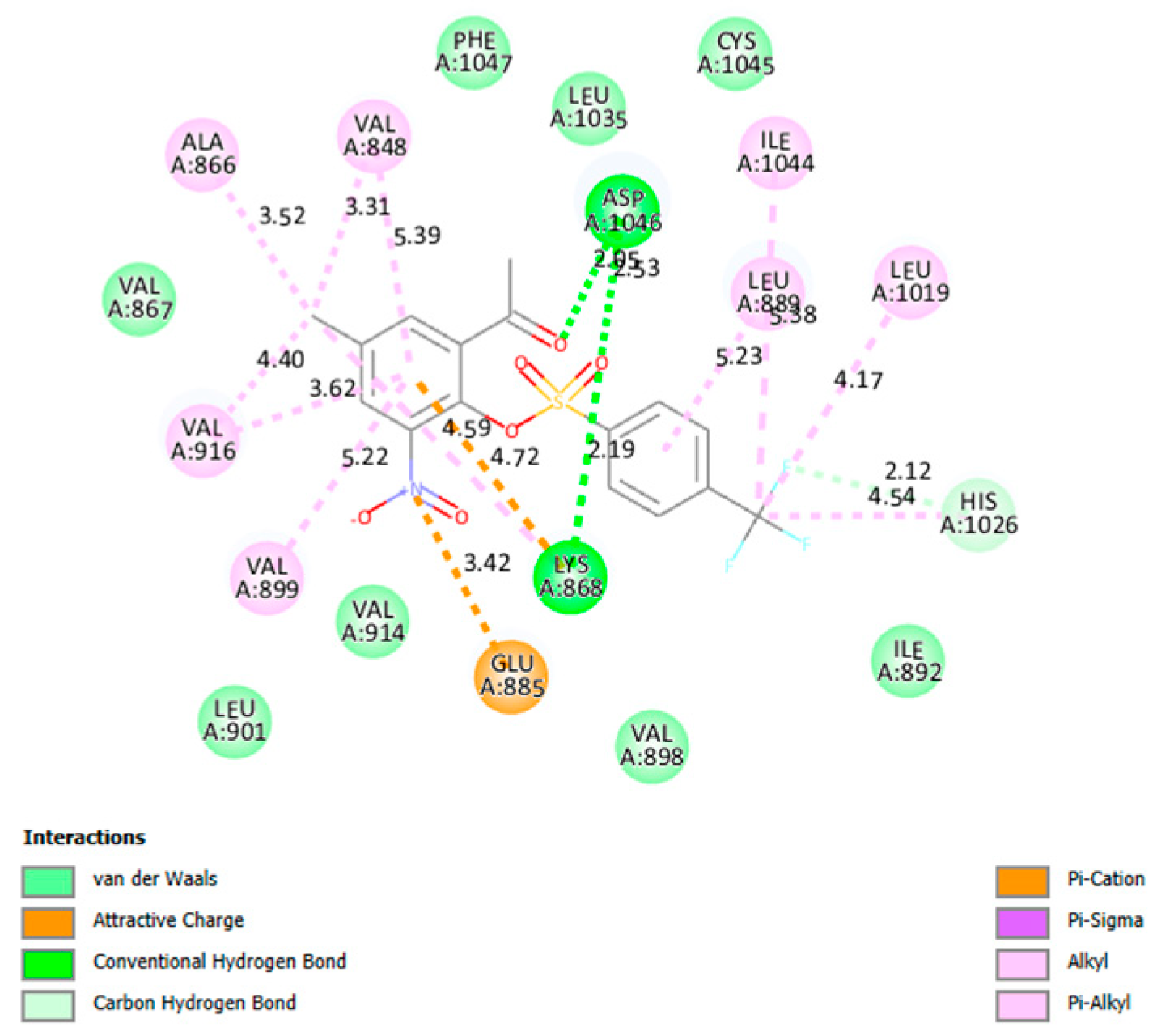
2.3.3. Molecular Docking of 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x into PTP1B
- (i)
- Docking into the Catalytic (PDB 2QBP) Site
- (ii)
- Docking into the Allosteric (PDB 1T49) Site
2.3.4. Docking into VEGFR-2 Catalytic Site (PDB 4ASD)
2.4. Pharmacokinetics Properties Prediction of 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x
3. Materials and Methods
3.1. Materials and Instrumentation
3.2. Typical Procedure for the Synthesis of 2a–x
3.2.1. 2-Acetyl-4-fluoro-6-nitrophenyl 2-fluorobenzenesulfonate (2a)
3.2.2. 2-Acetyl-4-fluoro-6-nitrophenyl 3-fluorobenzenesulfonate (2b)
3.2.3. 2-Acetyl-4-fluoro-6-nitrophenyl 4-fluorobenzenesulfonate (2c)
3.2.4. 2-Acetyl-4-fluoro-6-nitrophenyl 4-(trifluoromethyl)benzenesulfonate (2d)
3.2.5. 2-Acetyl-4-fluoro-6-nitrophenyl 4-(trifluoromethoxy)benzenesulfonate (2e)
3.2.6. 2-Acetyl-4-chloro-6-nitrophenyl 4-methoxybenzenesulfonate (2f)
3.2.7. 2-Acetyl-4-chloro-6-nitrophenyl 2-fluorobenzenesulfonate (2g)
3.2.8. 2-Acetyl-4-chloro-6-nitrophenyl 3-fluorobenzenesulfonate (2h)
3.2.9. 2-Acetyl-4-chloro-6-nitrophenyl 4-fluorobenzenesulfonate (2i)
3.2.10. 2-Acetyl-4-chloro-6-nitrophenyl 4-(trifluoromethyl)benzenesulfonate (2j)
3.2.11. 2-Acetyl-4-chloro-6-nitrophenyl 4-(trifluoromethoxy)benzenesulfonate (2k)
3.2.12. 2-Acetyl-4-chloro-6-nitrophenyl 4-methoxybenzenesulfonate (2l)
3.2.13. 2-Acetyl-4-bromo-6-nitrophenyl 2-fluorobenzenesulfonate (2m)
3.2.14. 2-Acetyl-4-bromo-6-nitrophenyl 3-fluorobenzenesulfonate (2n)
3.2.15. 2-Acetyl-4-bromo-6-nitrophenyl 4-fluorobenzenesulfonate (2o)
3.2.16. 2-Acetyl-4-bromo-6-nitrophenyl 4-(trifluoromethyl)benzenesulfonate (2p)
3.2.17. 2-Acetyl-4-bromo-6-nitrophenyl 4-(trifluoromethoxy)benzenesulfonate (2q)
3.2.18. 2-Acetyl-4-bromo-6-nitrophenyl 4-methoxybenzenesulfonate (2r)
3.2.19. 2-Acetyl-4-methyl-6-nitrophenyl 2-fluorobenzenesulfonate (2s)
3.2.20. 2-Acetyl-4-methyl-6-nitrophenyl 3-fluorobenzenesulfonate (2t)
3.2.21. 2-Acetyl-4-methyl-6-nitrophenyl 4-fluorobenzenesulfonate (2u)
3.2.22. 2-Acetyl-4-methyl-6-nitrophenyl 4-(trifluoromethyl)benzenesulfonate (2v)
3.2.23. 2-Acetyl-4-methyl-6-nitrophenyl 4-(trifluoromethoxy)benzenesulfonate (2w)
3.2.24. 2-Acetyl-4-methyl-6-nitrophenyl 4-methoxybenzenesulfonate (2x)
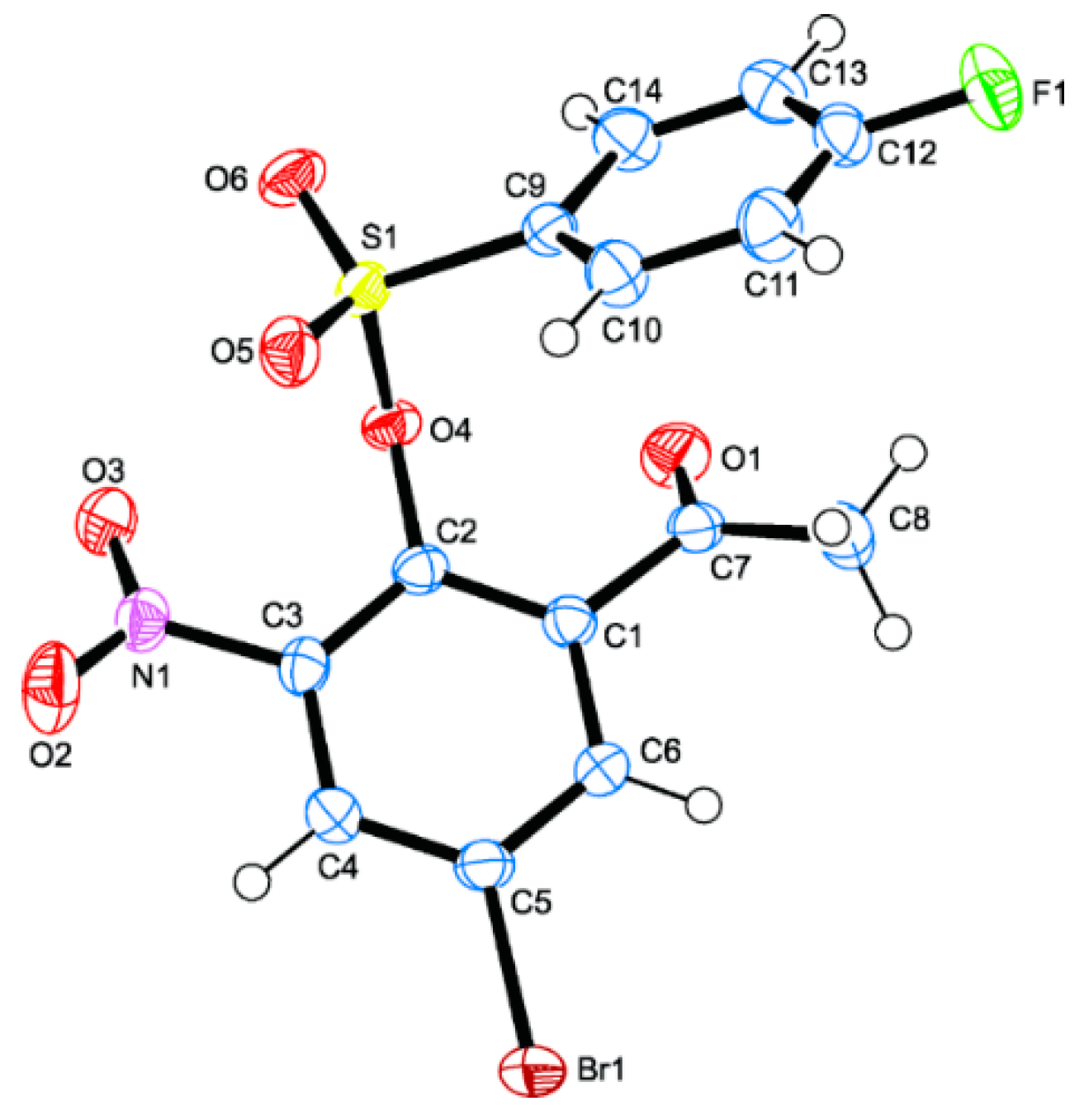
3.3. Single Crystal X-Ray Diffraction Analysis of 2o
3.4. Biology
3.4.1. In Vitro α-Glucosidase Inhibitory Assay of 2a–x
3.4.2. In Vitro α-Amylase Inhibitory Assay on 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x
3.5. In Vitro PTP1B Inhibitory Assay on 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x
3.6. Inhibition of VGEFR-2 Tyrosine Phosphorylation by 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x
3.7. Cytotoxicity Study on 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x
