
Cocatalytic Activity of the Furfuryl and Oxanorbornane-Substituted Guanidines in the Aldol Reaction Catalyzed by (S)-Proline


Abstract
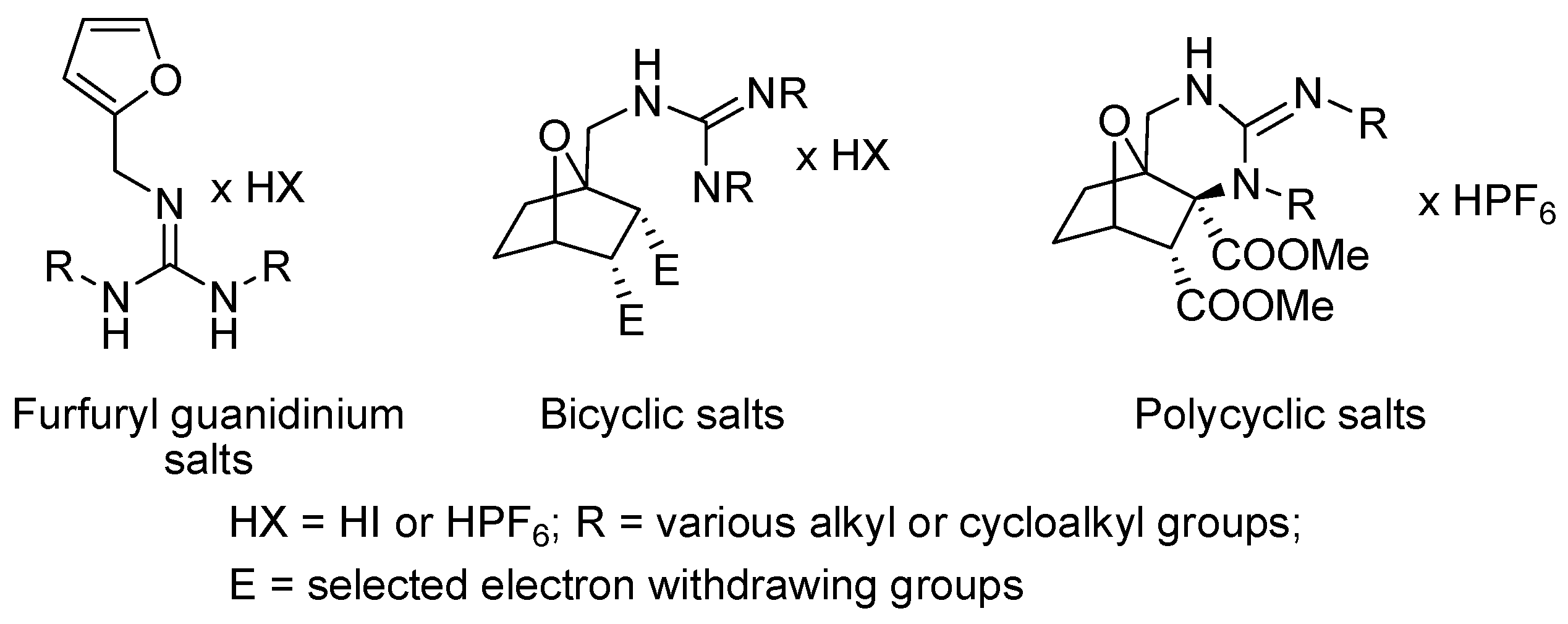
1. Introduction
2. Results and Discussion
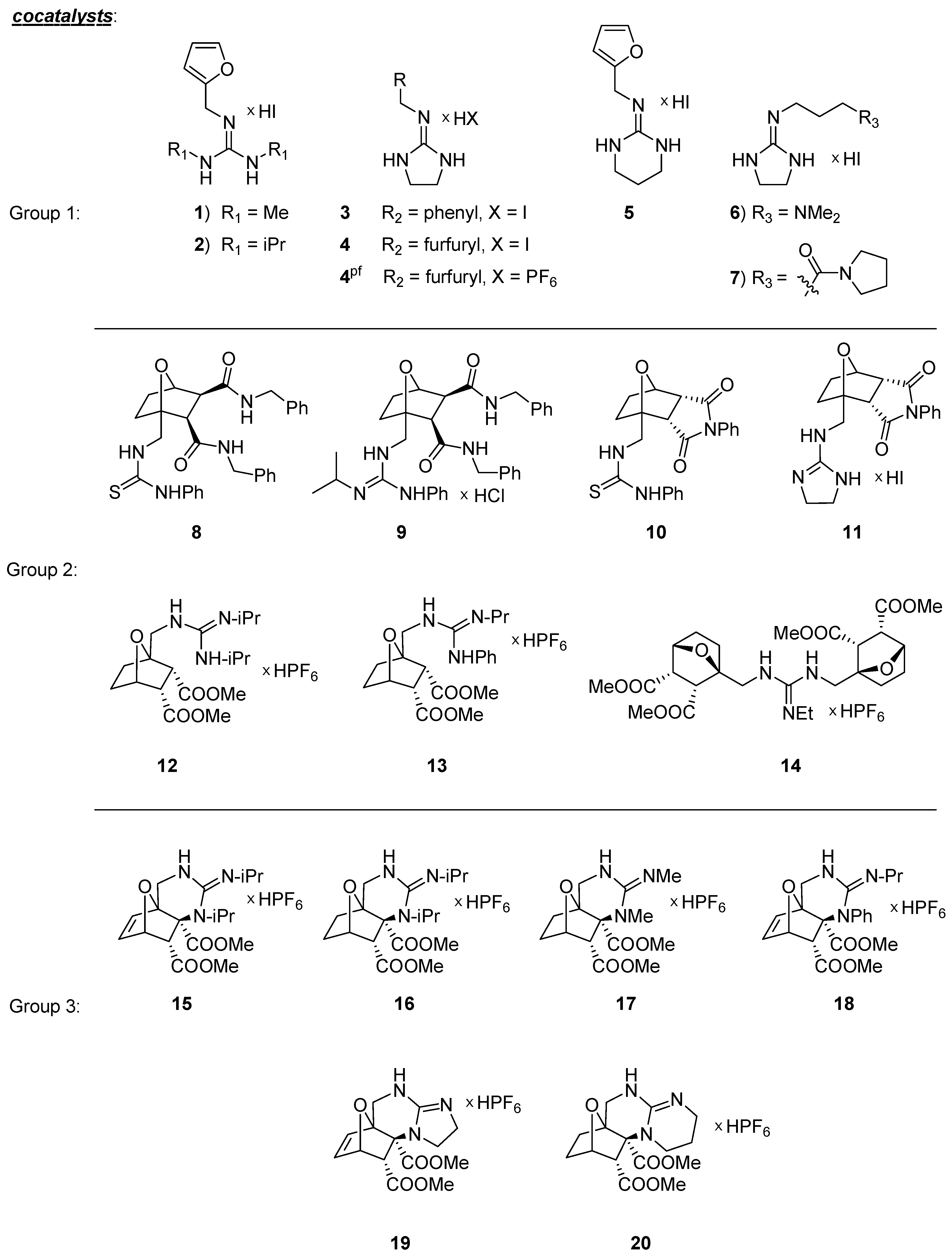
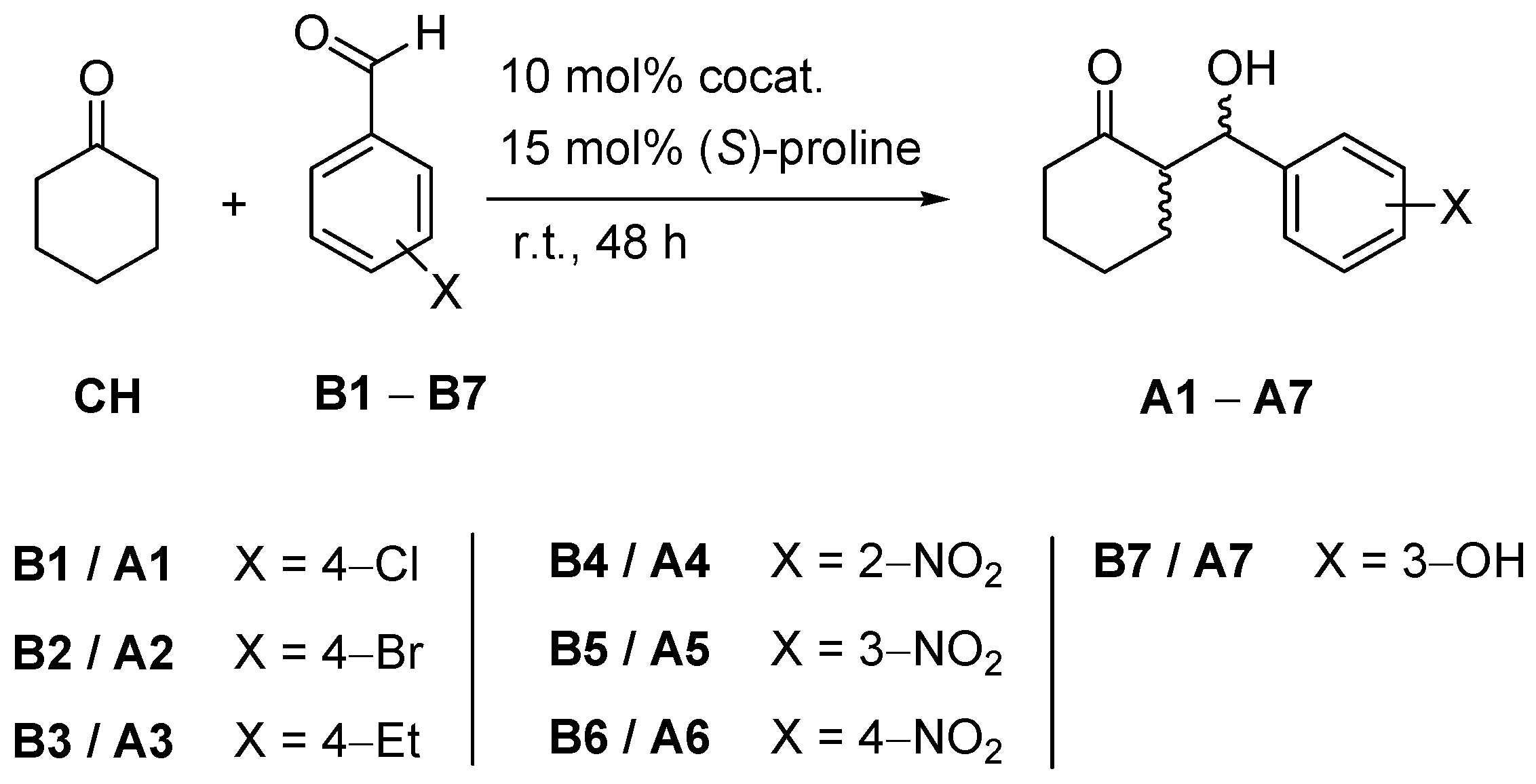
2.1. Cocatalytic Activity of the Guanidine Derivatives in the (S)-Proline-Catalyzed Aldol Reaction
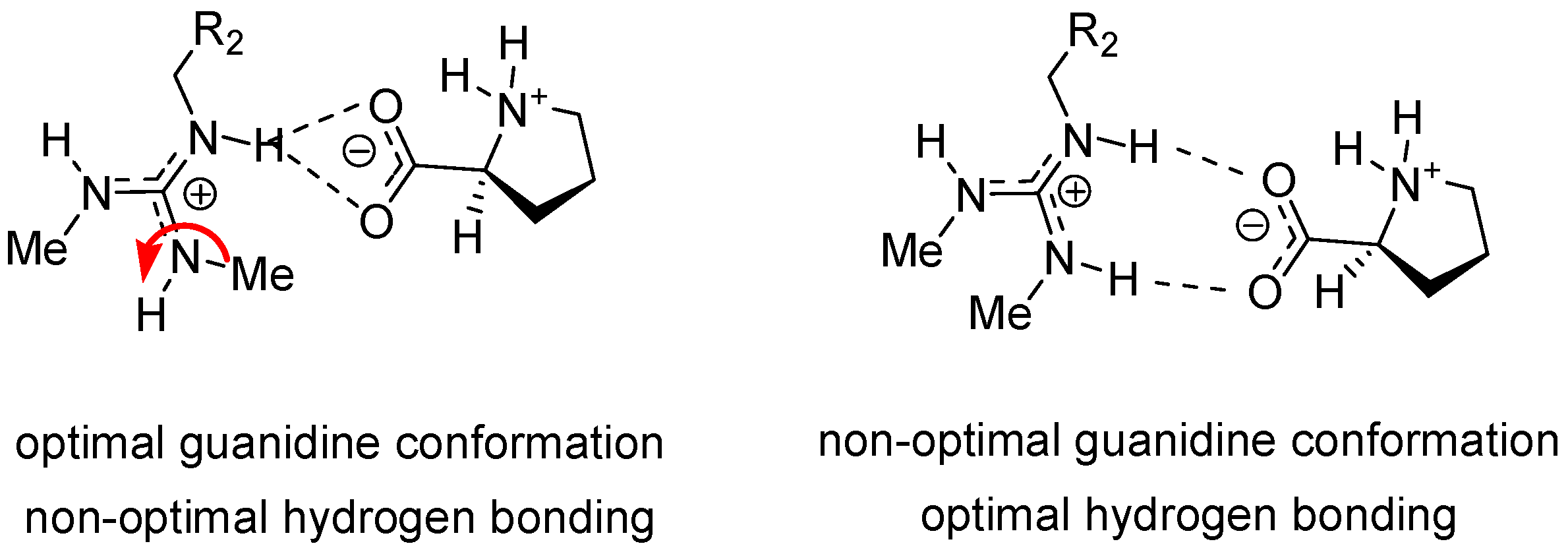
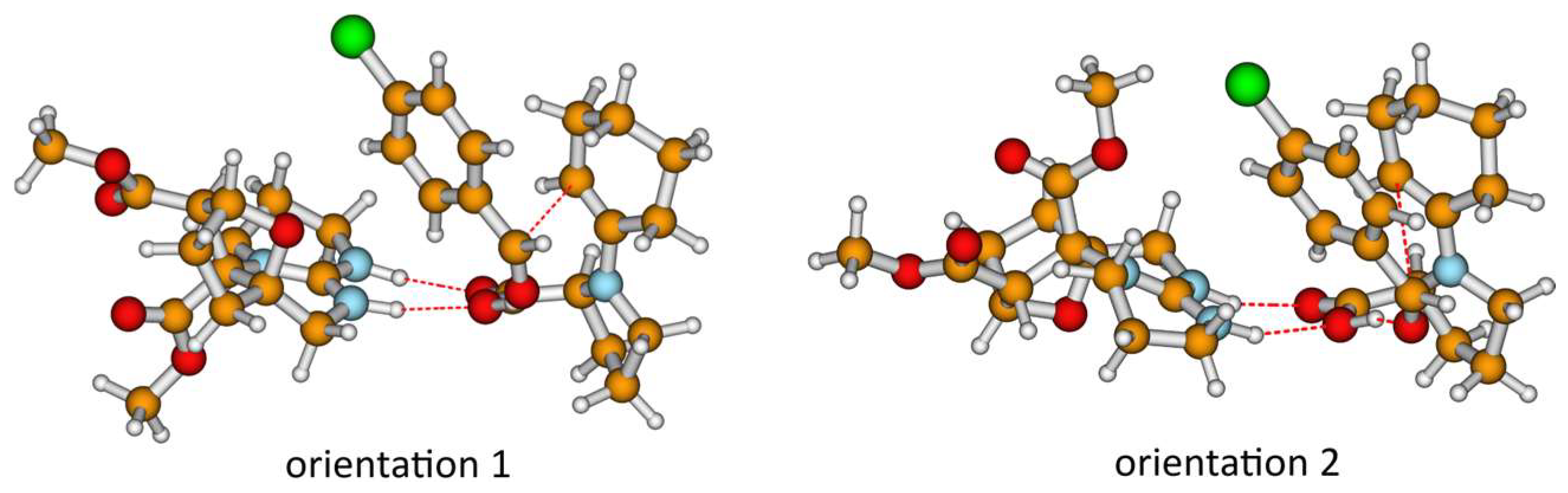
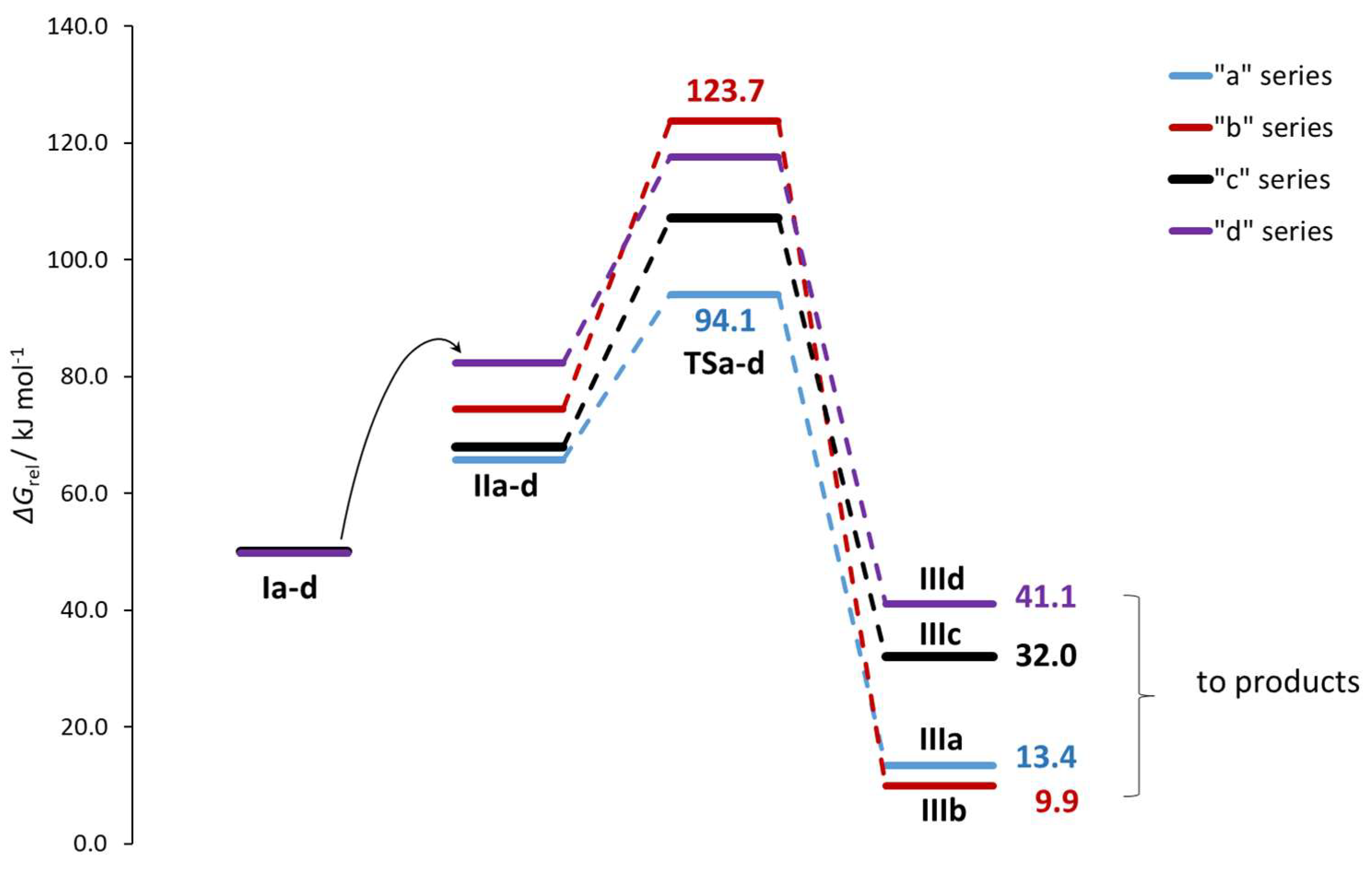
2.2. Modeling the Transition State Structures for the Formation of Enantiomers
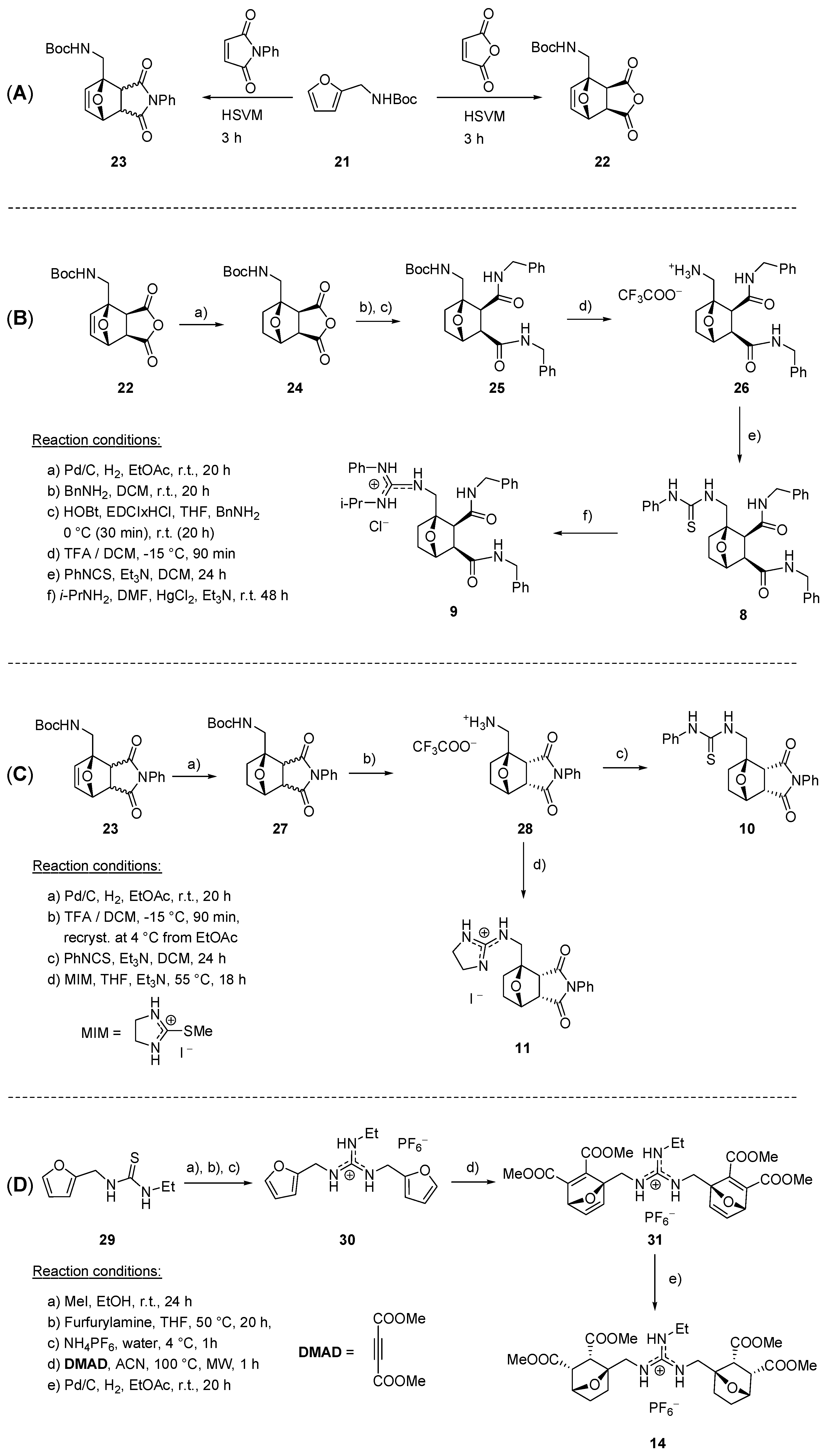
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, N.F.; Sprinkle, M.R. Relations between the Structure and Strength of Certain Organic Bases in Aqueous Solution. J. Am. Chem. Soc. 1932, 54, 3469–3485. [Google Scholar] [CrossRef]
- Schug, K.A.; Lindner, W. Noncovalent Binding Between Guanidinium and Anionic Groups: Focus on Biological- and Synthetic-Based Arginine/Guanidinium Interactions with Phosph[on]ate and Sulf[on]ate Residues. Chem. Rev. 2005, 105, 67–113. [Google Scholar] [CrossRef]
- Ishikawa, T. Guanidines in Organic Synthesis. In Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts; Ishikawa, T., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 93–144. ISBN 978-0-470-51800-7. [Google Scholar]
- Shaw, J.W.; Grayson, D.H.; Rozas, I. Synthesis of Guanidines and Some of Their Biological Applications. In Guanidines as Reagents and Catalysts I; Selig, P., Ed.; Top. Curr. Chem.; Springer Science and Business Media LLC: Dordrecht, The Netherlands, 2017; Volume 50, pp. 1–52. [Google Scholar] [CrossRef]
- Alonso-Moreno, C.; Antiñolo, A.; Carrillo-Hermosilla, F.; Otero, A. Guanidines: From classical approaches to efficient catalytic syntheses. Chem. Soc. Rev. 2014, 43, 3406–3425. [Google Scholar] [CrossRef] [PubMed]
