

Macrocephaly and Finger Changes: A Narrative Review
 , ,
, , 
Abstract
1. Introduction
2. Causes of Macrocephaly and Finger Changes
2.1. Skeletal Dysplasias
2.2. Inherited Metabolic Disorders
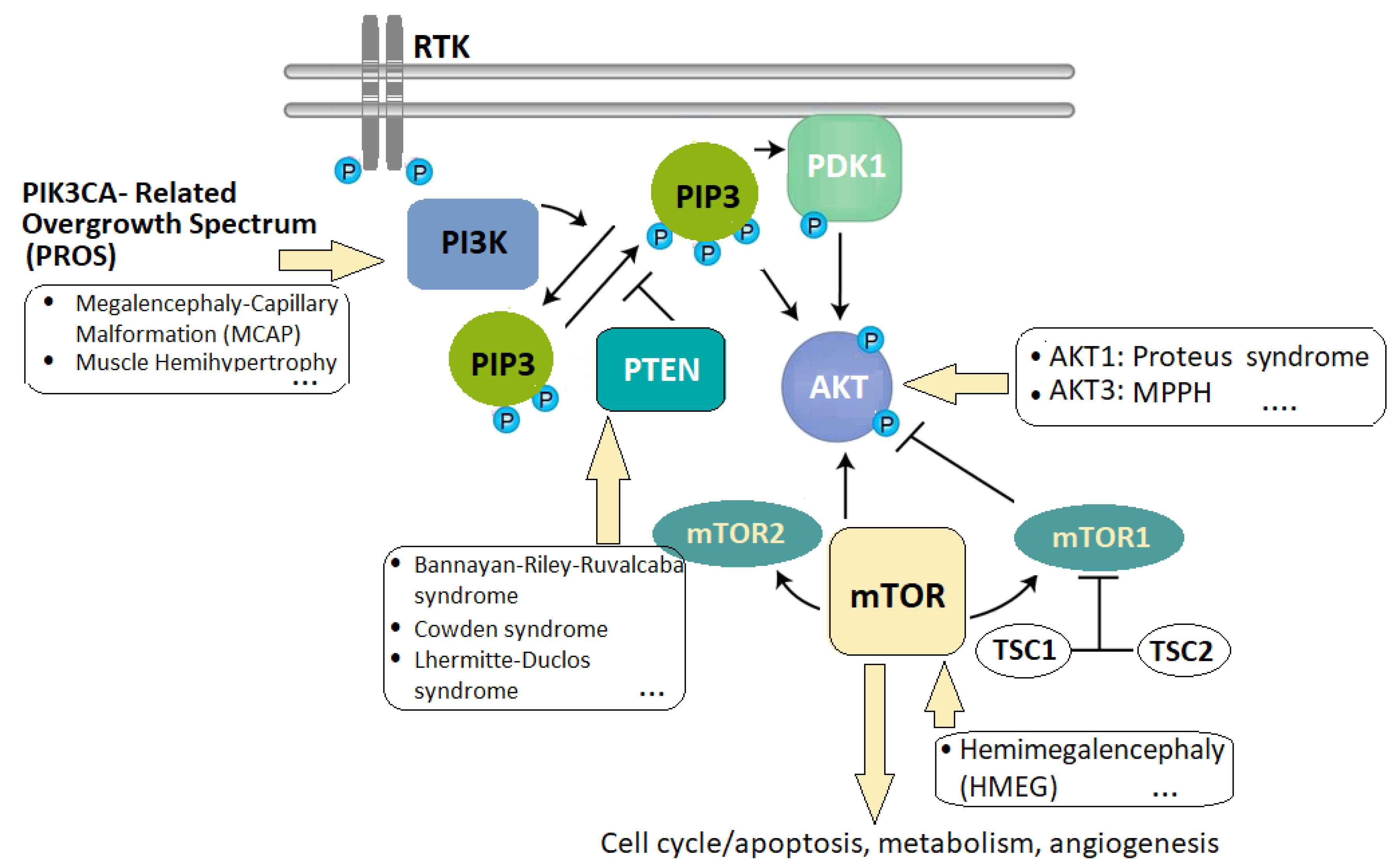
2.3. Overgrowth Syndromes
2.4. Congenital Infections
2.5. Autoimmune and Autoinflammatory Diseases
3. Clinical Approach and Therapies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACP5: Encoding the tartrate-resistant acid phosphatase 5 |
| AD: Autosomal dominant |
| AOS: Adams–Oliver syndrome |
| AR: Autosomal recessive |
| BMP: Bone morphogenetic protein |
| BRMUTD: Brain malformations with or without urinary tract defects (BRMUTD) |
| CFZS1: Carey–Fineman–Ziter syndrome |
| CFC: Cardiofaciocutaneous syndrome |
| CINCA/NOMID: Chronic infantile neurologic cutaneous articular syndrome/neonatal-onset multisystem inflammatory disease |
| GCPS: Greig cephalopolysyndactyly syndrome |
| CPRF: Cleft palate, psychomotor retardation, and distinctive facial feature |
| CLAPO syndrome: Capillary malformation of the lower lip, lymphatic malformation of the face and neck, asymmetry, and partial/generalized overgrowth |
| CLOVES syndrome: Congenital Lipomatous Overgrowth, Vascular Malformations, Epidermal Nevis, Spinal/Skeletal Anomalies/Scoliosis |
| CMDR: Craniometaphyseal dysplasia, autosomal recessive |
| CMV: Cytomegalovirus |
| CNS: Central nervous system |
| CNVs: Copy number variations |
| COGIS: Cohen–Gibson syndrome |
| DADA2: Adenosine deaminase-2 deficiency |
| DDVIBA: Developmental delay with variable impairment and behavioral abnormalities |
| EDHHACC: Ectodermal dysplasia, hyperhidrotic with hypothyroidism and agenesis of the corpus callosus |
| EGF: Epidermal growth factor |
| ERT: Enzyme replacement therapy |
| FB: Frontal bossing |
| FCAS: Familial cold-induced autoinflammatory syndrome |
| FGFR: Fibroblast growth factor receptors |
| GAGs: Glycosaminoglycans |
| GT: Gene therapy |
| HC: Hydrocephalus |
| HMEG: Hemimegaloencephaly |
| HSCT: Hematopoietic stem cell transplantation |
| ID: Intellectual disability |
| IGF-I: Insulin-like growth factor I |
| IHH-PTHrP: IHH (Indian Hedgehog), which stimulates PTH-related protein pathway |
| JIA: Juvenile idiopathic arthritis |
| LSDs: Lysosomal storage disorders |
| MC: Macrocephaly |
| MCAP: Macrocephaly-capillary malformation |
| ME: Megalencephaly |
| MDFPMR: Macrocephaly, dysmorphic facies, and psychomotor retardation |
| MiC: Microcephaly |
| MNKES: Muenke craniosynostosis syndrome |
| MPPH: Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome |
| MPPM: Megalencephaly-polymicrogyria-pigmentary mosaicism |
| MPS: Mucopolysaccharidoses |
| mTOR: Mammalian target of rapamycinNGS: next-generation sequencing |
| NS: Noonan syndrome |
| OMIM: Online Mendelian Inheritance in Man |
| PBD: Peroxisome Biogenesis Disorders |
| PCs: Pharmacological chaperones |
| PHTS: PTEN Hamartoma-tumor syndrome |
| PRAAS: Proteasome-associated autoinflammatory syndromes |
| PROS: PIK3CA-related overgrowth spectrum |
| PTEN: Phosphatase and tensin homologue deleted on chromosome 10 |
| SAVI: STING associated vasculopathy with onset in infancy |
| SGMRT: Singleton-Merten syndrome |
| SHH: Sonic hedgehog (pathway) |
| SIHIWES: Sifrim–Hitz–Weiss syndrome |
| SOGRI: Spanish Overgrowth Clinical Registry |
| SPENCD: Spondyloenchondrodysplasia |
| SRT: Substrate reduction therapy |
| SRTD8: Short-rib thoracic dysplasia 8 with or without polydactyly |
| STING: Stimulator of Interferon Genes |
| TBRS: Tatton–Brown–Rahman syndrome |
| THES: Trichohepatoenteric syndrome |
| TSC1, TSC2: Tuberous sclerosis complex 1, Tuberous sclerosis complex 2 |
| TSH: thyroid-stimulating hormone |
| UPD: Uniparental disomy |
| VEGF: Vascular endothelial growth factor |
| XL: X-linked disorder |
| XLD: X-linked dominant disorder |
References
- Wojcik, M.H.; Agrawal, P.B. Deciphering congenital anomalies for the next generation. Cold Spring Harb. Mol. Case Stud. 2020, 6, a005504. [Google Scholar] [CrossRef] [PubMed]
- Barbier, A.; Boivin, A.; Yoon, W.; Vallerand, D.; Platt, R.W.; Audibert, F.; Barrington, K.J.; Shah, P.S.; Nuyt, A.M. New reference curves for head circumference at birth, by gestational age. Pediatrics 2013, 131, e1158-67. [Google Scholar] [CrossRef]
- Accogli, A.; Geraldo, A.F.; Piccolo, G.; Riva, A.; Scala, M.; Balagura, G.; Salpietro, V.; Madia, F.; Maghnie, M.; Zara, F.; et al. Diagnostic Approach to Macrocephaly in Children. Front. Pediatr. 2022, 9, 794069. [Google Scholar] [CrossRef] [PubMed]
- Winden, K.D.; Yuskaitis, C.J.; Poduri, A. Megalencephaly and Macrocephaly. Semin. Neurol. 2015, 35, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Vanden Brande, L.; Alkan, S.; Barrea, C.; Leroy, P. Comment j’explore. Une macrocéphalie [How I explore a macrocephaly]. Rev. Med. Liege 2022, 77, 56–62. [Google Scholar] [PubMed]
- Guzik, A.; Perenc, L.; Drużbicki, M.; Podgórska-Bednarz, J. Abnormal cranium development in children and adolescents affected by syndromes or diseases associated with neurodysfunction. Sci. Rep. 2021, 11, 2908. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.G.; Samanta, D. Macrocephaly. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Renaud, D.L. Leukoencephalopathies associated with macrocephaly. Semin. Neurol. 2012, 32, 34–41. [Google Scholar] [CrossRef]
- Bastos, G.C.; Tolezano, G.C.; Krepischi, A.C.V. Rare CNVs and Known Genes Linked to Macrocephaly: Review of Genomic Loci and Promising Candidate Genes. Genes 2022, 13, 2285. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Praticò, A.D.; Rizzo, R.; Corsello, G.; Ruggieri, M.; Parano, E.; Falsaperla, R. A clinical review on megalencephaly: A large brain as a possible sign of cerebral impairment. Medicine 2017, 96, e6814. [Google Scholar] [CrossRef]
- Tully, H.M.; Dobyns, W.B. Infantile hydrocephalus: A review of epidemiology, classification and causes. Eur. J. Med. Genet. 2014, 57, 359–368. [Google Scholar] [CrossRef]
- Langner, S.; Fleck, S.; Baldauf, J.; Mensel, B.; Kühn, J.P.; Kirsch, M. Diagnosis and Differential Diagnosis of Hydrocephalus in Adults. RoFo 2017, 189, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Rekate, H.L. The definition and classification of hydrocephalus: A personal recommendation to stimulate debate. Cerebrospinal Fluid Res. 2008, 5, 2. [Google Scholar] [CrossRef]
- Schonstedt Geldres, V.; Stecher Guzmán, X.; Manterola Mordojovich, C.; Rovira, À. Imaging in the study of macrocephaly: Why?, when?, how? Radiologia Engl. Ed. 2022, 64, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Olney, A.H. Macrocephaly syndromes. Semin. Pediatr. Neurol. 2007, 14, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.A.; Dagli, A.; Battaglia, A. Genetic disorders associated with macrocephaly. Am. J. Med. Genet. A 2008, 146, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Manor, J.; Lalani, S.R. Overgrowth Syndromes-Evaluation, Diagnosis, and Management. Front. Pediatr. 2020, 8, 574857. [Google Scholar] [CrossRef] [PubMed]
- Keppler-Noreuil, K.M.; Parker, V.E.; Darling, T.N.; Martinez-Agosto, J.A. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am. J. Med. Genet. C Semin. Med. Genet. 2016, 172, 402–421. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Murray, A.; Hanks, S.; Douglas, J.; Armstrong, R.; Banka, S.; Bird, L.M.; Clericuzio, C.L.; Cormier-Daire, V.; Cushing, T.; et al. Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. Am. J. Med. Genet. A 2013, 161, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Macchiaiolo, M.; Panfili, F.M.; Vecchio, D.; Gonfiantini, M.V.; Cortellessa, F.; Caciolo, C.; Zollino, M.; Accadia, M.; Seri, M.; Chinali, M.; et al. A deep phenotyping experience: Up to date in management and diagnosis of Malan syndrome in a single center surveillance report. Orphanet J. Rare Dis. 2022, 17, 235. [Google Scholar] [CrossRef]
- Huybrechts, Y.; Mortier, G.; Boudin, E.; Van Hul, W. WNT Signaling and Bone: Lessons From Skeletal Dysplasias and Disorders. Front. Endocrinol. 2020, 11, 165. [Google Scholar] [CrossRef]
- Twigg, S.R.; Wilkie, A.O. A Genetic-Pathophysiological Framework for Craniosynostosis. Am. J. Hum. Genet. 2015, 97, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.; Singh, N.; Richtsmeier, J.T. Understanding craniosynostosis as a growth disorder. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 429–459. [Google Scholar] [CrossRef] [PubMed]
