
Astrocytic Regulation of Endocannabinoid-Dependent Synaptic Plasticity in the Dorsolateral Striatum

Abstract
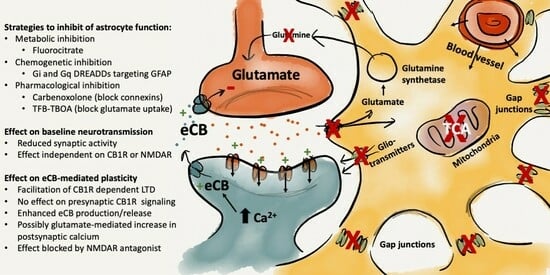
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
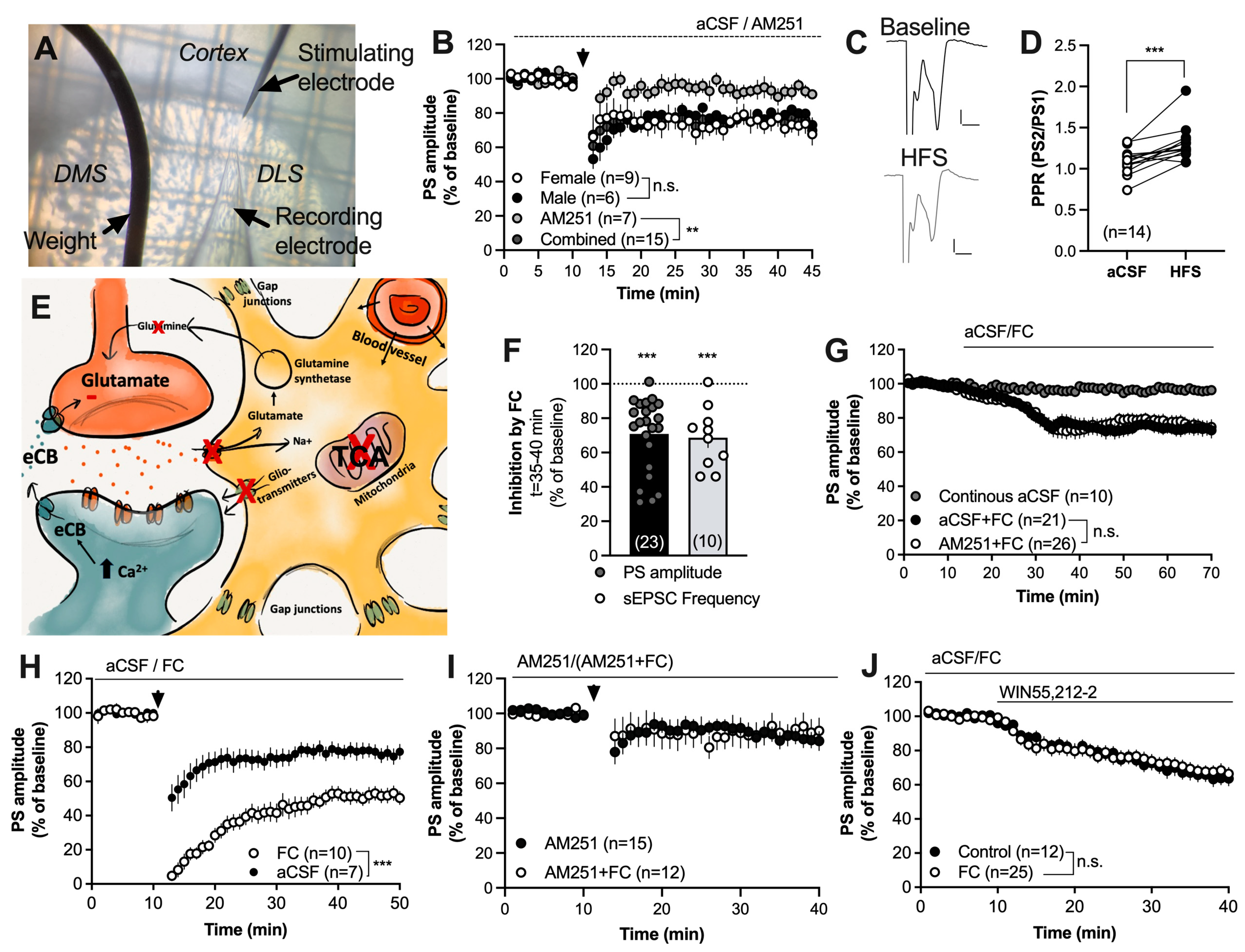
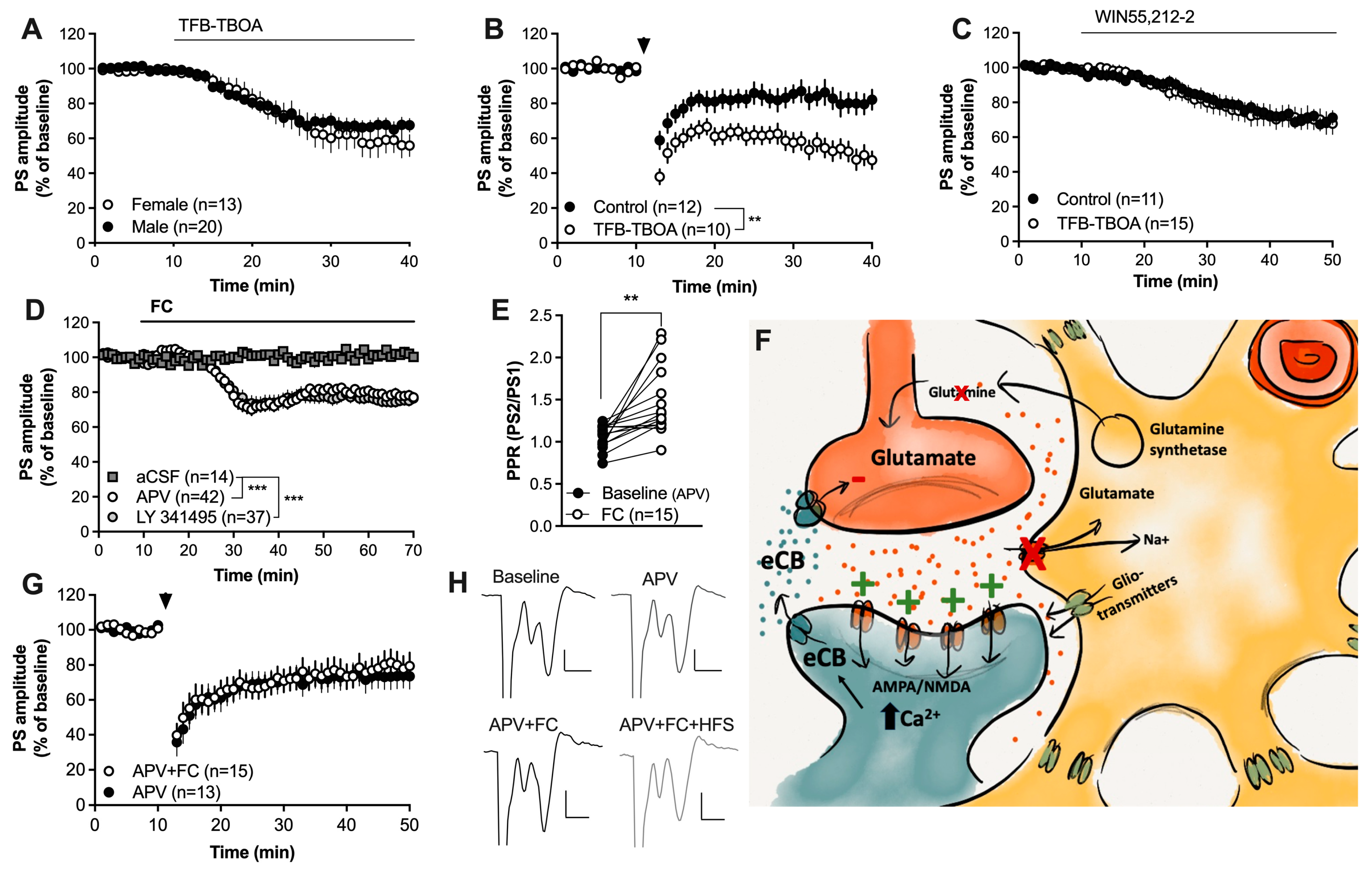
2.1. Impaired Astrocyte Function Enhances Endocannabinoid-Mediated