- Selig, P. Guanidine Organocatalysis. Synthesis 2013, 45, 0703–0718. [Google Scholar] [CrossRef]
- Fu, X.; Tan, C.-H. Mechanistic Considerations of Guanidine-catalyzed Reactions. Chem. Commun. 2011, 47, 8210–8222. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-C.; Leow, D.; Tan, C.-H. Recent Advances in Chiral Guanidine-Catalyzed Enantioselective Reactions. Chem. Asian J. 2019, 14, 3803–3822. [Google Scholar] [CrossRef]
- Do Espírito Santo, R.D.; Capitão, R.M.; González, E.R.P. Guanidines as Catalysts for Direct and Indirect CO2 Capture and Activation. In Guanidines as Reagents and Catalysts II. In Guanidines as Reagents and Catalysts II; Selig, P., Ed.; Top. Curr. Chem.; Springer Science and Business Media LLC: Dordrecht, The Netherlands, 2017; Volume 51, pp. 27–74. [Google Scholar] [CrossRef]
- Horvath, A. Catalysis and Regioselectivity in the Michael Addition of Azoles. Kinetic vs. Thermodynamic Control. Tetrahedron Lett. 1996, 37, 4423–4426. [Google Scholar] [CrossRef]
- Kiesewetter, M.K.; Scholten, M.D.; Kirn, N.; Weber, R.L.; Hedrick, J.L.; Waymouth, R.M. Cyclic Guanidine Organic Catalysts: What Is Magic About Triazabicyclodecene? J. Org. Chem. 2009, 74, 9490–9496. [Google Scholar] [CrossRef]
- Van Aken, E.; Vynberg, H.; van Bolhuis, F. Nitroalkanes in C-C Bond Forming Reactions: A Crystal Structure of a Complex of a Guanidine Catalyst and a Nitroalkane Substrate. J. Chem. Soc. Chem. Commun. 1992, 1992, 629–630. [Google Scholar] [CrossRef]
- Best, M.D.; Tobey, S.L.; Anslyn, E.V. Abiotic Guanidinium Containing Receptors for Anionic Species. Coord. Chem. Rev. 2003, 240, 3–15. [Google Scholar] [CrossRef]
- Schuchardt, U.; Serchelli, R.; Vargas, R.M. Transesterification of Vegetable Oils: A Review. J. Braz. Chem. Soc. 1998, 9, 199–210. [Google Scholar] [CrossRef]