- Goos, J.A.C.; Mathijssen, I.M.J. Genetic Causes of Craniosynostosis: An Update. Mol. Syndromol. 2019, 10, 6–23. [Google Scholar] [CrossRef]
- Ferguson, J.W.; Atit, R.P. A tale of two cities: The genetic mechanisms governing calvarial bone development. Genesis 2019, 57, e23248. [Google Scholar] [CrossRef] [PubMed]
- Matošević, M.; Lamot, L.; Antičević, D. Camptodactyly and clinodactyly—New understanding of known deformities. Acta Clin. Croat. 2022, 60, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.M. Objective assessment of tapering of the fingers in adults. PLoS ONE 2022, 17, e0279202. [Google Scholar] [CrossRef]
- Al-Qattan, M.M. The Classification of VACTERL Association into 3 Groups According to the Limb Defect. Plastic and reconstructive surgery. Glob. Open 2021, 9, e3360. [Google Scholar] [CrossRef]
- Miraoui, H.; Ringe, J.; Haupl, T.; Marie, P.J. Increased EFG- and PDGF-alpha-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis. Hum. Molec. Genet. 2010, 19, 1678–1689. [Google Scholar] [CrossRef]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028191. [Google Scholar] [CrossRef]
- Koch, C.A.; Chrousos, G.P.; Chandra, R.; Evangelista, R.S.; Gilbert, J.C.; Nobuhara, K.; Zhuang, Z.; Vortmeyer, A.O. Two-hit model for tumorigenesis of nevoid basal cell carcinoma (Gorlin) syndrome-associated hepatic mesenchymal tumor. Am. J. Med. Genet. 2002, 109, 74–76. [Google Scholar] [CrossRef]
- Kansal, A.; Brueton, L.; Lahiri, A.; Lester, R. Hypoplastic thumb in Gorlin’s syndrome. J. Plast. Reconstr. Aesthetic Surg. 2007, 60, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J.; Goltz, R.W. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N. Engl. J. Med. 1960, 262, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Kimonis, V.E.; Goldstein, A.M.; Pastakia, B.; Yang, M.L.; Kase, R.; DiGiovanna, J.J.; Bale, A.E.; Bale, S.J. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am. J. Med. Genet. 1997, 69, 299–308. [Google Scholar] [CrossRef]
- Lile, H.A.; Rogers, J.F.; Gerald, B. The basal cell nevus syndrome. Am. J. Roentgen. Radium. Ther. Nucl. Med. 1968, 103, 214–217. [Google Scholar] [CrossRef]
- Demurger, F.; Ichkou, A.; Mougou-Zerelli, S.; Le Merrer, M.; Goudefroye, G.; Delezoide, A.-L.; Quelin, C.; Manouvrier, S.; Baujat, G.; Fradin, M.; et al. New insights into genotype-phenotype correlation for GLI3 mutations. Europ. J. Hum. Genet. 2015, 23, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Tunovic, S.; Barañano, K.W.; Barkovich, J.A.; Strober, J.B.; Jamal, L.; Slavotinek, A.M. Novel KIF7 missense substitutions in two patients presenting with multiple malformations and features of acrocallosal syndrome. Am. J. Hum. Genet. 2015, 167, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Logan, C.V.; Mougou-Zerelli, S.; Lee, J.H.; Silhavy, J.L.; Brancati, F.; Iannicelli, M.; Travaglini, L.; Romani, S.; Illi, B.; et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010, 42, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Nyboe, D.; Kreiborg, S.; Kirchhoff, M.; Hove, H.B. Familial craniosynostosis associated with a microdeletion involving the NFIA gene. Clin. Dysmorph. 2015, 24, 109–112. [Google Scholar] [CrossRef]
- Turnpenny, P.D.; Wright, M.J.; Sloman, M.; Caswell, R.; van Essen, A.J.; Gerkes, E.; Pfundt, R.; White, S.M.; Shaul-Lotan, N.; Carpenter, L.; et al. Missense mutations of the Pro65 residue of PCGF2 cause a recognizable syndrome associated with craniofacial, neurological, cardiovascular, and skeletal features. Am. J. Hum. Genet. 2018, 103, 786–793, Erratum in Am. J. Hum. Genet. 2018, 103, 1054. [Google Scholar] [CrossRef]
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A.F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am. J. Hum. Genet. 2016, 99, 934–941. [Google Scholar] [CrossRef]
- Pasetti, M.; Mazzoleni, F.; Novelli, G.; Iascone, M.; Bozzetti, A.; Selicorni, A. Temporomandibular joint ankylosis as part of the clinical spectrum of Carey-Fineman-Ziter syndrome? Am. J. Med. Genet. A 2016, 170, 2191–2195. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.M.; Rauch, F.; Travers, R.; Roy, M.; Montes, J.; Chabot, G.; Glorieux, F.H. Osteopathia striata with cranial sclerosis: Clinical, radiological, and bone histological findings in an adolescent girl. Am. J. Med. Genet. 2004, 129, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Dumić, M.; Vuković, J.; Cvitkovic, M.; Medica, I. Twins and their mildly affected mother with Weaver syndrome. Clin. Genet. 1993, 44, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.S.A.; Tuysuz, B.; Shen, Y.; Bhalla, S.K.; Jones, S.J.M.; Gibson, W.T. A novel mutation in EED associated with overgrowth. J. Hum. Genet. 2015, 60, 339–342, Erratum in J. Hum. Genet. 2017, 62, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Cooney, E.; Bi, W.; Schlesinger, A.E.; Vinson, S.; Potocki, L. Novel EED mutation in patient with Weaver syndrome. Am. J. Med. Genet. 2017, 173, 541–545. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Loveday, C.; Yost, S.; Clarke, M.; Ramsay, E.; Zachariou, A.; Elliott, A.; Wylie, H.; Ardissone, A.; Rittinger, O.; et al. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am. J. Hum. Genet. 2017, 100, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Gueneau, L.; Fish, R.J.; Shamseldin, H.E.; Voisin, N.; Mau-Them, F.T.; Preiksaitiene, E.; Monroe, G.R.; Lai, A.; Putoux, A.; Allias, F.; et al. KIAA1109 variants are associated with a severe disorder of brain development and arthrogryposis. Am. J. Hum. Genet. 2018, 102, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Fabre, A.; Bourgeois, P.; Coste, M.E.; Roman, C.; Barlogis, V.; Badens, C. Management of syndromic diarrhea/tricho-hepato-enteric syndrome: A review of the literature. Intractable Rare Dis. Res. 2017, 6, 152–157. [Google Scholar] [CrossRef]
- Goulet, O.; Vinson, C.; Roquelaure, B.; Brousse, N.; Bodemer, C.; Cézard, J.P. Syndromic (phenotypic) diarrhea in early infancy. Orphanet J. Rare Dis. 2008, 3, 6. [Google Scholar] [CrossRef]
- Bourgeois, P.; Esteve, C.; Chaix, C.; Béroud, C.; Lévy, N.; THES Clinical Consortium; Fabre, A.; Badens, C. Tricho-Hepato-Enteric Syndrome mutation update: Mutations spectrum of TTC37 and SKIV2L, clinical analysis and future prospects. Hum. Mutat. 2018, 39, 774–789. [Google Scholar] [CrossRef]
- Kinnear, C.; Glanzmann, B.; Banda, E.; Schlechter, N.; Durrheim, G.; Neethling, A.; Nel, E.; Schoeman, M.; Johnson, G.; van Helden, P.D.; et al. Exome sequencing identifies a novel TTC37 mutation in the first reported case of Trichohepatoenteric syndrome (THE-S) in South Africa. BMC Med. Genet. 2017, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Amor, D.J.; Stephenson, S.E.M.; Mustapha, M.; Mensah, M.A.; Ockeloen, C.W.; Lee, W.S.; Tankard, R.M.; Phelan, D.G.; Shinawi, M.; de Brouwer, A.P.M.; et al. Pathogenic Variants in GPC4 Cause Keipert Syndrome. Am. J. Hum. Genet. 2019, 104, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Bowen, P.; Lef, C.S.; Zellweger, H.; Linderberg, R. A familial syndrome of multiple congenital defects. Bull. Johns Hopkins Hosp. 1964, 114, 402–414. [Google Scholar] [PubMed]
- Passarge, E.; McAdams, A.J. Cerebro-hepato-renal syndrome. A newly recognized hereditary disorder of multiple congenital defects, including sudanophilic leukodystrophy, cirrhosis of the liver, and polycystic kidneys. J. Pediatr. 1967, 71, 691–702. [Google Scholar] [CrossRef]
- Vetrini, F.; McKee, S.; Rosenfeld, J.A.; Suri, M.; Lewis, A.M.; Nugent, K.M.; Roeder, E.; Littlejohn, R.O.; Holder, S.; Zhu, W.; et al. De novo and inherited TCF20 pathogenic variants are associated with intellectual disability, dysmorphic features, hypotonia, and neurological impairments with similarities to Smith-Magenis syndrome. Genome Med. 2019, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, A.O.; Byren, J.C.; Hurst, J.A.; Jayamohan, J.; Johnson, D.; Knight, S.J.; Lester, T.; Richards, P.G.; Twigg, S.R.; Wall, S.A. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics 2010, 126, e391–e400. [Google Scholar] [CrossRef] [PubMed]
- Saal, H.M.; Harbison, M.D.; Netchine, I. Silver-Russell Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2024. [Google Scholar]
- Sarig, O.; Nahum, S.; Rapaport, D.; Ishida-Yamamoto, A.; Fuchs-Telem, D.; Qiaoli, L.; Cohen-Katsenelson, K.; Spiegel, R.; Nousbeck, J.; Israeli, S.; et al. Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis syndrome is caused by a POC1A mutation. Am. J. Hum. Genet. 2012, 91, 337–342. [Google Scholar] [CrossRef]