3.8. Molecular Docking Studies of 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x into α-Glucosidase and into α-Amylase
3.9. Prediction of Pharmacokinetic Properties for 2a, 2e, 2f, 2g, 2j, 2k, 2v and 2x
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Artasensi, A.; Pedretti, A.; Vistoli, G.; Fumagalli, L. Type 2 diabetes mellitus: A review of multi-target drugs. Molecules 2020, 25, 1987. [Google Scholar] [CrossRef]
- Gong, L.; Feng, D.; Wang, T.; Ren, Y.; Liu, Y.; Wang, J. Inhibitors of α-amylase and α-glucosidase: Potential linkage for whole cereal foods on prevention of hyperglycemia. Food Sci. Nutr. 2020, 8, 6320–6337. [Google Scholar] [CrossRef]
- Kashtoh, H.; Baek, K.-H. New insights into the latest advancement in α-amylase inhibitors of plant origin with anti-diabetic effects. Plants 2023, 12, 2944. [Google Scholar] [CrossRef]
- Lam, T.-P.; Tran, N.-V.N.; Pham, L.-H.D.; Lai, N.V.-T.; Dang, B.-T.N.; Truong, N.-L.N.; Nguyen-Vo, S.-K.; Hoang, T.-L.; Mai, T.T.; Tran, T.-D. Flavonoids as dual-target inhibitors against α-glucosidase and α-amylase: A systematic review of in vitro studies. Nat. Prod. Bioprospecting 2024, 14, 4. [Google Scholar]
- Mai, T.T.; Phan, M.-H.; Thai, T.T.; Lam, T.-P.; Lai, N.V.-T.; Nguyen, T.-T.; Nguyen, T.-V.-P.; Vo, C.-V.T.; Thai, K.-M.; Tran, T.-D. Discovery of novel flavonoid derivatives as potential dual inhibitors against α-glucosidase and α-amylase: Virtual screening, synthesis, and biological evaluation. Mol. Divers. 2024, 28, 1629–1650. [Google Scholar]
- Liu, G.; Trevillyan, J.M. Protein tyrosine phosphatase 1B as a target for the treatment of impaired glucose tolerance and type II diabetes. Curr. Opin. Investig. Drugs 2002, 3, 1608–1616. [Google Scholar]
- Norrisa, K.; Norrisa, F.; Konod, D.H.; Vestergaardc, H.; Pedersenc, O.; Theofilopoulosd, A.N.; Mollerb, N.P.H. Expression of protein-tyrosine phosphatases in the major insulin target tissues. FEBS Lett. 1997, 415, 243–248. [Google Scholar] [CrossRef]
- Teimouri, M.; Hosseini, H.; ArabSadeghabadi, Z.; Babaei-Khorzoughi, R.; Gorgani-Firuzjaee, S.; Meshkani, R. The role of protein tyrosine phosphatase 1B (PTP1B) in the pathogenesis of type 2 diabetes mellitus and its complications. J. Physiol. Biochem. 2022, 78, 307–322. [Google Scholar] [CrossRef]
- Qhobosheane, M.A.; Legoabe, L.J.; Josselin, N.; Bach, S.; Ruchaud, S.; Beteck, R.M. Synthesis and evaluation of C3 substituted chalcone-based derivatives of 7-azaindole as protein kinase inhibitors. Chem. Biol. Drug Des. 2020, 96, 1395–1407. [Google Scholar] [CrossRef]
- Villalta, S.A.; Lang, J.; Kubeck, S.; Kabre, B.; Szot, G.L.; Calderon, B.; Wasserfall, C.; Atkinson, M.A.; Brekken, R.A.; Pullen, N.; et al. Inhibition of VEGFR-2 reverses type 1 diabetes in NOD mice by abrogating insulitis and restoring islet function. Diabetes 2013, 62, 2870–2878. [Google Scholar] [CrossRef]