- Avila, A.; Chinchilla, R.; Fiser, B.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Michael Addition of Isobutyraldehyde to Nitroalkenes Organocatalyzed by Chiral Primary Amine-Guanidines. Tetrahedron Asymm. 2014, 25, 462–467. [Google Scholar] [CrossRef]
- Morita, S.; Yoshimura, T.; Matsuo, J. Intermolecular Domino Michael/aldol Reactions of α,β-Unsaturated Esters, Aromatic Aldehydes, and Various Nucleophiles Promoted with a Catalytic Amount of a Guanidine Base in DMSO. Tetrahedron 2021, 94, 132329. [Google Scholar] [CrossRef]
- Selig, P.; Turočkin, A.; Raven, W. Guanidine-Catalyzed γ-Selective Morita–Baylis–Hillman Reactions on α,γ-Dialkyl-Allenoates: Access to Densely Substituted Heterocycles. Synlett 2013, 24, 2535–2539. [Google Scholar] [CrossRef]
- Howard-Jones, A.; Murphy, P.J.; Thomas, D.A.; Caulkett, P.W.R. Synthesis of a Novel C2-Symmetric Guanidine Base. J. Org. Chem. 1999, 64, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Kurzmeier, H.; Schmidtchen, F.P. Abiotic Anion Receptor Functions. A Facile and Dependable Access to Chiral Guanidinium Anchor Groups. J. Org. Chem. 1990, 55, 3749–3755. [Google Scholar] [CrossRef]
- Nájera, C.; Yus, M. Chiral Guanidines in Michael Reactions. In Guanidines as Reagents and Catalysts I; Springer: Cham, Switzerland, 2015; pp. 95–128. [Google Scholar]
- Kondoh, A.; Oishi, M.; Tezuka, H.; Terada, M. Development of Chiral Organosuperbase Catalysts Consisting of Two Different Organobase Functionalities. Angew. Chem. Int. Ed. 2020, 59, 7472–7477. [Google Scholar] [CrossRef]
- Al-Taie, Z.S.; Anetts, S.R.; Christensen, J.; Coles, S.J.; Horton, P.N.; Evans, D.M.; Jones, L.F.; de Kleijne, F.F.J.; Ledbetter, S.M.; Mehdar, Y.T.H.; et al. Proline Derived Guanidine Catalysts Forge Extensive H-bonded Architectures: A Solution and Solid State Study. RSC Adv. 2020, 10, 22397. [Google Scholar] [CrossRef] [PubMed]
- Betancort, J.M.; Sakthivel, K.; Thayumanavan, R.; Barbas, C.F., III. Catalytic Enantioselective Direct Michael Additions of Ketones to Alkylidene Malonates. Tetrahedron Lett. 2001, 42, 4441–4444. [Google Scholar] [CrossRef]