- James, P.A.; Aftimos, S.; Oei, P. Severe musculoskeletal phenotype associated with an unbalanced t(6;10) translocation: Clarification of the locus for this phenotype on distal 6p. Am. J. Med. Genet. 2003, 119, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, R.; Berg, J.; Craft, E.; Foster, A.; Gibbons, R.J.; Hobson, E.; Lachlan, K.; Naik, S.; Sampson, J.R.; Sharif, S.; et al. Refining the Primrose syndrome phenotype: A study of five patients with ZBTB20 de novo variants and a review of the literature. Am. J. Med. Genet. 2019, 179, 344–349. [Google Scholar] [CrossRef]
- Schaaf, C.P.; Koster, J.; Katsonis, P.; Kratz, L.; Shchelochkov, O.A.; Scaglia, F.; Kelley, R.I.; Lichtarge, O.; Waterham, H.R.; Shinawi, M. Desmosterolosis--phenotypic and molecular characterization of a third case and review of the literature. Am. J. Med. Genet. 2011, 155, 1597–1604. [Google Scholar] [CrossRef]
- Lucignani, G.; Guarnera, A.; Rossi-Espagnet, M.C.; Moltoni, G.; Antonelli, A.; Figà Talamanca, L.; Carducci, C.; Calo Carducci, F.I.; Napolitano, A.; Gandolfo, C.; et al. From Fetal to Neonatal Neuroimaging in TORCH Infections: A Pictorial Review. Children 2022, 9, 1210. [Google Scholar] [CrossRef]
- Caksen, H.; Odabaş, D.; Anlar, O. Congenital cytomegalovirus infection associated with finger anomaly. J. Paediatr. Child Health 2002, 38, 105. [Google Scholar] [CrossRef] [PubMed]
- Bacino, C.A. ROR2-Related Robinow Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Mishra, R.; Jain, V.; Gupta, D.; Saxena, R.; Kulshreshtha, S.; Ramprasad, V.L.; Verma, I.C.; Dua Puri, R. Robinow Syndrome and Brachydactyly: An Interplay of High-Throughput Sequencing and Deep Phenotyping in a Kindred. Mol. Syndromol. 2020, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Noureldine, M.H.A.; Taher, A.T.; Haydar, A.A.; Berjawi, A.; Khamashta, M.A.; Uthman, I. Rheumatological complications of beta-thalassaemia: An overview. Rheumatology 2018, 57, 19–27. [Google Scholar] [CrossRef]
- Karakas, S.; Tellioglu, A.M.; Bilgin, M.; Omurlu, I.K.; Caliskan, S.; Coskun, S. Craniofacial Characteristics of Thalassemia Major Patients. Eurasian J. Med. 2016, 48, 204–208. [Google Scholar] [CrossRef]
- Stark, Z.; Savarirayan, R. Osteopetrosis. Orphanet J. Rare Dis. 2009, 4, 5. [Google Scholar] [CrossRef]
- Tenorio, J.; Mansilla, A.; Valencia, M.; Martínez-Glez, V.; Romanelli, V.; Arias, P.; Castrejón, N.; Poletta, F.; Guillén-Navarro, E.; Gordo, G.; et al. A new overgrowth syndrome is due to mutations in RNF125. Hum. Mutat. 2014, 35, 1436–1441. [Google Scholar] [CrossRef]
- Xuan, J.Y.; Hughes-Benzie, R.M.; MacKenzie, A.E. A small interstitial deletion in the GPC3 gene causes Simpson-Golabi-Behmel syndrome in a Dutch-Canadian family. J. Med. Genet. 1999, 36, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Temtamy, S.A.; Salam, M.A.; Aboul-Ezz, E.H.; Hussein, H.A.; Helmy, S.A.; Shalash, B.A. New autosomal recessive multiple congenital abnormalities/mental retardation syndrome with craniofacial dysmorphism absent corpus callosum, iris colobomas and connective tissue dysplasia. Clin. Dysmorphol. 1996, 5, 231–240. [Google Scholar] [CrossRef]
- Li, M.; Shuman, C.; Fei, Y.L.; Cutiongco, E.; Bender, H.A.; Stevens, C.; Wilkins-Haug, L.; Day-Salvatore, D.; Yong, S.L.; Geraghty, M.T.; et al. GPC3 mutation analysis in a spectrum of patients with overgrowth expands the phenotype of Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. 2001, 102, 161–168. [Google Scholar] [CrossRef]
- Ortega-Recalde, O.; Beltrán, O.I.; Gálvez, J.M.; Palma-Montero, A.; Restrepo, C.M.; Mateus, H.E.; Laissue, P. Biallelic HERC1 mutations in a syndromic form of overgrowth and intellectual disability. Clin. Genet. 2015, 88, e1–e3. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Schneider, T.; Rio, M.; Moutton, S.; Siquier-Pernet, K.; Verny, F.; Boddaert, N.; Desguerre, I.; Munich, A.; Rosa, J.L.; et al. A nonsense variant in HERC1 is associated with intellectual disability, megalencephaly, thick corpus callosum and cerebellar atrophy. Eur. J. Hum. Genet. 2016, 24, 455–458. [Google Scholar] [CrossRef]
- Aggarwal, S.; Bhowmik, A.D.; Ramprasad, V.L.; Murugan, S.; Dalal, A. A splice site mutation in HERC1 leads to syndromic intellectual disability with macrocephaly and facial dysmorphism: Further delineation of the phenotypic spectrum. Am. J. Med. Genet. 2016, 170, 1868–1873. [Google Scholar] [CrossRef]
- Devriendt, K.; D’Espallier, L.; Fryns, J.-P. Mental retardation, distinct craniofacial dysmorphism, and central nervous system malformation: Confirmation of a syndrome. Am. J. Med. Genet. 1996, 33, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.; Hao, M.; Luu, M. PIK3CA vascular overgrowth syndromes: An update. Curr. Opin. Pediatr. 2020, 32, 539–546. [Google Scholar] [CrossRef]
- Mirzaa, G.M.; Conway, R.L.; Gripp, K.W.; Lerman-Sagie, T.; Siegel, D.H.; deVries, L.S.; Lev, D.; Kramer, N.; Hopkins, E.; Graham, J.M., Jr.; et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: Two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am. J. Med. Genet. A 2012, 158, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Lin, A.E. Costello syndrome: A Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet. Med. 2012, 14, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Møller, R.S.; Weckhuysen, S.; Chipaux, M.; Marsan, E.; Taly, V.; Bebin, E.M.; Hiatt, S.M.; Prokop, J.W.; Bowling, K.M.; Mei, D.; et al. Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol. Genet. 2016, 2, e118. [Google Scholar] [CrossRef] [PubMed]
- Suarez, E.; Bertoli, M.J.; Eloy, J.D.; Shah, S.P. Case report and review of literature of a rare congenital disorder: Adams-Oliver syndrome. BMC Anesthesiol. 2021, 21, 117. [Google Scholar] [CrossRef]
- Morishita, K.; Petty, R.E. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. S5), v19–v25. [Google Scholar] [CrossRef]
- Galimberti, C.; Madeo, A.; Di Rocco, M.; Fiumara, A. Mucopolysaccharidoses: Early diagnostic signs in infants and children. Ital. J. Pediatr. 2018, 44 (Suppl. S2), 133. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. Heritable Disorders of Connective Tissue, 4th ed.; C.V. Mosby Co.: St. Louis, MO, USA, 1972. [Google Scholar]
- Litjens, T.; Morris, C.P.; Robertson, E.F.; Peters, C.; von Figura, K.; Hopwood, J.J. An N-acetylgalactosamine-4-sulfatase mutation (delta G238) results in a severe Maroteaux-Lamy phenotype. Hum. Mutat. 1992, 1, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Umeki, I.; Niihori, T.; Abe, T.; Kanno, S.; Okamoto, N.; Mizuno, S.; Kurosawa, K.; Nagasaki, K.; Yoshida, M.; Ohashi, H.; et al. Delineation of LZTR1 mutation-positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1-PPP1CB complexes. Hum. Genet. 2019, 138, 21–35. [Google Scholar] [CrossRef] [PubMed]
- van Der Burgt, I.; Brunner, H. Genetic heterogeneity in Noonan syndrome: Evidence for an autosomal recessive form. Am. J. Med. Genet. 2000, 94, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Dreger, C.K.; Olins, A.L.; Olins, D.E.; Shultz, L.D.; Lucke, B.; Karl, H.; Kaps, R.; Muller, D.; Vaya, A.; et al. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly). Nat. Genet. 2002, 31, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Sperling, K.; Olins, A.L.; Olins, D.E. The granulocyte nucleus and lamin B receptor: Avoiding the ovoid. Chromosoma 2007, 116, 227–235. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, I.; de Almeida, S.; Tiziani, V.; Raposo Do Amaral, C.M.; Gowrishankar, K.; Passos-Bueno, M.R.; Reichenberger, E.J. A novel autosomal recessive GJA1 missense mutation linked to craniometaphyseal dysplasia. PLoS ONE 2013, 8, e73576. [Google Scholar] [CrossRef] [PubMed]
- Kara, B.; Ekinci, Z.; Sahin, S.; Gungor, M.; Gunes, A.S.; Ozturk, K.; Adrovic, A.; Cefle, A.; Inanç, M.; Gul, A.; et al. Monogenic lupus due to spondyloenchondrodysplasia with spastic paraparesis and intracranial calcification: Case-based review. Rheumatol. Int. 2020, 40, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Briggs, T.A.; Rice, G.I.; Adib, N.; Ades, L.; Barete, S.; Baskar, K.; Baudouin, V.; Cebeci, A.N.; Clapuyt, P.; Coman, D.; et al. Spondyloenchondrodysplasia Due to Mutations in ACP5: A Comprehensive Survey. J. Clin. Immunol. 2016, 36, 220–234. [Google Scholar] [CrossRef]