- Fountas, A.; Diamantopoulos, L.N.; Tsatsoulis, A. Tyrosine kinase inhibitors and diabetes: A novel treatment paradigm? Trends Endocrinol. Metabol. 2015, 26, 643–656. [Google Scholar] [CrossRef]
- Sun, D.; Nakao, S.; Xie, F.; Zandi, S.; Bagheri, A.; Kanavi, M.R.; Samiei, S.; Soheili, Z.-S.; Frimmel, S.; Zhang, Z.; et al. Molecular imaging reveals elevated VEGFR-2 expression in retinal capillaries in diabetes: A novel biomarker for early diagnosis. FASEB J. 2014, 28, 3942–3951. [Google Scholar] [CrossRef]
- Kropp, M.; Golubnitschaja, O.; Mazurakova, A.; Koklesova, L.; Sargheini, N.; Vo, T.-T.K.S.; De Clerck, E.; Polivka, J.; Potuznik, P.; Polivka, J.; et al. Diabetic retinopathy as the leading cause of blindness and early predictor of cascading complications–risks and mitigation. EPMA J. 2023, 14, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: Medicinal chemistry perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Abudawood, M. Diabetes and cancer: A comprehensive review. J. Res. Med. Sci. 2019, 24, 94. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Chang, J.; Partis, R.A.; Mueller, R.A.; Chrest, F.J.; Passaniti, A. The α-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth. Cancer Res. 1995, 55, 2920–2926. [Google Scholar]
- Ahmadpourmir, H.; Attar, H.; Asil, J.; Soheili, V.; Taghizadeh, S.F.; Shakeri, A. Natural-derived acetophenones: Chemistry and pharmacological activities. Nat. Prod. Bioprospecting 2024, 14, 28. [Google Scholar] [CrossRef]
- Parry, R.; Nishino, S.; Spain, J. Naturally-occurring nitro compounds. Nat. Prod. Rep. 2011, 28, 152–167. [Google Scholar] [CrossRef]
- Olender, D.; Zwawiak, J.; Zaprutko, L. Multidirectional efficacy of biologically active nitro compounds included in medicines. Pharmaceuticals 2018, 11, 54. [Google Scholar] [CrossRef]
- Ray, S.; Kreitler, D.F.; Gulick, A.M.; Murkin, A.S. The nitro group as a masked electrophile in covalent enzyme inhibition. ACS Chem. Biol. 2018, 13, 1470–1473. [Google Scholar] [CrossRef]
- Noriega, S.; Cardoso-Ortiz, J.; López-Luna, A.; Cuevas-Flores, M.D.R.; De La Torre, J.A.F. The diverse biological activity of recently synthesized nitro compounds. Pharmaceuticals 2022, 152, 717. [Google Scholar] [CrossRef]
- Taslimi, P. In vitro inhibitory effects of some acetophenone derivatives on some metabolic enzymes and molecular docking. Arch. Pharm. 2020, 353, e2000210. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Lv, S.; El-kott, A.F.; El-kenawy, A.E. Anti-human lung cancer activities and survey of glutathione reductase and glutathione S transferase inhibition properties with molecular modeling studies of 2′-hydroxy-5′-methyl-3′-nitroacetophenone: A pre-clinical trial study. Arch. Med. Sci. 2021. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Kouznetsov, V.V. Traveling across life sciences with acetophenone– A simple ketone that has special multipurpose missions. Molecules 2023, 28, 370. [Google Scholar] [CrossRef]