- List, B.; Pojarliev, P.; Martin, H.J. Efficient Proline-Catalyzed Michael Additions of Unmodified Ketones to Nitro Olefins. Org. Lett. 2001, 3, 2423–2425. [Google Scholar] [CrossRef]
- Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C.F., III. Organocatalytic Direct Asymmetric Aldol Reactions in Water. J. Am. Chem. Soc. 2006, 128, 734–735. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Castañeda, A.; Poladura, B.; Rodríguez-Solla, H.; Concellón, C.; del Amo, V. Direct Aldol Reactions Catalyzed by a Heterogeneous Guanidinium Salt/Proline System under Solvent-Free Conditions. Org. Lett. 2011, 13, 3032–3035. [Google Scholar] [CrossRef]
- Schmid, M.B.; Zeitler, K.; Gschwind, R.M. Stabilization of Proline Enamine Carboxylates by Amine Bases. Chem. Eur. J. 2012, 18, 3362–3370. [Google Scholar] [CrossRef]
- Companyó, X.; Valero, G.; Crovetto, L.; Moyano, A.; Rios, R. Highly Enantio- and Diastereoselective Organocatalytic Desymmetrization of Prochiral Cyclohexanones by Simple Direct Aldol Reaction Catalyzed by Proline. Chem. Eur. J. 2009, 15, 6564–6568. [Google Scholar] [CrossRef]
- Emma, M.G.; Tamburrini, A.; Martinelli, A.; Lombardo, M.; Quintavalla, A.; Trombini, C. A Simple and Efficient Protocol for Proline-Catalysed Asymmetric Aldol Reaction. Catalysts 2020, 10, 649. [Google Scholar] [CrossRef]
- Reis, Ö.; Eymur, S.; Reis, B.; Demir, A.S. Direct Enantioselective Aldol Reactions Catalyzed by a Proline–Thiourea Host–Guest Complex. Chem. Commun. 2009, 2009, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.-S.; Yang, C.; Li, X.; Cheng, J.-P. Computational Study on the pKa Shifts in Proline Induced by Hydrogen-Bond-Donating Cocatalysts. J. Org. Chem. 2014, 79, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Glasovac, Z.; Trošelj, P.; Jušinski, I.; Margetić, D.; Eckert-Maksić, M. Synthesis of Highly Basic Hexasubstituted Biguanides by Environmentally Friendly Methods. Synlett 2013, 24, 2540–2544. [Google Scholar] [CrossRef]