- Hong, S.W.; Huh, K.H.; Lee, J.K.; Kang, J.H. Craniofacial anomalies associated with spondyloenchondrodysplasia: Two case reports. Medicine 2018, 97, e13644. [Google Scholar] [CrossRef]
- Feigenbaum, A.; Müller, C.; Yale, C.; Kleinheinz, J.; Jezewski, P.; Kehl, H.G.; MacDougall, M.; Rutsch, F.; Hennekam, R.C. Singleton-Merten syndrome: An autosomal dominant disorder with variable expression. Am. J. Med. Genet. A 2013, 161, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Rauch, F.; Fahiminiya, S.; Majewski, J.; Carrot-Zhang, J.; Boudko, S.; Glorieux, F.; Mort, J.S.; Bächinger, H.P.; Moffatt, P. Cole-Carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am. J. Hum. Genet. 2015, 96, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Bursztejn, A.C.; Briggs, T.A.; del Toro Duany, Y.; Anderson, B.H.; O’Sullivan, J.; Williams, S.G.; Bodemer, C.; Fraitag, S.; Gebhard, F.; Leheup, B.; et al. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: Overlap between Aicardi-Goutières and Singleton-Merten syndromes. Br. J. Dermatol. 2015, 173, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Mankin, H.J.; Jupiter, J.; Trahan, C.A. Hand and foot abnormalities associated with genetic diseases. Hand 2011, 6, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Gregory, L.C.; Shah, P.; Sanner, J.R.F.; Arancibia, M.; Hurst, J.; Jones, W.D.; Spoudeas, H.; Le Quesne Stabej, P.; Williams, H.J.; Ocaka, L.A.; et al. Mutations in MAGEL2 and L1CAM Are Associated With Congenital Hypopituitarism and Arthrogryposis. J. Clin. Endocrinol. Metab. 2019, 104, 5737–5750. [Google Scholar] [CrossRef] [PubMed]
- Volpi, S.; Picco, P.; Caorsi, R.; Candotti, F.; Gattorno, M. Type I interferonopathies in pediatric rheumatology. Pediatr. Rheumatol. Online J. 2016, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.A.; Kim, E.K.; Now, H.; Nguyen, N.T.; Kim, W.J.; Yoo, J.Y.; Lee, J.; Jeong, Y.M.; Kim, C.H.; Kim, O.H.; et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 266–274. [Google Scholar] [CrossRef]
- Karalis, V.; Bateup, H.S. Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 2021, 43, 143–158. [Google Scholar] [CrossRef]
- Welzel, T.; Kuemmerle-Deschner, J.B. Diagnosis and Management of the Cryopyrin-Associated Periodic Syndromes (CAPS): What Do We Know Today? J. Clin. Med. 2021, 10, 128. [Google Scholar] [CrossRef]
- Brennenstuhl, H.; Nashawi, M.; Schröter, J.; Baronio, F.; Beedgen, L.; Gleich, F.; Jeltsch, K.; von Landenberg, C.; Martini, S.; Simon, A.; et al. Unified Registry for Inherited Metabolic Disorders (U-IMD) Consortium and the European Registry for Hereditary Metabolic Disorders (MetabERN). Phenotypic diversity, disease progression, and pathogenicity of MVK missense variants in mevalonic aciduria. J. Inherit. Metab. Dis. 2021, 44, 1272–1287. [Google Scholar] [CrossRef]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Cirstea, I.C.; Kutsche, K.; Dvorsky, R.; Gremer, L.; Carta, C.; Horn, D.; Roberts, A.E.; Lepri, F.; Merbitz-Zahradnik, T.; König, R.; et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat. Genet. 2010, 42, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Yoo, H.-W. Noonan syndrome and RASopathies: Clinical features, diagnosis and management. J. Genet. Med. 2019, 16, 1–9. [Google Scholar] [CrossRef]
- Sukalo, M.; Tilsen, F.; Kayserili, H.; Müller, D.; Tüysüz, B.; Ruddy, D.M.; Wakeling, E.; Ørstavik, K.H.; Snape, K.M.; Trembath, R.; et al. DOCK6 mutations are responsible for a distinct autosomal-recessive variant of Adams-Oliver syndrome associated with brain and eye anomalies. Hum. Mutat. 2015, 36, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Park, S.; Gavazzi, F.; Adang, L.A.; Ayuk, L.A.; Van Eyck, L.; Seabra, L.; Barrea, C.; Battini, R.; Belot, A.; et al. Genetic and phenotypic spectrum associated with IFIH1 gain-of-function. Hum. Mutat. 2020, 41, 837–849. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Xia, Y.; Yang, J. Systemic inflammation and chronic kidney disease in a patient due to the RNASEH2B defect. Pediatr. Rheumatol. Online J. 2021, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Goutières, F.; Aicardi, J.; Barth, P.G.; Lebon, P. Aicardi-Goutières syndrome: An update and results of interferon-alpha studies. Ann. Neurol. 1998, 44, 900–907. [Google Scholar] [CrossRef]
- Tolmie, J.L.; Shillito, P.; Hughes-Benzie, R.; Stephenson, J.B. The Aicardi-Goutières syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis). J. Med. Genet. 1995, 32, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Walor, D.M.; Berdon, W.E.; Westra, S.J. ‘Hair-on-end’ skull changes resembling thalassemia caused by marrow expansion in uncorrected complex cyanotic heart disease. Pediatr. Radiol. 2005, 35, 698–701. [Google Scholar] [CrossRef]
- Boros, C.A.; Spence, D.; Blaser, S.; Silverman, E.D. Hydrocephalus and macrocephaly: New manifestations of neonatal lupus erythematosus. Arthritis Rheum. 2007, 57, 261–266. [Google Scholar] [CrossRef]
- Tirosh, I.; Spielman, S.; Barel, O.; Ram, R.; Stauber, T.; Paret, G.; Rubinsthein, M.; Pessach, I.M.; Gerstein, M.; Anikster, Y.; et al. Whole exome sequencing in childhood-onset lupus frequently detects single gene etiologies. Pediatr. Rheumatol. Online J. 2019, 17, 52. [Google Scholar] [CrossRef]
- Palmucci, S.; Attinà, G.; Lanza, M.L.; Belfiore, G.; Cappello, G.; Foti, P.V.; Milone, P.; Di Bella, D.; Barone, R.; Fiumara, A.; et al. Imaging findings of mucopolysaccharidoses: A pictorial review. Insights Imaging 2013, 4, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Bedair, E.M.A.; Helmy, A.N.; Yakout, K.; Soliman, A.T. Review of radiologic skeletal changes in thalassemia. Pediatr. Endocrinol. 2008, 6 (Suppl. S1), 123–126. [Google Scholar]
- Rizzuto, V.; Koopmann, T.T.; Blanco-Álvarez, A.; Tazón-Vega, B.; Idrizovic, A.; Díaz de Heredia, C.; Del Orbe, R.; Pampliega, M.V.; Velasco, P.; Beneitez, D.; et al. Usefulness of NGS for Diagnosis of Dominant Beta-Thalassemia and Unstable Hemoglobinopathies in Five Clinical Cases. Front. Physiol. 2021, 12, 628236. [Google Scholar] [CrossRef]
- Caimi, G.; Canino, B.; Lo Presti, R.; Urso, C.; Hopps, E. Clinical conditions responsible for hyperviscosity and skin ulcers complications. Clin. Hemorheol. Microcirc. 2017, 67, 25–34. [Google Scholar] [CrossRef]
- Brioude, F.; Toutain, A.; Giabicani, E.; Cottereau, E.; Cormier-Daire, V.; Netchine, I. Overgrowth syndromes—Clinical and molecular aspects and tumour risk. Nat. Rev. Endocrinol. 2019, 15, 299–311. [Google Scholar] [CrossRef]
- Ivars, M.; Boixeda, P.; Triana, P.; Martinez-Glez, V.; Rodríguez-Laguna, L.; Agra, N.; López-Gutiérrez, J.C. Clinical overlap between CLAPO syndrome and macrocephaly-capillary malformation syndrome. J. Dtsch. Dermatol. Ges. 2020, 18, 479–482. [Google Scholar] [CrossRef]
- Rodriguez-Laguna, L.; Ibañez, K.; Gordo, G.; Garcia-Minaur, S.; Santos-Simarro, F.; Agra, N.; Vallespín, E.; Fernández-Montaño, V.E.; Martín-Arenas, R.; Del Pozo, Á.; et al. CLAPO syndrome: Identification of somatic activating PIK3CA mutations and delineation of the natural history and phenotype. Genet. Med. 2018, 20, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Keppler-Noreuil, K.M.; Rios, J.J.; Parker, V.E.; Semple, R.K.; Lindhurst, M.J.; Sapp, J.C.; Alomari, A.; Ezaki, M.; Dobyns, W.; Biesecker, L.G. PIK3CA-related overgrowth spectrum (PROS): Diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am. J. Hum. Genet. Part A 2015, 167, 287–295. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Riller, Q.; Rieux-Laucat, F. RASopathies: From germline mutations to somatic and multigenic diseases. Biomed. J. 2021, 44, 422–432. [Google Scholar] [CrossRef]
- Brems, H.; Park, C.; Maertens, O.; Pemov, A.; Messiaen, L.; Upadhyaya, M.; Claes, K.; Beert, E.; Peeters, K.; Mautner, V.; et al. Glomus tumors in neurofibromatosis type 1: Genetic, functional, and clinical evidence of a novel association. Cancer Res. 2009, 69, 7393–7401. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.R.; Sloan, J.L.; Yao, L.; Mannes, A.J.; Moshyedi, A.; Lee, C.C.; Sciot, R.; De Smet, L.; Mautner, V.F.; Legius, E. Diagnosis, management, and complications of glomus tumours of the digits in neurofibromatosis type 1. J. Med. Genet. 2010, 47, 525–532. [Google Scholar] [CrossRef] [PubMed]
- İncecik, F.; Hergüner, M.Ö.; Ballı, T.; Altunbaşak, Ş. Pseudoarthrosis of the hand in neurofibromatosis type 1: A case report. Turk J. Pediatr. 2013, 55, 335–336. [Google Scholar] [PubMed]