- Shangari, N.; Chan, T.S.; O’Brien, P.J. Sulfation and glucuronidation of phenols: Implications in coenyzme Q metabolism. Methods Enzymol. 2005, 400, 342–359. [Google Scholar]
- Gao, S.; Siddiqui, N.; Etim, I.; Du, T.; Zhang, Y.; Liang, D. Developing nutritional component chrysin as a therapeutic agent: Bioavailability and pharmacokinetics consideration, and ADME mechanisms. Biomed. Pharmacother. 2021, 142, 112080. [Google Scholar] [CrossRef]
- Gwaltney, L., II; Imade, H.M.; Barr, K.J.; Li, Q.; Gehrke, L.; Credo, R.B.; Warner, R.B.; Lee, J.Y.; Kovar, P.; Wang, J.; et al. Novel sulfonate analogues of combretastatin A-4: Potent antimitotic agents. Bioorg. Med. Chem. Lett. 2001, 11, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, A.; Bursal, E. An in vitro and in silico study on the synthesis and characterization of novel bis(sulfonate) derivatives as tyrosinase and pancreatic lipase inhibitors. J. Mol. Struct. 2022, 1259, 132734. [Google Scholar] [CrossRef]
- Huang, T.-J.; Chuang, H.; Liang, Y.-C.; Lin, H.-H.; Horng, J.-C.; Kuo, Y.-C.; Chen, C.-W.; Tsai, F.Y.; Yen, S.-C.; Chou, S.-C.; et al. Design, synthesis, and bioevaluation of paeonol derivatives as potential anti-HBV agents. Eur. J. Med. Chem. 2015, 90, 428–435. [Google Scholar] [CrossRef]
- Xie, D.; Yang, Z.; Hu, X.; Wen, Y. Synthesis, antibacterial and insecticidal activities of novel capsaicin derivatives containing a sulfonic acid esters moiety. Front. Chem. 2022, 10, 929050. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Nkoana, J.K.; Gildenhuys, S.; Elhenawy, A.A. Structure and biological property studies of the fluorinated sulfonic esters derived from 2-hydroxy-4-(hydroxy/methoxy)acetophenone as inhibitors of biochemical targets linked to type 2 diabetes mellitus. J. Fluor. Chem. 2024, 273, 110233. [Google Scholar] [CrossRef]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kissb, L.; Soloshonoke, V.A.; Han, J. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- Chandra, G.; Singh, D.V.; Mahato, G.K.; Patel, S. Fluorine- a small magic bullet atom in the drug development: Perspective to FDA approved and COVID19 recommended drugs. Chem. Pap. 2023, 77, 4085–4106. [Google Scholar] [CrossRef]
- Biffinger, J.C.; Kim, H.W.; DiMagno, S.G. The polar hydrophobicity of fluorinated compounds. ChemBioChem 2004, 5, 622–627. [Google Scholar] [CrossRef]
- Jersche, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest. Manag. Sci. 2010, 66, 10–27. [Google Scholar]