- Faraguna, F.; Racar, M.; Glasovac, Z.; Jukić, A. Correlation Method for Conversion Determination of Biodiesel Obtained from Different Alcohols by 1H NMR Spectroscopy. Energy Fuels 2017, 31, 3943–3948. [Google Scholar] [CrossRef]
- Racar, M.; Faraguna, F.; Glasovac, Z.; Jukić, A. Experimental Modeling and Optimization of Biodiesel Production from Waste Cooking Oil and Ethanol using N,N′,N″-Tris(3-dimethylaminopropyl)-guanidine as Catalyst. Renew. Energy 2020, 146, 2374–2379. [Google Scholar] [CrossRef]
- Barešić, L.; Margetić, D.; Glasovac, Z. Anion-controlled synthesis of novel guanidine-substituted oxanorbornanes. Int. J. Mol. Sci. 2022, 23, 16036. [Google Scholar] [CrossRef] [PubMed]
- Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I.A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Gilli, P.; Pretto, L.; Bertolasi, V.; Gilli, G. Predicting Hydrogen-Bond Strengths from Acid-Base Molecular Properties. The pKa Slide Rule: Toward the Solution of a Long-Lasting Problem. Acc. Chem. Res. 2009, 42, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, C. How to Improve Guanidinium Cations for Oxoanion Binding in Aqueous Solution? The design of Artificial Peptide Receptors. Coord. Chem. Rev. 2006, 250, 3053–3067. [Google Scholar] [CrossRef]
- Mara, G.; Freire, C.M.S.S.; Neves, I.M.; Marrucho, J.; Coutinho, A.P.; Fernandes, A.M. Hydrolysis of Tetrafluoroborate and Hexafluorophosphate Counter Ions in Imidazolium-Based Ionic Liquids. J. Phys. Chem. A 2010, 114, 3744–3749. [Google Scholar] [CrossRef] [PubMed]
- Giron, R.G.P.; Ferguson, G.S. Tetrafluoroborate and Hexafluorophosphate Ions are not Interchangeable: A Density Functional Theory Comparison of Hydrogen Bonding. ChemistrySelect 2017, 2, 10895–10901. [Google Scholar] [CrossRef]