- Denayer, E.; Legius, E. Legius Syndrome and its Relationship with Neurofibromatosis Type 1. Acta Derm. Venereol. 2020, 100, adv00093. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.B.; Moraes, R.A.; Pereira, L.B. Cardiofaciocutaneous syndrome and the dermatologist’s contribution to diagnosis. Cutis 2017, 99, E4–E7. [Google Scholar] [PubMed]
- Kimonis, V.E.; Singh, K.E.; Zhong, R.; Pastakia, B.; Digiovanna, J.J.; Bale, S.J. Clinical and radiological features in young individuals with nevoid basal cell carcinoma syndrome. Genet. Med. 2013, 15, 79–83. [Google Scholar] [CrossRef]
- Thomas-Sohl, K.A.; Vaslow, D.F.; Maria, B.L. Sturge-Weber syndrome: A review. Pediatr. Neurol. 2004, 30, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.R.S.; Duis, J.; Kulungowski, A.M.; Annam, A.; Siegele, B.; Nakano, T.A. A Challenging Diagnosis: PTEN Hamartoma Tumor Syndrome Presenting as Isolated Soft-tissue Vascular Anomalies. J. Vasc. Anom. 2021, 2, e011. [Google Scholar] [CrossRef]
- Giorgianni, A.; Pellegrino, C.; De Benedictis, A.; Mercuri, A.; Baruzzi, F.; Minotto, R.; Tabano, A.; Balbi, S. Lhermitte-Duclos disease. A case report. Neuroradiol. J. 2013, 26, 655–660. [Google Scholar] [CrossRef]
- Mirzaa, G.; Graham, J.M., Jr.; Keppler-Noreuil, K. PIK3CA-Related Overgrowth Spectrum. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 2013. [Google Scholar]
- Damian, L.; Lebovici, A.; Pamfil, C.; Belizna, C.; Vulturar, R. Rheumatoid Arthritis and CLOVES Syndrome: A Tricky Diagnosis. Diagnostics 2020, 10, 467. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lin, L.; Xue, Y.; Wang, Y.; Liu, Z.; Ou, Z.; Wu, S.; Lan, X.; Zhang, Y.; Yuan, F.; et al. MED12-Related Disease in a Chinese Girl: Clinical Characteristics and Underlying Mechanism. Front. Genet. 2020, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Humayun, M.; Haider, I.; Ayub, M. Lujan-Fryns Syndrome (LFS): A Unique Combination of Hypernasality, Marfanoid Body Habitus, and Neuropsychiatric Issues, Presenting as Acute-Onset Dysphagia. Clin. Med. Insights Case Rep. 2016, 9, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Isikay, S.; Yilmaz, K. Congenital cytomegalovirus infection and finger anomaly. BMJ Case Rep. 2013, 16, bcr2013009486. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wu, H.; Yin, H.; Chang, J.; Wang, L.; Wang, R.; Ma, W.; Li, Y.; Guan, J.; Liu, J.; et al. Management of hydrocephalus associated with autoimmune diseases: A series of 19 cases. Autoimmunity 2017, 50, 422–427. [Google Scholar] [CrossRef]
- Meyts, I.; Aksentijevich, I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J. Clin. Immunol. 2018, 38, 569–578. [Google Scholar] [CrossRef]
- d’Angelo, D.M.; Di Filippo, P.; Breda, L.; Chiarelli, F. Type I Interferonopathies in Children: An Overview. Front. Pediatr. 2021, 9, 631329. [Google Scholar] [CrossRef]
- Crow, Y.J. Type I interferonopathies: A novel set of inborn errors of immunity. Ann. N. Y. Acad. Sci. 2011, 1238, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Alsohime, F.; Martin-Fernandez, M.; Temsah, M.H.; Alabdulhafid, M.; Le Voyer, T.; Alghamdi, M.; Qiu, X.; Alotaibi, N.; Alkahtani, A.; Buta, S.; et al. JAK Inhibitor Therapy in a Child with Inherited USP18 Deficiency. N. Eng. J. Med. 2020, 382, 256–265. [Google Scholar] [CrossRef]
- Mauro, A.; Omoyinmi, E.; Sebire, N.J.; Barnicoat, A.; Brogan, P. De Novo PTEN Mutation in a Young Boy with Cutaneous Vasculitis. Case Rep. Pediatr. 2017, 2017, 9682803. [Google Scholar] [CrossRef]
- Heindl, M.; Händel, N.; Ngeow, J.; Kionke, J.; Wittekind, C.; Kamprad, M.; Rensing-Ehl, A.; Ehl, S.; Reifenberger, J.; Loddenkemper, C.; et al. Autoimmunity, intestinal lymphoid hyperplasia, and defects in mucosal B-cell homeostasis in patients with PTEN hamartoma tumor syndrome. Gastroenterology 2012, 142, 1093–1096.e6. [Google Scholar] [CrossRef]
- Kamien, B.; Ronan, A.; Poke, G.; Sinnerbrink, I.; Baynam, G.; Ward, M.; Gibson, W.T.; Dudding-Byth, T.; Scott, R.J. A Clinical Review of Generalized Overgrowth Syndromes in the Era of Massively Parallel Sequencing. Mol. Syndromol. 2018, 9, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H. Recent Advances and Contradictions in the Study of the Individual Roles of Ubiquitin Ligases That Regulate RIG-I-Like Receptor-Mediated Antiviral Innate Immune Responses. Front. Immunol. 2020, 11, 1296. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; Pineda-Alvarez, D.E.; Raam, M.S.; Bous, S.M.; Keaton, A.A.; Vélez, J.I.; Cummings, D.A. Analysis of component findings in 79 patients diagnosed with VACTERL association. Am. J. Med. Genet. A 2010, 152, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Iafolla, A.K.; McConkie-Rosell, A.; Chen, Y.T. VATER and hydrocephalus: Distinct syndrome? Am. J. Med. Genet. 1991, 38, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Duran, I.; Nevarez, L.; Sarukhanov, A.; Wu, S.; Lee, K.; Krejci, P.; Weis, M.; Eyre, D.; Krakow, D.; Cohn, D.H. HSP47 and FKBP65 cooperate in the synthesis of type I procollagen. Hum. Molec. Genet. 2015, 24, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Malm, D.; Nilssen, Ø. Alpha-mannosidosis. Orphanet J. Rare Dis. 2008, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Proud, V.K.; Braddock, S.R.; Cook, L.; Weaver, D.D. Weaver syndrome: Autosomal dominant inheritance of the disorder. Am. J. Med. Genet. 1998, 79, 305–310. [Google Scholar] [CrossRef]
- Fan, Y.; Yin, W.; Hu, B.; Kline, A.D.; Zhang, V.W.; Liang, D.; Sun, Y.; Wang, L.; Tang, S.; Powis, Z.; et al. De novo mutations of CCNK cause a syndromic neurodevelopmental disorder with distinctive facial dysmorphism. Am. J. Hum. Genet. 2018, 103, 448–455. [Google Scholar] [CrossRef]
- Kerzendorfer, C.; Whibley, A.; Carpenter, G.; Outwin, E.; Chiang, S.-C.; Turner, G.; Schwartz, C.; El-Khamisy, S.; Raymond, F.L.; O’Driscoll, M. Mutations in Cullin 4B result in a human syndrome associated with increased camptothecin-induced topoisomerase I-dependent DNA breaks. Hum. Molec. Genet. 2010, 19, 1324–1334. [Google Scholar] [CrossRef]
- Chong, J.X.; Yu, J.-H.; Lorentzen, P.; Park, K.M.; Jamal, S.M.; Tabor, H.K.; Rauch, A.; Sifuentes Saenz, M.; Boltshauser, E.; Patterson, K.E.; et al. Gene discovery for Mendelian conditions via social networking: De novo variants in KDM1A cause developmental delay and distinctive facial features. Genet. Med. 2016, 18, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, F.; Brundage, E.; Scheuerle, A.; Lanpher, B.; Erickson, R.P.; Powis, Z.; Robinson, H.B.; Trapane, P.L.; Stachiw-Hietpas, D.; et al. Genomic duplication resulting in increased copy number of genes encoding the sister chromatid cohesion complex conveys clinical consequences distinct from Cornelia de Lange (Letter). J. Med. Genet. 2009, 46, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Disciglio, V.; Lo Rizzo, C.; Mencarelli, M.A.; Mucciolo, M.; Marozza, A.; Di Marco, C.; Massarelli, A.; Canocchi, V.; Baldassarri, M.; Ndoni, E.; et al. Interstitial 22q13 deletions not involving SHANK3 gene: A new contiguous gene syndrome. Am. J. Med. Genet. 2014, 164, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Wambach, J.A.; Wegner, D.J.; Patni, N.; Kircher, M.; Willing, M.C.; Baldridge, D.; Xing, C.; Agarwal, A.K.; Vergano, S.A.S.; Patel, C.; et al. Bi-allelic POLR3A Loss-of-Function Variants Cause Autosomal-Recessive Wiedemann-Rautenstrauch Syndrome. Am. J. Hum. Genet. 2018, 103, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xie, Y.A.; Abouzeid, H.; Gordon, C.T.; Fiorentino, A.; Sun, Z.; Lehman, A.; Osman, I.S.; Dharmat, R.; Riveiro-Alvarez, R.; et al. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am. J. Hum. Genet. 2017, 100, 592–604. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Hirth, L.; Singh, S.; Schilling, S.; Müller, E.; Goedde, H.W. Dermatoglyphic findings in patients with fragile X-chromosome. Clin. Genet. 1985, 27, 118–121. [Google Scholar] [CrossRef]
- Lachiewicz, A.M.; Dawson, D.V.; Spiridigliozzi, G.A. Physical characteristics of young boys with fragile X syndrome: Reasons for difficulties in making a diagnosis in young males. Am. J. Med. Genet. 2000, 92, 229–236. [Google Scholar] [CrossRef]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X syndrome: A review of clinical and molecular diagnoses. Ital. J. Pediatr. 2017, 43, 39. [Google Scholar] [CrossRef]