- Sheikhi, N.; Bahraminejad, M.; Saeedi, M.; Mirfazli, S.S. A review: FDA-approved fluorine-containing small molecules from 2015 to 2022. Eur. J. Med. Chem. 2023, 260, 115758. [Google Scholar] [CrossRef]
- Xu, P.; Zhu, L.; Zhang, D.; Li, Z.; Ge, R.; Tian, Q. Design and synthesis of fluorine aromatic scaffolds containing drugs approved by the US FDA from 2002 to 2022. Results Chem. 2024, 7, 101446. [Google Scholar] [CrossRef]
- Bőhm, H.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef]
- De Freitas, R.F.; Schapira, M. A systematic analysis of atomic protein–ligand interactions in the PDB. Med. Chem. Commun. 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. Fluorine in medicinal chemistry: In perspective to COVID-19. ACS Omega 2022, 7, 18206–18212. [Google Scholar] [CrossRef] [PubMed]
- Petkowski, J.J.; Seager, S.; Bains, W. Reasons why life on Earth rarely makes fluorine-containing compounds and their implications for the search for life beyond. Earth. Sci. Rep. 2024, 14, 15575. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Zhu, W. Nonbonding interactions of organic halogens in biological systems: Implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. [Google Scholar] [CrossRef]
- Sun, S.; Fu, J. Methyl-containing pharmaceuticals: Methylation in drug design. Bioorganic Med. Chem. Lett. 2018, 28, 3283–3289. [Google Scholar] [CrossRef]
- Norton, R.S.; Leung, E.W.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of 19F-NMR in fragment-based drug discovery. Molecules 2016, 21, 860. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakov, A.; Roos, M.; Kwon, B.; Hong, M. Two-dimensional 19F–13C correlation NMR for 19F resonance assignment of fluorinated proteins. J. Biomol. NMR 2020, 74, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Buchholza, C.R.; Pomerantz, W.C.K. 19F NMR viewed through two different lenses: Ligand-observed and protein-observed 19F NMR applications for fragment-based drug discovery. RSC Chem. Biol. 2021, 2, 1312–1330. [Google Scholar] [CrossRef]
- Gimenez, D.; Phelan, A.; Murphy, C.D.; Cobb, S.L. 19F NMR as a tool in chemical biology. Beilstein J. Org. Chem. 2021, 17, 293–318. [Google Scholar] [CrossRef]
- Blundell, C.D.; Nowak, T.; Watson, M.J. Chapter two- Measurement, interpretation and use of free ligand solution conformations in drug discovery. Prog. Med. Chem. 2016, 55, 45–147. [Google Scholar]
- Ali, A.; Khalid, M.; Ur Rehman, M.F.; Haq, S.; Ali, A.; Tahir, M.N.; Ashfaq, M.; Rasool, F.; Braga, A.C. Efficient synthesis, SC-XRD, and theoretical studies of O-benzenesulfonylated pyrimidines: Role of noncovalent interaction influence in their supramolecular network. ACS Omega 2020, 5, 15115–15128. [Google Scholar] [CrossRef]