- Glasovac, Z.; Eckert-Maksić, M. Effect of Intramolecular Hydrogen Bonds on the Gas-Phase Basicity of Guanidines. Aust. J. Chem. 2014, 67, 1056–1062. [Google Scholar] [CrossRef]
- Glasovac, Z.; Eckert-Maksić, M.; Kaljurand, I.; Saame, J.; Leito, I. Gas phase Basicity of Biguanides—Comparison of the Equilibrium and the Kinetic Methods. Int. J. Mass Spectrom. 2019, 435, 61–68. [Google Scholar] [CrossRef]
- Yu, L.-J.; Blyth, M.T.; Coote, M.L. Re-Examination of Proline-Catalyzed Intermolecular Aldol Reactions: An Ab Initio Kinetic Modelling Study. Top. Catal. 2022, 65, 354–365. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Houk, K.N.; Martin, H.J.; List, B. Quantum Mechanical Predictions of the Stereoselectivities of Proline-Catalyzed Asymmetric Intermolecular Aldol Reactions. J. Am. Chem. Soc. 2003, 125, 2475–2479. [Google Scholar] [CrossRef]
- List, B.; Lerner, A.R.; Barbas, C.F., III. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Allemann, C.; Gordillo, R.; Clemente, F.R.; Cheong, P.H.-Y.; Houk, K.N. Theory of Asymmetric Organocatalysis of Aldol and Related Reactions: Rationalizations and Predictions. Acc. Chem. Res. 2004, 37, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Bahmanyar, S.; Houk, K.N. The Origin of Stereoselectivity in Proline-catalyzed Intramolecular Aldol Reactions. J. Am. Chem. Soc. 2001, 123, 12911–12912. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, N.; Endo, T. Synthesis of Five- and Six-membered Cyclic Guanidines by Guanylation with Isothiouronium Iodides and Amines Under Mild Conditions. Synth. Commun. 2017, 47, 442–448. [Google Scholar] [CrossRef]
- Maji, S.K.; Banerjee, R.; Velmurugan, D.; Razak, A.; Fun, H.K.; Banerjee, A. Peptide Design Using ω-Amino Acids: Unusual Turn Structures, Nucleated by an N-Terminal Single γ-Aminobutyric Acid Residue in Short Model Peptides. J. Org. Chem. 2002, 67, 633–639. [Google Scholar] [CrossRef]