- Sousa, S.B.; Ramos, F.; Garcia, P.; Pais, R.P.; Paiva, C.; Beales, P.L.; Moore, G.E.; Saraiva, J.M.; Hennekam, R.C. Intellectual disability, coarse face, relative macrocephaly, and cerebellar hypotrophy in two sisters. Am. J. Med. Genet. A 2014, 164, 10–14. [Google Scholar] [CrossRef]
- Knoblauch, H.; Tennstedt, C.; Brueck, W.; Hammer, H.; Vulliamy, T.; Dokal, I.; Lehmann, R.; Hanefeld, F.; Tinschert, S. Two brothers with findings resembling congenital intrauterine infection-like syndrome (pseudo-TORCH syndrome). Am. J. Med. Genet. 2003, 120, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, M.E.; Schot, R.; Buta, S.; Oudesluijs, G.; Tinschert, S.; Speer, S.D.; Li, Z.; van Unen, L.; Heijsman, D.; Goldmann, T.; et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 2016, 213, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, S.M.; Adam, L.; Atef, H.; Häberli, D.; Bramer, W.M.; Minder, B.; Döring, Y.; Laine, J.E.; Muka, T.; Rössler, J.; et al. A systematic review of the safety and efficacy of currently used treatment modalities in the treatment of patients with PIK3CA-related overgrowth spectrum. J. Vasc. Surg. Venous Lymphat. Disord. 2022, 10, 527–538.e2. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Espejo-Mojica, A.J.; Sánchez, O.F.; Ramírez, C.M.; Reyes, L.H.; Cruz, J.C.; Alméciga-Díaz, C.J. Lysosomal storage diseases: Current therapies and future alternatives. J. Mol. Med. 2020, 98, 931–946. [Google Scholar] [CrossRef]
- Crow, Y.J.; Stetson, D.B. The type I interferonopathies: 10 years on. Nat. Reviews. Immunol. 2022, 22, 471–483. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Upper Extremity Changes | Syndromes | Head Enlargement Type | References |
|---|---|---|---|
| Polydactyly | VACTERL-H syndrome | MC | [28] |
| Apert syndrome | MC | [29] | |
| Short-rib thoracic dysplasia 8 | MC | [30] | |
| Bardet–Biedl syndrome | MC | [30] | |
| Gorlin syndrome | MC, HC, FB | [31,32,33,34,35] | |
| GCPS | MC | [36] | |
| Acro-callosal syndrome | MC | [37] | |
| Joubert syndrome 2 | MC, FB | [38] | |
| Oligodactyly | VACTERL-H syndrome | MC | [28] |
| Digit malposition | BRMUTD (First finger insertion) | MC, HC | [39] |
| Finger tapering | Turnpenny—Fry syndrome | FB | [40] |
| Sifrim–Hitz–Weiss syndrome | MC | [41] | |
| Carey–Fineman–Ziter syndrome | MC | [42] | |
| Camptodactyly | Osteopathia striata with cranial sclerosis | MC | [43] |
| MC | [19] | ||
| Sotos syndrome | MC | [19,44] | |
| Weaver syndrome | MC | [45,46] | |
| Cohen–Gibson syndrome | MC | [47] | |
| Rahman syndrome | MC | [48] | |
| Alcuraya–Kucinskas syndrome | MC | [38] | |
| Joubert syndrome 2 | MC, FB | [49,50,51,52] | |
| Trichohepatoenteric syndrome | FB | [49,50,51,52] | |
| Clinodactyly | Keipert syndrome | MC | [53] |
| Osteopathia striata with cranial sclerosis | MC | [43] | |
| Peroxisome biogenesis disorder (Zellweger syndrome) | MC | [54,55] | |
| DDVIBA | MC | [56] | |
| Muenke craniosynostosis syndrome | MC | [57,58] | |
| Silver–Russel syndrome | MC | [59] | |
| SOFT syndrome | MC | [60] | |
| Larsen-like syndrome | MC | [61] | |
| CPRF | MC, FB | [48] | |
| Alcuraya–Kucinskas syndrome | MC | [62] | |
| Desmosterolosis | MC, FB | [42] | |
| Brachydactyly | Congenital CMV infection | MC | [63,64] |
| Robinow syndrome | MC | [65,66] | |
| Beta thalassemia | MC | [67,68] | |
| Pycnodysostosis | MC | [69] | |
| Pfeiffer syndrome | MC | [23] | |
| Gorlin syndrome | MC, FB | [31,32,33,34,35] | |
| Keipert syndrome | MC | [53] | |
| Simpson–Golabi–Behmel | MC | [70,71] | |
| Muenke craniosynostosis syndrome | MC | [57] | |
| SOFT syndrome | MC | [59] | |
| Temtamy syndrome | MC, FB | [72] | |
| Retinitis pigmentosa with or without skeletal anomalies | MC, FB | [42] | |
| Syndactyly | VACTERL-H syndrome | MC | [28] |
| Pfeiffer syndrome | MC | [23] | |
| Apert syndrome | MC, ME | [29] | |
| Gorlin syndrome | MC | [31,32,33,34,35] | |
| Simpson–Golabi–Behmel (second to third fingers) | MC | [73] | |
| GCPS (cutaneous syndactyly) | MC | [36] | |
| Arachnodactyly | MDFPMR | MC, FB | [74,75,76] |
| EDHHACC | MC, FB | [77] | |
| Divergent fingers | Achondroplasia (trident hand) | MC | [3] |
| CLOVES syndrome (wide spacing between digits) | MC, ME | [78,79] | |
| Digital webbing | Pfeiffer syndrome | MC | [23] |
| Apert syndrome | MC, ME | [29] | |
| Polyphalangy | VACTERL-H syndrome (triphalangeal thumb) | HC | [28] |
| Osteopathia striata with cranial sclerosis | MC | [43] | |
| Broad terminal phalanges (spatulate fingers) | Keipert syndrome | MC | [53] |
| Osteopathia striata with cranial sclerosis | MC | [43] | |
| Costello syndrome | MC | [80] | |
| Phalangeal hypoplasia | Robinow syndrome | MC | [65,66] |
| Joubert syndrome | MC | [30] | |
| Gorlin syndrome (First finger) | MC, HC, FB | [31,32,33,34,35] | |
| Smith–Kingsmore syndrome | MC, FB | [81] | |
| Simpson–Golabi–Behmel syndrome | MC | [73] | |
| Adams–Oliver syndrome | MC, HC | [82] | |
| Retinitis pigmentosa with or without skeletal anomalies | MC, FB | [42] | |
| Phalangeal form changes | Mucopolysaccharidoses are bullet-shaped (proximal pointing) | MC, HC | [83,84,85,86] |
| Muenke craniosynostosis syndrome (thimble-like middle phalanges) | |||
| MC | [57] | ||
| Brachymetacarpia (short metacarpal or metatarsal) | Noonan syndrome 2 (fifth finger) | MC | [87,88] |
| Gorlin syndrome (fourth finger) | MC, HC, FB | [31,32,33,34,35] | |
| Pelger–Huet anomaly | MC | [89,90] | |
| Bone changes | |||
| Radial hypo/aplasia | VACTERL-H syndrome | HC | [28] |
| Radioulnar synostosis | VACTERL-H syndrome | HC | [28] |
| Ulnar deviation | Peroxisome biogenesis disorder (Zellweger syndrome) | MC | [54,55] |
| Costello syndrome | MC | [80] | |
| Bone sclerosis | Craniometaphyseal dysplasia AR | MC | [91] |
| Dysostosis multiplex | Mucopolysaccharidoses | MC, HC | [83,84,85,86] |
| Skeletal dysplasia | SPENCDI | MiC/MC/FB | [70,92,93,94,95] |
| Singleton–Merten syndrome | MiC/MC/FB | ||
| Enchondromas | SPENCDI | MiC/MC/FB | [92,93,94] |
| Acroosteolysis | Picnodysostosis | MC | [69] |
| Cole–Carpenter syndrome | HC, FB | [96] | |
| Singleton–Merten syndrome | MiC/MC/FB | [95,97] | |
| Joint changes | |||
| Joint contractures | Mucopolysaccharidosis | MC, HC | [83,84,85,86] |
| Gangliosidosis type I | MC | [8,98] | |
| Peroxisome biogenesis disorder (Zellweger syndrome) | MC | [54,55] | |
| Noonan syndrome type 2 | MC | [87,88] | |
| L1 syndrome | HC | [99] | |
| Desmosterolosis | MC | [48] | |
| Carey–Fineman–Ziter | MC | [42] | |
| Singleton–Merten syndrome | MiC/MC, FB | [95,100,101] | |
| Joint hypermobility | Osteogenesis imperfecta | MC | [96] |
| Osteopathia striata with cranial sclerosis | MC, FB | [43] | |
| Pycnodysostosis | MC | [69] | |
| Pretzel syndrome | ME | [102] | |
| Cohen–Gibson syndrome | MC | [45,46] | |
| Tatton–Brown–Rahman syndrome | MC | [17] | |
| MDFPMR | MC, FB | [74,75,76] | |
| Small joint arthritis | Cryopyrinopathies: CINCA/NOMID, Muckle–Wells etc. | FB, HC | [103] |
| MVK deficiency | FB | [104] | |
| Beta-thalassemia | MC | [67,68] | |
| Villonodular synovitis, multiple sites | Noonan syndrome | MC | [105,106] |
| Carpal tunnel syndrome | Mucopolysaccharidoses | MC, HC | [83,84,85,86] |
| Nail changes | |||
| Nail dystrophy | Nail-patella syndrome | MC | [98] |
| Robinow syndrome | MC | [65,66] | |
| Costello syndrome | MC | [80] | |
| Adams–Oliver syndrome | MC, HC | [82] | |
| SOFT syndrome | MC | [59] | |
| Primrose syndrome | MC | [61] | |
| Single nail common to more digits | Apert syndrome | MC, FB, ME | [29] |
| Skin changes | |||
| Thickened skin | Gangliosidosis type I | MC | [8,98] |
| Dry, hyperkeratotic skin | Cardiofaciocutaneous syndrome | MC | [107] |
| Wrinkled skin | Costello syndrome | MC | [80] |
| Pitted hands | Gorlin syndrome | MC, HC, FB | [31,32,33,34,35] |
| Deep palmar creases | Smith–Kingsmore syndrome | MC | [81] |
| Single palmar creases | Adams–Oliver 2 syndrome | MC | [108] |
| Vascular, lymphatic, and other changes | |||
| Finger necrosis/amputation | Interferonopathies | MiC/MC, FB | [109,110,111,112] |
| Acrocyanosis | Congenital heart disease | MC | [113] |
| Aicardi–Goutières | MC/MiC | [111,112] | |
| Hand and feet edema | Mucopolysaccharidosis VI | MC | [83,84,85,86] |
| Noonan syndrome | MC | [105,106] | |
| Raynaud’s phenomenon | Neonatal SLE | MC | [114,115] |
| Interferonopathies | MiC/MC | [109,110] | |
| Soft tissue masses | PROS | MC, HC, FB | [31,32,33,34,35] |
| Disease | Inheritance | Causes/Mutations | Main Craniofacial and Hand/Finger Changes | References |
|---|---|---|---|---|