- Mokoena, T.P.; Maluleka, M.M.; Mampa, R.M.; Mphahlele, M.J.; Monchusi, B.A. Synthesis, crystal structures, spectroscopic characterization and in vitro evaluation of the 4-sulfono-3-methoxycinnamaldehydes as potential α-glucosidase and/or α-amylase inhibitors. J. Mol. Struct. 2023, 1271, 134119. [Google Scholar] [CrossRef]
- Henary, E.; Casa, S.; Dost, T.L.; Sloop, J.C.; Henary, M. The role of small molecules containing fluorine atoms in medicine and imaging applications. Pharmaceuticals 2024, 17, 281. [Google Scholar] [CrossRef]
- Sheen, A.J. Is there a role for α-glucosidase inhibitors in the prevention of type 2 diabetes mellitus? Drugs 2003, 63, 933–951. [Google Scholar] [CrossRef]
- Vieira, R.; Souto, S.B.; Sánchez-López, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Silva, A.M.; Fortuna, A.; García, M.L.; et al. Sugar-lowering drugs for type 2 diabetes mellitus and metabolic syndrome- Review of classical and new compounds: Part-I. Pharmaceuticals 2019, 12, 152. [Google Scholar] [CrossRef]
- Csermely, P.; Agoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P. α-Glucosidase inhibitors and their use in clinical practice. Arch. Med. Sci. 2012, 8, 899–906. [Google Scholar] [CrossRef]
- Sekar, N.; Li, J.; Shechter, Y. Vanadium salts as insulin substitutes: Mechanisms of action, a scientific and therapeutic tool in diabetes mellitus research. Crit. Rev. Biochem. Mol. Biol. 1996, 31, 339–359. [Google Scholar] [CrossRef]
- Wind, S.; Schmid, U.; Freiwald, M.; Marzin, K.; Lotz, R.; Ebner, T.; Stopfer, P.; Dallinger, C. Clinical pharmacokinetics and pharmacodynamics of nintedanib. Clin. Pharmacokinet. 2019, 58, 1131–1147. [Google Scholar] [CrossRef]
- Aziz, M.A.; Serya, R.A.T.; Lasheen, D.S.; Abdel-Aziz, A.K.; Esmat, A.; Mansour, A.M.; Singab, A.N.B.; Abouzid, K.A.M. Discovery of potent VEGFR-2 inhibitors based on furopyrimidine and thienopyrimidne scaffolds as cancer targeting agents. Sci. Rep. 2016, 6, 24460. [Google Scholar] [CrossRef] [PubMed]
- Ur Rehman, N.; Rafiq, K.; Khan, A.; Ahsan Halim, S.; Ali, L.; Al-Saady, N.; Hilal Al-Balushi, A.; Al-Busaidi, H.K.; Al-Harrasi, A. α-Glucosidase inhibition and molecular docking studies of natural brominated metabolites from marine macro brown alga Dictyopteris hoytii. Mar. Drugs 2019, 17, 666. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Begum, A.; Numao, S.; Park, K.H.; Withers, S.G.; Brayer, G.D. Acarbose rearrangement mechanism implied by the kinetic and structural analysis of human pancreatic α-amylase in complex with analogues and their elongated counterparts. Biochemistry 2005, 44, 347–3357. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randall, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows an update. J. Appl. Crystallogr. 2012, 245, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-2017/1. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; More, G.K.; Maluleka, M.M.; Choong, Y.S. Bio-evaluation of the 2-nitrochalcones as potential anti-lung cancer agents, inducers of apoptosis and inhibitors of protein kinase (VEGFR-2). Med. Chem. Res. 2023, 32, 2380–2393. [Google Scholar] [CrossRef]