- Barešić, L.; Margetić, D.; Glasovac, Z. Cycloaddition of Thiourea- and Guanidine-Substituted Furans to Dienophiles: A Comparison of the Environmentally-Friendly Methods. Chem. Proc. 2021, 3, 57. [Google Scholar] [CrossRef]
- Saroj, S.; Janni, D.S.; Reddy, U.C.; Muraleedharan, K.M. Functionalizable oxanorbornane-based head-group in the design of new Non-ionic amphiphiles and their drug delivery properties. Mater. Sci. Eng. C 2020, 112, 110857. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16. Rev C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Schaftenaar, G.; Vlieg, E.; Vriend, G. Molden 2.0: Quantum Chemistry Meets Proteins. J. Comput.-Aided. Mol. Des. 2017, 31, 789–800. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A Pre- and Post-processing Program for Molecular and Electronic Structures. J. Comput.-Aided. Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Chankeshwara, S.V.; Chakraborti, A.K. Catalyst-Free Chemoselective N-tert-Butyloxycarbonylation of Amines in Water. Org. Lett. 2006, 8, 3259–3262. [Google Scholar] [CrossRef] [PubMed]
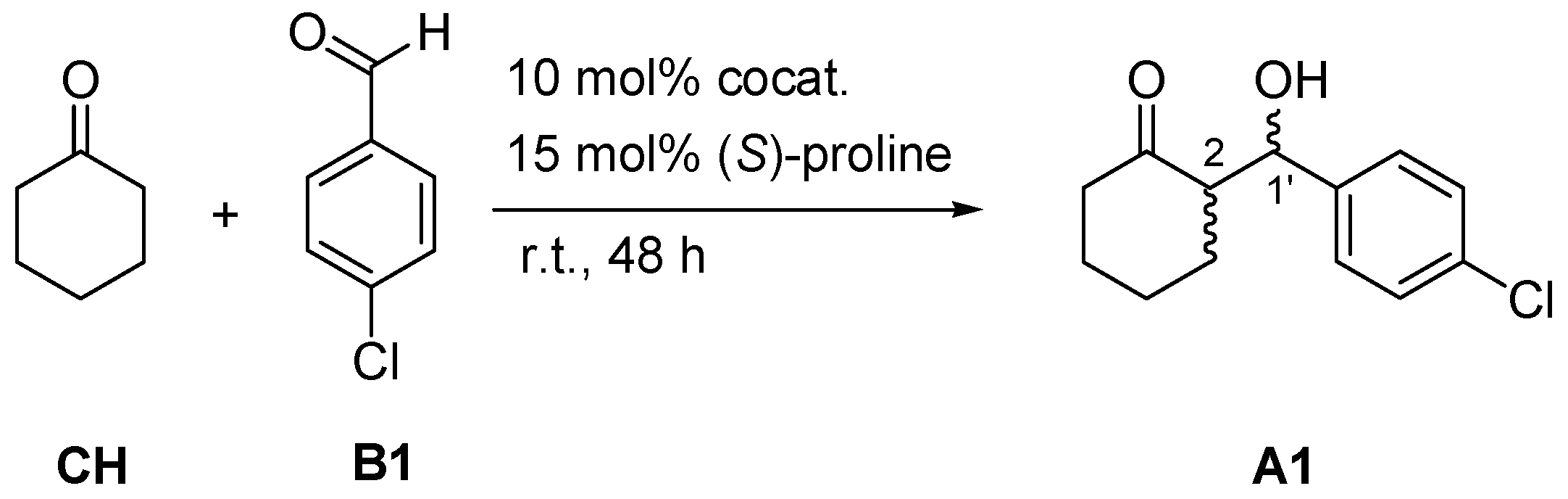


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | cocat | anti/syn b | Conv. (%) b | Yield (%) c |
|---|---|---|---|---|
| 1 | - | 67:33 | 73 | 53 |
| 2 | 1 | 84:16 | 97 | 66 |
| 3 | 2 | 83:17 | 88 | 61 |
| 4 | 3 | 85:15 | 97 | 66 |
| 5 | 4 | 89:11 | 98 | 72 |
| 6 | 4pf | 92:8 | 98 | 83 |
| 7 | 5 | 89:11 | 98 | 75 |
| 8 | 6 | 61:39 | 98 | 75 |
| 9 | 7 | 77:23 | 98 | 72 |
| 10 | 8 | 69:31 | 78 | 54 |
| 11 | 9 | 70:30 | 73 | 55 |
| 12 | 10 | 79:21 | 93 | 75 |
| 13 | 11 | 83:17 | 97 | 82 |
| 14 | 12 | 75:25 | 90 | 73 |
| 15 | 13 | 79:21 | 92 | 84 |
| 16 | 14 | 88:12 | 90 | 90 |
| 17 | 15 | 69:31 | 87 | 58 |
| 18 | 16 | 73:27 | 84 | 75 |
| 19 | 17 | 75:25 | 90 | 75 |
| 20 | 18 | 76:24 | 94 | 80 |
| 21 | 19 | 84:16 | 97 | 62 |