| Acquired conditions | ||||
| Rickets | Vitamin D deficiency | MC, FB, broad front, delayed closure of fontanelles, craniotabes, thickened wrists | [3] | |
| Congenital infection | Cytomegalovirus infection | HC, cortical development abnormalities, brachydactyly | [63,64] | |
| Bone marrow disorders | ||||
| Beta-thalassemia OMIM # 613985 | AR (AD) | HBB | Prominent frontal and maxillary bones, sausage-like digits, small joints arthritis, fractures, signs of hypovitaminosis D | [67,68] |
| Congenital cyanotic heart diseases | MC with “hair-on-end” radiological skull changes, acrocyanosis | [113] | ||
| Skeletal dysplasias | ||||
| Achondroplasia OMIM # 100800 | AD | FGFR3 | MC, short fingers with divergent ring and middle fingers (trident hand) | [98] |
| Pycnodysostosis OMIM # 265800 | AR | CTSK | MC, acro-osteolysis of the terminal phalanges, short fingers, joint hypermobility | [69] |
| Cole–Carpenter syndrome | AR | P4HB | HC, frontal bossing, craniosynostosis, ocular proptosis, frequent fractures, wide metacarpal and phalangeal epiphyses, cystic appearance, acro-osteolysis | [96] |
| VACTERL-H, VACTERL association, X-linked, with or without hydrocephalus; VACTERLX, OMIM # 314390 | XLR | SHH signaling, GLI3 TBX-SALL4-SALL1-WNT pathway FGF8-FGF10 pathway | HC, radial hypo/aplasia, triphalangeal thumb, polydactyly, oligodactyly, syndactyly, radioulnar synostosis | [28,149,150] |
| Robinow syndrome OMIM # 268310 | AR/AD | ROR2, WNT5A, DVL1, DVL3 | MC, FB, limb shortening, brachydactyly, phalangeal and nail hypoplasia | [65,66] |
| Osteopathia striata with cranial sclerosis (OSCS) OMIM # 300373 | XLD | AMER1 | MC, FB, long, slender fingers, fifth finger clinodactyly, camptodactyly, finger contractures, duplicate phalanges, spatulate distal phalanges | [43] |
| Pfeiffer syndrome, OMIM # 101600 | AD | FGFR1 FGFR2 | Cloverleaf skull, high forehead, syndactyly, brachydactyly, digital webbing | [23] |
| Apert syndrome OMIM # 101200 | AD | FGFR2 | ME, high, broad forehead, single nail common to digits 2 to 4, symmetric osseous and/or cutaneous syndactyly, polydactyly | [29] |
| Muenke craniosynostosis Syndrome (MNKES), OMIM # 602849 | AD | FGFR3 | MC, plagiocephaly, brachycephaly, premature suture closure, midface retrusion, hypertelorism, clinodactyly, brachydactyly, mild hand and feet anomalies | [23] |
| Craniometaphyseal dysplasia, autosomal recessive (CMDR) OMIM #218400 | AR | GJA1 | MC, coarse facial features, metacarpal and phalangeal sclerosis | [91] |
| Osteogenesis Imperfecta type X | AR | SERPINH1 | MC, high forehead, triangular face, midface hypoplasia, hyperextensibility of the fingers | [151] |
| Keipert syndrome OMIM # 301026 | XLR | GPC4 | MC, hypertelorism, flat midface, prominent lips, brachydactyly, clinodactyly, broad terminal phalanges | [53] |
| Craniometaphyseal dysplasia, autosomal recessive (CMDR) OMIM #218400 | AR | GJA1 | MC, coarse facial features, metacarpal and phalangeal sclerosis | [91] |
| Ciliopathies | ||||
| Short-rib thoracic dysplasia 8 with or without polydactyly (SRTD8) OMIM # 615503 | AR | WDR60 | MC, polydactyly, skin changes | [30] |
| Bardet–Biedl syndrome OMIM # 617119 | AR | IFT74 | MC (or MiC), polydactyly | [30] |
| Joubert syndrome OMIM # 213300 | AR | INPP5E | MC, prominent forehead, high rounded eyebrows, missing digital phalanges | [30] |
| Inherited metabolic disorders | ||||
| Mucopolysaccharidoses MPS I (Hurler syndrome) OMIM # 607014 MPS II (Hunter syndrome), OMIM # 309900 MPS VI (Maroteaux-Lamy), OMIM # 253200 MPS VII (Sly syndrome), OMIM # 253220 | MPS I: AR, MPS II: X-linked, MPS VI: AR, MPS VII: AR | MPS I: IDUA MPS II: IDS MPS VI: ARSB, MPS VII: GUSB | MPS I: large head with bulging frontal bones (MC, HC), carpal tunnel syndrome with weakness in the hand and fingers, phalanges are bullet-shaped with proximal pointing of the second to fifth metacarpals. MPS II: MC, claw hands, stiffness, joint contractures, carpal tunnel syndrome, joint hypermobility, dysostosis multiplex. MPS VI: coarse dysmorphic features, HC, edema of the hands and feet, dysostosis multiplex. MPS VII (mild form): MC, mild craniofacial dysmorphism, dysostosis multiplex | [83,84,85,86] |
| Gangliosidosis type I OMIM # 230500 | AR | GLB1 | MC, claw hands, thickened subcutaneous tissues | [8,98] |
| Alpha-mannosidosis type I (Hurler-like disease) OMIM # 248500 | AR | MAN2B1 | HC, large head with prominent forehead, dysostosis multiplex | [152] |
| Peroxisome biogenesis disorder (cerebrohepatorenal/ Zellweger syndrome) OMIM # 214100 | AR | PEX1, PEX3, PEX6, PEX16, PEX2, PEX12, PEX14, | MC (or MiC), dysmorphic features (large anterior fontanel, prominent high forehead), finger flexion; long fingers, deviated to the ulnar side, the thumbs were not held in apposition | [54,55] |
| RASopathies | ||||
| Costello syndrome OMIM # 218040 | AD | HRAS | Coarse facies, wrinkled skin, splayed spatulate fingers, abnormal nails, ulnar deviation | [80] |
| Cardio-facio-cutaneous syndrome OMIM # 115150 | AD | BRAF, MAP2K1, MAP2K2, KRAS | Joint contractures, dry, hyperkeratotic scaly skin, ulnar deviation, deep palmar creases | [107] |
| Noonan syndrome (NS) NS 1, OMIM # 163950 NS 3, OMIM # 609942 NS 4, OMIM # 610733 NS 5, OMIM # 611553 NS 6, OMIM # 613224 | AD | PTPN11, KRAS, SOS1, RAF1, NRAS, | Broad forehead, dolichocephaly, polyarticular pigmented villonodular synovitis, peripheral lymphedema | [105,106] |
| Noonan syndrome (NS) 2 OMIM # 605275 | AR | LZTR1 | Broad forehead, fifth brachymetapody, arthrogryposis | [87,88] |
| Overgrowth syndromes | ||||
| Gorlin–Goltz syndrome/ Gorlin syndrome/ [Nevoid basal cell carcinoma syndrome (NBCCS)] OMIM # 109400 | AD | PTCH1 | MC/ relative MC, mild HC, FB, pitted hands, brachydactyly, short fourth metacarpal, polydactyly, 2–3 syndactyly, short thumb terminal phalanx | [31,32,33,34,35] |
| PTEN hamartoma tumor syndrome: Cowden’s syndrome, Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos syndrome OMIM # 158350 | AD | PTEN | MC/ME, HC, asymmetric soft-tissue masses, increased fat deposition, enlarged vessels MC/ME, HC, cerebellar signs, papilledema | [133] [134] |
| Smith–Kingsmore syndrome (SKS), Minds syndrome OMIM # 616638 | AD | MTOR | MC, FB, tall forehead, midface hypoplasia, short proximal and distal phalanges, deep palmar creases | [81] |
| CLOVES syndrome, OMIM # 612918 Proteus syndrome, OMIM # 176920 MCAP syndrome, OMIM # 602501 MPPH syndrome 1 OMIM # 603387 | AD, somatic mutations, arise randomly in one cell during embryonic development | PI3K/AKT/mTOR pathway (CCND2, PIK3R2, AKT3, PIK3CA, MCC, NSD1) | MC, ME and capillary malformations, asymmetric overgrowth of the extremities, wide spacing between digits, lymphatic anomalies | [78,79] |
| Sotos syndrome OMIM # 117550 | AD | NSD1 | MC, high broad forehead, long face, prominent chin, advanced bone age, camptodactyly | [19] |
| Weaver syndrome OMIM # 277590 | AD | EZH2, NSD1 | MC, camptodactyly of the fingers and/or toes, hyperextensibility of the fingers, finger contractures, thin, deep-set nails, boutonniere deformity in adults | [19,44,153] |
| Malan syndrome OMIM # 277590 | AD | NFIX | MC, long and narrow triangular face, prognathia, aortopathy, advanced bone age, scoliosis, long hands, ID | [20] |
| * Polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome, Pretzel syndrome (PMSE) OMIM # 611087 | AR | STRAD-alpha (LYK5) | ME, cognitive delay, hyperextensible fingers | [102] |
| Simpson–Golabi–Behmel syndrome (SGBS1) OMIM # 312870 | X-linked | GPC3, GPC4 | MC, pre- and postnatal overgrowth, coarse facies, index finger hypoplasia, syndactyly second to third fingers, brachydactyly, broad hands, polydactyly | [70,71,73] |
| Cohen–Gibson syndrome (COGIS) OMIM # 617561 | AD | EED | MC, broad forehead, long fingers, broad thumbs, camptodactyly, joint laxity of the small joints of the hand | [45,46] |
| Rahman syndrome (RMNS) OMIM # 617537 | AD | HIST1H1E (H1-4) | Increased height and/or head circumference early in life, camptodactyly | [47] |
| Tatton–Brown–Rahman syndrome (TBRS) OMIM #615879 | AD | DNMT3A | MC, round facies, bushy eyebrows, prominent maxillary incisors, joint hyperlaxity | [17] |
| Intellectual developmental disorder with hypertelorism and distinctive facies OMIM # 618147 | AD | CCNK | MC, high anterior hairline, tapered fingers | [154] |