- Nkoana, J.K.; Mphahlele, M.J.; More, G.K.; Elhenawy, A.A. Synthesis and in vitro exploration of the 8-carbo substituted 5-methoxyflavones as anti-breast and anti-lung cancer agents targeting protein kinases (VEGFR-2 & EGFR). Bioorganic Chem. 2024, 153, 107875. [Google Scholar]
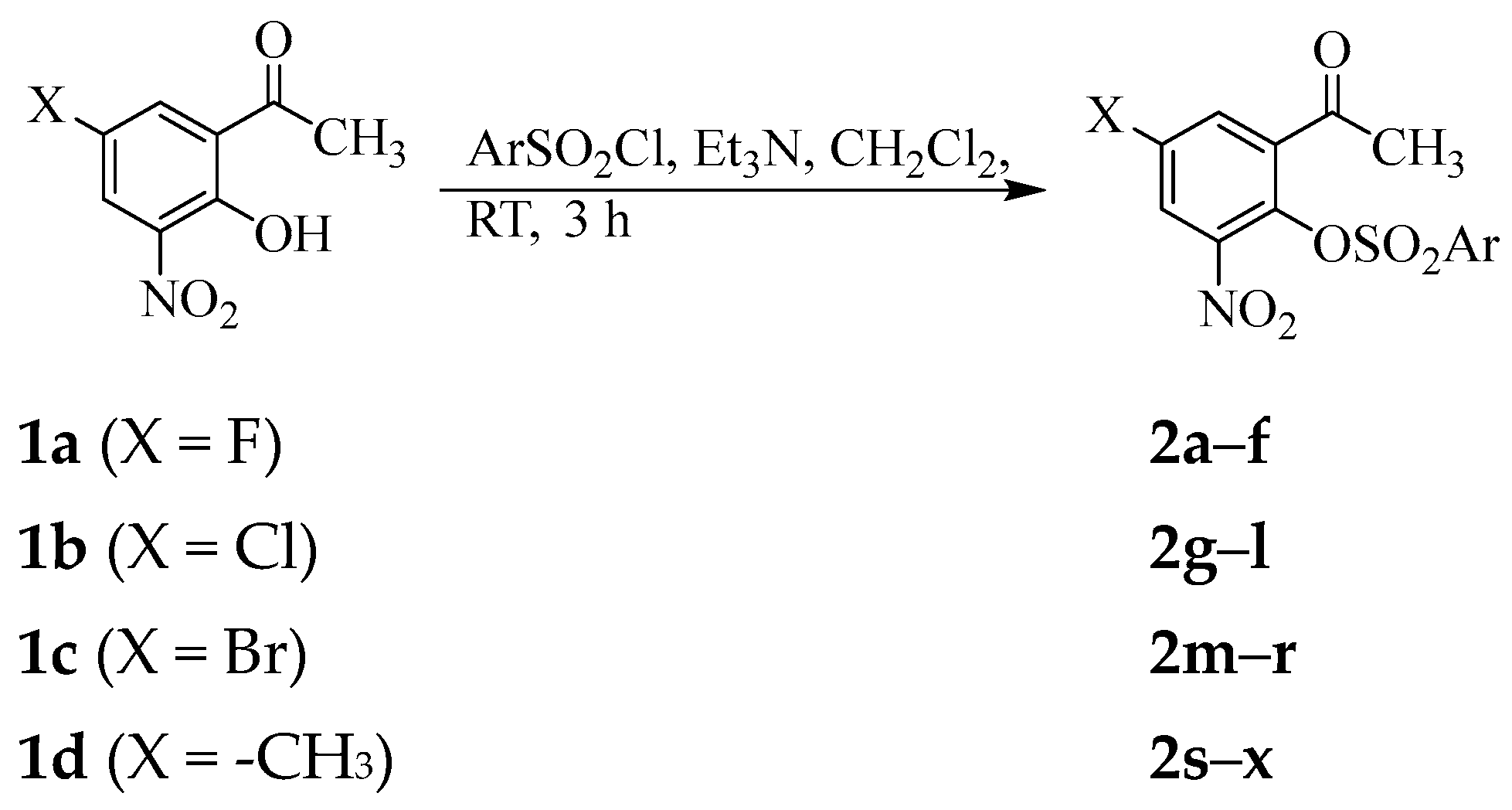


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ar | X (Compound Number) | |||
|---|---|---|---|---|
| -C6H4(2-F) | F (2a) | Cl (2g) | Br (2m) | -CH3 (2s) |
| -C6H4(3-F) | F (2b) | Cl (2h) | Br (2n) | -CH3 (2t) |
| -C6H4(4-F) | F (2c) | Cl (2i) | Br (2o) | -CH3 (2u) |
| -C6H4(4-CF3) | F (2d) | Cl (2j) | Br (2p) | -CH3 (2v) |
| -C6H4(4-OCF3) | F (2e) | Cl (2k) | Br (2q) | -CH3 (2w) |
| -C6H4(4-OCH3) | F (2f) | Cl (2l) | Br (2r) | -CH3 (2x) |
| Compound | IC50 (µM ± SD) | |||
|---|---|---|---|---|
| α-Glucosidase | α-Amylase | PTP1B | VEGFR-2 | |
| 1a | 16.9 ± 0.064 | - | - | - |
| 1b | 27.2 ± 0.195 | - | - | - |
| 1c | 11.9 ± 0.010 | - | - | - |
| 1d | 8.2 ± 0.015 | - | - | - |
| 2a | 5.6 ± 0.038 | 7.9 ± 0.054 | 6.3 ± 0.029 | 28.7 ± 0.27 |
| 2b | 8.6 ± 0.025 | - | - | - |
| 2c | 27.4 ± 0.044 | - | - | - |
| 2d | 6.4 ± 0.012 | 18.1 ± 0.185 | - | - |
| 2e | 3.1 ± 0.043 | 15.2 ± 0.019 | 23.1 ± 0.006 | 28.5 ± 0.23 |
| 2f | 5.3 ± 0.159 | 3.1 ± 0.110 | 5.9 ± 0.018 | 28.3 ± 031 |
| 2g | 4.2 ± 0.054 | 7.0 ± 0.040 | 16.9 ± 0.016 | 34.4 ± 0.26 |
| 2h | 12.4 ± 0.041 | - | - | - |
| 2i | 10.6 ± 0.039 | - | - | - |
| 2j | 5.4 ± 0.030 | 7.3 ± 0.008 | 19.6 ± 0.042 | 20.8 ± 0.35 |
| 2k | 3.9 ± 0.031 | 9.8 ± 0.021 | 20.1 ± 0.065 | 26.4 ± 0.20 |
| 2l | 8.5 ± 0.043 | - | - | - |
| 2m | 7.9 ± 0.044 | - | - | - |
| 2n | 18.2 ± 0.041 | - | - | - |
| 2o | 20.5 ± 0.058 | - | - | - |
| 2p | 8.0 ± 0.060 | - | - | - |
| 2q | 8.7 ± 0.075 | - | - | - |
| 2r | 13.2 ± 0.043 | - | - | - |
| 2s | 18.1 ± 0.048 | - | - | - |
| 2t | 13.1 ± 0.045 | - | - | - |
| 2u | 11.2 ± 0.045 | - | - | - |
| 2v | 3.0 ± 0.014 | 6.0 ± 0.020 | 7.6 ± 0.070 | 16.3 ± 0.29 |
| 2w | 6.0 ± 0.034 | 16.7 ± 0.035 | - | - |
| 2x | 3.6 ± 0.029 | 11.9 ± 0.022 | 21.5 ± 0.066 | 24.2 ± 0.22 |
| Acarbose | 6.4 ± 0.134 | 5.4 ± 0.131 | - | - |
| Na3VO4 | - | - | 6.3 ± 0.050 | - |
| Nintedanib | - | - | - | 1.4 ± 0.42 |
| Compounds | IC50 (µM ± SD) | ||
|---|---|---|---|
| MCF-7 | A549 | Vero | |
| 2a | 4.96 ± 0.05 | 7.75 ± 0.11 | 39.21 ± 0.13 |
| 2e | 8.18 ± 0.03 | 7.42 ± 0.09 | 20.78 ± 0.14 |
| 2f | 4.38 ± 0.07 | 1.91 ± 0.01 | 24.03 ± 1.01 |
| 2g | 9.92 ± 0.09 | 6.15 ± 0.06 | 33.93 ± 0.15 |
| 2j | 2.58 ± 0.02 | 2.52 ± 0.02 | 38.86 ± 0.16 |
| 2k | 34.53 ± 0.12 | 1.89 ± 0.05 | 33.39 ± 0.13 |