| 22 | 20 | 92:8 | 97 | 88 |
| Entry | benzaldehyde | Conditions b | anti/syn c | Conv. (%) c | Yield (%) d | e.e. /% e |
|---|---|---|---|---|---|---|
| 1 | B1 | 10 °C | 94:6 | 97 | 93 | 95 (80) |
| 2 | B1 | 0 °C | 96:4 | 86 | 70 | 99 |
| 3 | B1 | MeOH | 79:21 | 93 | 80 | 33 |
| 4 | B1 f | 44:56 | 99 | 48 | rac. | |
| 5 | B1 g | 0 °C | 95:5 | 80 | 63 | 99.5 |
| 6 | B2 | 85:15 | 98 | 95 | 95 | |
| 7 | B3 | 83:17 | 66 | 53 | 84 | |
| 8 | B4 | 94:6 | 99 | 93 | 98 | |
| 9 | B4 | 0 °C | 98:2 | 99 | 97 | >99.5 |
| 10 | B5 | 87:13 | 99 | 97 | 89 | |
| 11 | B6 | 86:14 | 99 | 96 | 92 | |
| 12 | B6 | 0 °C | 94:6 | 99 | 97 | 95 |
| 13 | B7 | 79:21 | 89 | 65 | 55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barešić, L.; Marijanović, M.; Dokli, I.; Margetić, D.; Glasovac, Z. Cocatalytic Activity of the Furfuryl and Oxanorbornane-Substituted Guanidines in the Aldol Reaction Catalyzed by (S)-Proline. Int. J. Mol. Sci. 2024, 25, 5570. https://doi.org/10.3390/ijms25105570
Barešić L, Marijanović M, Dokli I, Margetić D, Glasovac Z. Cocatalytic Activity of the Furfuryl and Oxanorbornane-Substituted Guanidines in the Aldol Reaction Catalyzed by (S)-Proline. International Journal of Molecular Sciences. 2024; 25(10):5570. https://doi.org/10.3390/ijms25105570
Chicago/Turabian StyleBarešić, Luka, Monika Marijanović, Irena Dokli, Davor Margetić, and Zoran Glasovac. 2024. "Cocatalytic Activity of the Furfuryl and Oxanorbornane-Substituted Guanidines in the Aldol Reaction Catalyzed by (S)-Proline" International Journal of Molecular Sciences 25, no. 10: 5570. https://doi.org/10.3390/ijms25105570
APA StyleBarešić, L., Marijanović, M., Dokli, I., Margetić, D., & Glasovac, Z. (2024). Cocatalytic Activity of the Furfuryl and Oxanorbornane-Substituted Guanidines in the Aldol Reaction Catalyzed by (S)-Proline. International Journal of Molecular Sciences, 25(10), 5570. https://doi.org/10.3390/ijms25105570