| Brain malformations with or without urinary tract defects (BRMUTD) OMIM # 613735 | AD | NFIA | MC, ventriculomegaly, overgrowth, bilateral proximally placed first fingers | [39] |
| Developmental Delay with Variable Intellectual Impairment and Behavioral Abnormalities (DDVIBA) OMIM # 618430 | AD | TCF20 | MC, brachycephaly, FB, Tapering fingers, fifth finger clinodactyly | [56] |
| Other inherited causes | ||||
| L1 syndrome OMIM # 307000 | XLR | L1CAM | HC, arthrogryposis, adducted thumbs, developmental delay | [99] |
| Adams–Oliver syndrome (AOS1) OMIM # 100300 | AD/ AR | NOTCH1, ARHGAP31, DOCK6, EOGT, DLL4, or RBPJ | Encephalocele, ventriculomegaly, slight ventricular dilation, periventricular leukomalacia, short or missing phalanges, dysplastic or absent nails | [82] |
| Adams–Oliver syndrome-2 (AOS2) OMIM # 614219 | AR | DOCK6 | MC (or MiC), mild facial dysmorphism, low hair line, shortened digits, single palmar creases | [108] |
| Macrocephaly, dysmorphic facies, and psychomotor retardation (MDFPMR) OMIM # 617011 | AR | HERC1 | MC, FB, somatic overgrowth apparent at birth, seizures, joint laxity, and long fingers; large hands with arachnodactyly | [74,75,76] |
| Silver–Russell Syndrome OMIM # 180860 | hypomethylation on ch. 11p15.5 or maternal UPD for ch. 7 | IGF2, CDKN1C, PLAG1, HMGA2, H19 | FB or prominent forehead, fifth finger clinodactyly | [58] |
| Turnpenny–Fry syndrome OMIM # 618371 | AD | PCGF2 | FB, short tapering fingers | [40] |
| Greig cephalopolysyndactyly syndrome (GCPS)OMIM # 175700 | AD | GLI3 | MC, high, prominent forehead, preaxial polydactyly, abnormally wide thumb or big toe, cutaneous syndactyly | [36] |
| Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis (SOFT syndrome)OMIM # 614813 | AR | POC1A | MC (present during early childhood), protruding forehead, short rectangular fingers and hypoplastic fingernails, clinodactyly, brachydactyly | [59] |
| Acro-callosal Syndrome (ACLS), Joubert syndrome 12, OMIM # 200990 | AR | KIF7 | MC, prominent forehead, postaxial polydactyly of the hands, and preaxial polydactyly of the feet | [37] |
| Intellectual developmental disorder, X-linked syndromic, Cabezas type OMIM # 300354 | XLR | CUL4B | MC/relative MC, short thumbs, and little fingers with adduction, brachydactyly | [155] |
| Ectodermal Dysplasia, Hypohidrotic, with Hypothyroidism and Agenesis of The Corpus Callosum OMIM # 225040 | may be XL | may represent a contiguous gene syndrome | MC, FB, long slender fingers | [77] |
| Primrose syndrome (PRIMS) OMIM # 259050 | AD | ZBTB20 | MC, dystrophic/abnormal fingernails, and toenails | [61] |
| Larsen-like syndrome OMIM # 608545 | Isolated cases | location: 6p25 | MC, brachycephaly, prominent forehead, cylindrical fingers, clinodactyly (fourth and fifth fingers) | [60] |
| Cleft palate, psychomotor retardation, and distinctive facial feature (CPRF) OMIM # 616728 | AD | KDM1A | MC (in some patients), brachycephaly, FB, tapered fingers, fifth finger clinodactyly, short thumbs | [156] |
| Chromosome 5p13 duplication syndrome OMIM # 613174 | Isolated cases | microduplications 5p13 | MC, turricephaly, FB, broad forehead, large hands, long fingers | [157] |
| Phelan–McDermid syndrome (PHMDS) OMIM # 606232 | AD | SHANK3 | MC, dolichocephaly, asymmetric face, dysplastic toenails | [158] |
| Sifrim–Hitz–Weiss syndrome (SIHIWES) OMIM # 617159 | AD | CHD4 | MC, trigonocephaly, coarse facies, tapered fingers, fusion of the wrist bones | [41] |
| Alkuraya–Kucinskas syndrome (ALKKUCS) OMIM # 617822 | AR | KIAA1109 | MC, plagiocephaly, overlapping fingers, camptodactyly, clenched hands adducted thumbs, clinodactyly | [48] |
| Desmosterolosis OMIM # 602398 | AR | DHCR24 | MC relative (sometimes microcephaly), FB, arthrogryposis, fifth finger clinodactyly, | [62] |
| Wiedemann–Rautenstrauch syndrome (WDRTS) OMIM #264090 | AR | POLR3A | MC/relative MC, FB, triangular face, long fingers, large hands | [159] |
| Joubert syndrome 2 (JBTS2) OMIM # 608091 | AR | TMEM216 | MC, dolichocephaly, FB, postaxial polydactyly, camptodactyly | [38] |
| Temtamy syndrome (TEMTYS) OMIM #218340 | AR | C12ORF57 | MC, FB, long face, brachydactyly (second to fifth fingers), bulbous thumbs | [72] |
| Carey–Fineman–Ziter syndrome (CFZS1) OMIM # 254940 | AR | MYMK | MC, (sometimes microcephaly), plagiocephaly, tapering fingers, distal contractures | [42] |
| Retinitis pigmentosa with or without skeletal anomalies OMIM #250410 | AR | CWC27 | MC (in some patients), FB, brachydactyly, shortening of distal phalanges | [160] |
| Pelger–Huet anomaly OMIM #169400 | AD | LBR | MC with prominent forehead, short metacarpals in several fingers | [89,90] |
| Fragile X syndrome, OMIM # 300624 | XLD | FMR1 | MC, coarse facies, large forehead, long face prominent jaw, hyperextensibility finger joints, dermatoglyphic findings, double-jointed thumbs | [161,162,163,164] |
| Spinocerebellar ataxia, autosomal recessive 20, OMIM # 616354 | AR | SNX14 | Relative MC, clinodactyly, camptodactyly, brachydactyly | [165] |
| Autoimmune | ||||
| Systemic lupus erythematosus | MC in 8% neonatal SLE, HC; Raynaud’s phenomenon, vasculitis, rashes | [114,115] | ||
| Juvenile idiopathic arthritis | HC (rarely); small joints arthritis | [140] | ||
| Systemic sclerosis | HC (rarely), Raynaud’s phenomenon, sclerodactyly | [140]. | ||
| Autoinflammatory | ||||
| Cryopyrinopathies [CINCA/NOMID (OMIM # 607115), Muckle-Wells (OMIM # 191900), FCAS (OMIM # 120100)] | AD | NLRP3 | Urticarial-like rashes, aseptic meningitis, FB, MC, oligoarthritis | [103] |
| * Mevalonate kinase deficiency (MVK, mevalonic aciduria, hyper IgD syndrome) OMIM # 610377 | AR | MVK | FB, dolichocephaly, triangular facies; rash, edema and arthralgia may occur during febrile crisis | [104] |
| Spondyloenchondrodysplasia with immune features OMIM # 607944 | AR | ACP5 | Cranio-facial deformities, hand anomalies, enchondromas | [92,93,94] |
| Aicardi–Goutières syndrome (AGS) AGS1: OMIM # 225750 AGS2: OMIM # 610181 AGS3: OMIM # 610329 AGS4: OMIM # 610333 AGS5: OMIM # 612952 AGS6: OMIM # 615010 AGS7: OMIM # 615846 AGS8: OMIM # 619486 AGS9: OMIM # 619487 | AD (for several cases with TREX1gene mutation), AR | TREX1, RNASEH2B, RNASEH2C, RNASEH2A, SAMHD1, ADAR, IFIH-1, LSM11, RNU7-1 | MiC/MC, pseudo-TORCH syndrome, dysmorphic features; acrocyanosis, autoamputation of the fingers, chilblain-like lesions | [109,110,111,112] |
| Singleton–Merten syndrome (SGMRT) OMIM # 182250 | AD | IFIH1 | Broad forehead, high hairline, acro-osteolysis, skeletal dysplasia, aortic calcifications, psoriasis, glaucoma | [95,97] |
| * Atypical Singleton–Merten syndrome SGMRT2, OMIM # 616298 | AD | DDX58 | Similar to SGMRT1, arthritis of the hands, metacarpophalangeal contractures; possibly calcified ligaments of the interphalangeal and metacarpophalangeal joints, mild distal erosions | [95,100,101] |
| * Tenorio syndrome OMIM # 616260 | AD | RNF125 | MC, overgrowth, large forehead, mild HC, Sjogren’s syndrome features | [70] |
| USP18 deficiency, OMIM # 617397 | AR | USP18 | Pseudo-TORCH syndrome 2; HC, brain malformation, metaphyseal X-ray changes resembling intrauterine infections | [142,144,166,167] |
| * Trichohepatoenteric syndrome (THES) THES1: OMIM # 222470 THES2: OMIM # 614602 | AR | TTC37 (SKIC3) SKIV2L (SKIC2) | Prominent forehead and cheeks, broad nasal root, trichorrhexis nodosa, skin changes, diarrhea; café-au-lait spots on the lower limbs, camptodactyly | [49,50,51,52,142] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazea, C.; Vulturar, R.; Chiș, A.; Encica, S.; Horvat, M.; Belizna, C.; Damian, L.-O. Macrocephaly and Finger Changes: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 5567. https://doi.org/10.3390/ijms25105567
Lazea C, Vulturar R, Chiș A, Encica S, Horvat M, Belizna C, Damian L-O. Macrocephaly and Finger Changes: A Narrative Review. International Journal of Molecular Sciences. 2024; 25(10):5567. https://doi.org/10.3390/ijms25105567
Chicago/Turabian StyleLazea, Cecilia, Romana Vulturar, Adina Chiș, Svetlana Encica, Melinda Horvat, Cristina Belizna, and Laura-Otilia Damian. 2024. "Macrocephaly and Finger Changes: A Narrative Review" International Journal of Molecular Sciences 25, no. 10: 5567. https://doi.org/10.3390/ijms25105567
APA StyleLazea, C., Vulturar, R., Chiș, A., Encica, S., Horvat, M., Belizna, C., & Damian, L.-O. (2024). Macrocephaly and Finger Changes: A Narrative Review. International Journal of Molecular Sciences, 25(10), 5567. https://doi.org/10.3390/ijms25105567