| 2v | 8.58 ± 0.07 | 5.50 ± 0.01 | 48.02 ± 0.18 |
| 2x | 11.01 ± 0.07 | 1.01 ± 0.01 | 56.77 ± 0.18 |
| Doxorubicin | 0.59 ± 0.03 | 0.71 ± 0.09 | 0.94 ± 0.04 |
| Nintedanib | 0.40 ± 0.03 | 0.78 ± 0.04 | 0.24 ± 0.02 |
| Property | 2a | 2e | 2f | 2g | 2j | 2k | 2v | 2x |
|---|---|---|---|---|---|---|---|---|
| XLogP3 | 2.73 | 3.82 | 2.61 | 3.26 | 4.05 | 4.34 | 3.78 | 2.87 |
| mLogP | 2.44 | 2.14 | 1.78 | 2.56 | 3.05 | 2.26 | 2.78 | 1.63 |
| TPSA (Å) | 114.64 | 123.87 | 123.87 | 114.64 | 114.64 | 123.87 | 114.64 | 123.87 |
| Solubility (Log S) | −3.82 | −4.73 | −3.73 | −4.25 | −4.95 | −5.16 | −4.65 | −3.87 |
| GI Absorption | High | Low | High | High | Low | Low | Low | High |
| Cytochrome P450 enzyme inhibitor * | 2C19 3A4 | 2C19, 2C9, 2D6, 3A4 | 2C19, 2C9, 2D6, 3A4 | 2C19, 2C9, 3A4 | 2C19, 2C9, 3A4 | 2C19, 2C9, 3A4 | 1A2, 2C19, 2C9,3A4 | 1A2, 2C19,2C9, 2D6, 3A4 |
| Saturation (fraction Csp3) | 0.07 | 0.13 | 0.13 | 0.07 | 0.13 | 0.13 | 0.19 | 0.19 |
| MW(g/mol) | 357.29 | 423.29 | 369.32 | 373.74 | 423.75 | 439.75 | 403.33 | 365.36 |
| H-bond acceptor | 8 | 11 | 8 | 7 | 9 | 10 | 9 | 7 |
| H-bond donor | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rotatable bonds | 5 | 7 | 6 | 5 | 6 | 7 | 6 | 6 |
| Lipinski’s violation | none | none | none | none | none | none | none | none |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olomola, T.O.; Nkoana, J.K.; More, G.K.; Gildenhuys, S.; Mphahlele, M.J. Enzyme (α-Glucosidase, α-Amylase, PTP1B & VEGFR-2) Inhibition and Cytotoxicity of Fluorinated Benzenesulfonic Ester Derivatives of the 5-Substituted 2-Hydroxy-3-nitroacetophenones. Int. J. Mol. Sci. 2024, 25, 11862. https://doi.org/10.3390/ijms252211862
Olomola TO, Nkoana JK, More GK, Gildenhuys S, Mphahlele MJ. Enzyme (α-Glucosidase, α-Amylase, PTP1B & VEGFR-2) Inhibition and Cytotoxicity of Fluorinated Benzenesulfonic Ester Derivatives of the 5-Substituted 2-Hydroxy-3-nitroacetophenones. International Journal of Molecular Sciences. 2024; 25(22):11862. https://doi.org/10.3390/ijms252211862
Chicago/Turabian StyleOlomola, Temitope O., Jackson K. Nkoana, Garland K. More, Samantha Gildenhuys, and Malose J. Mphahlele. 2024. "Enzyme (α-Glucosidase, α-Amylase, PTP1B & VEGFR-2) Inhibition and Cytotoxicity of Fluorinated Benzenesulfonic Ester Derivatives of the 5-Substituted 2-Hydroxy-3-nitroacetophenones" International Journal of Molecular Sciences 25, no. 22: 11862. https://doi.org/10.3390/ijms252211862
APA StyleOlomola, T. O., Nkoana, J. K., More, G. K., Gildenhuys, S., & Mphahlele, M. J. (2024). Enzyme (α-Glucosidase, α-Amylase, PTP1B & VEGFR-2) Inhibition and Cytotoxicity of Fluorinated Benzenesulfonic Ester Derivatives of the 5-Substituted 2-Hydroxy-3-nitroacetophenones. International Journal of Molecular Sciences, 25(22), 11862. https://doi.org/10.3390/ijms252211862