Synaptic Plasticity at Excitatory Synapses in the Striatum
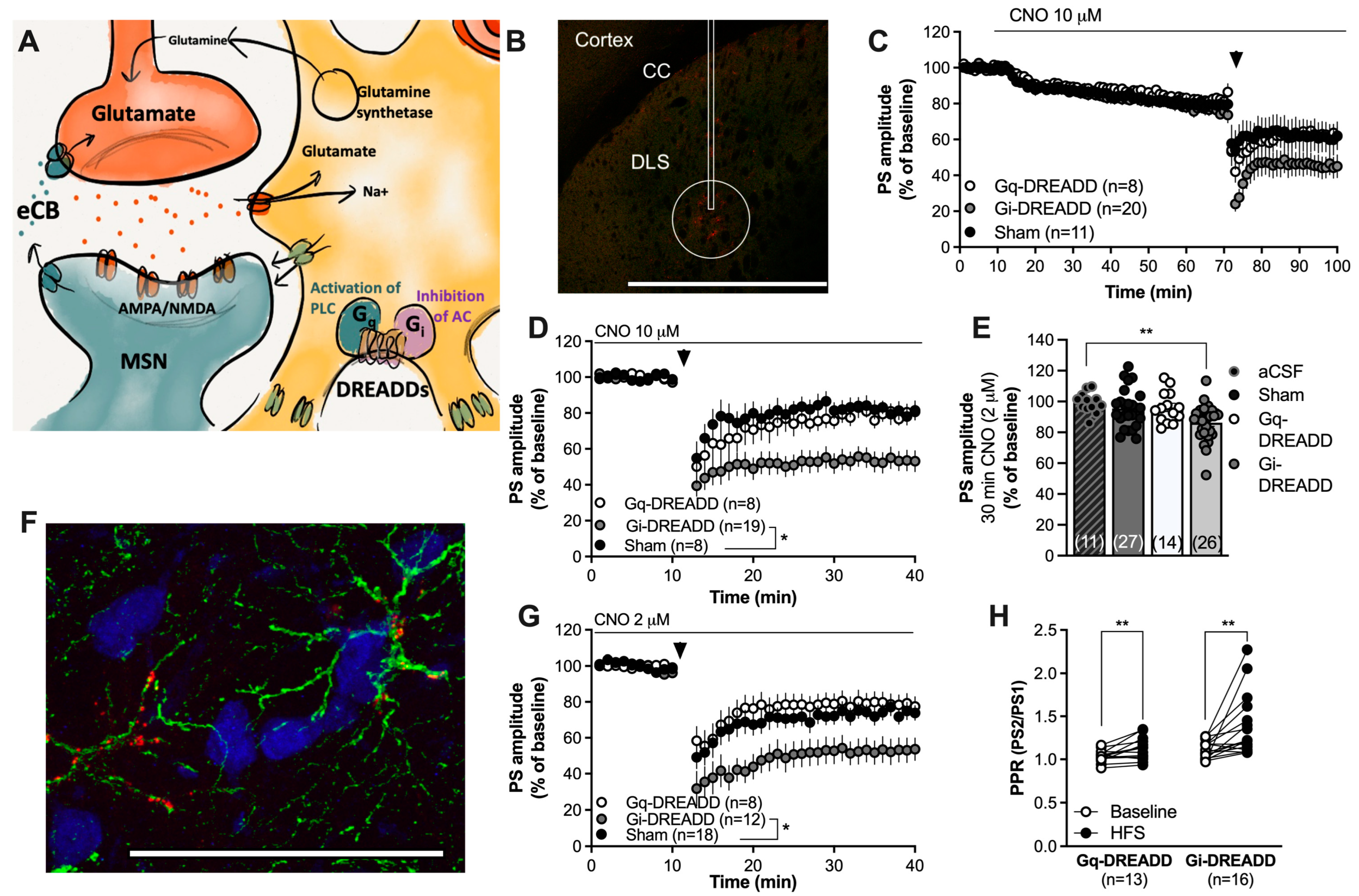
2.2. Activation of Gi-Coupled DREADDs Targeting Astrocytes Facilitates HFS-LTD
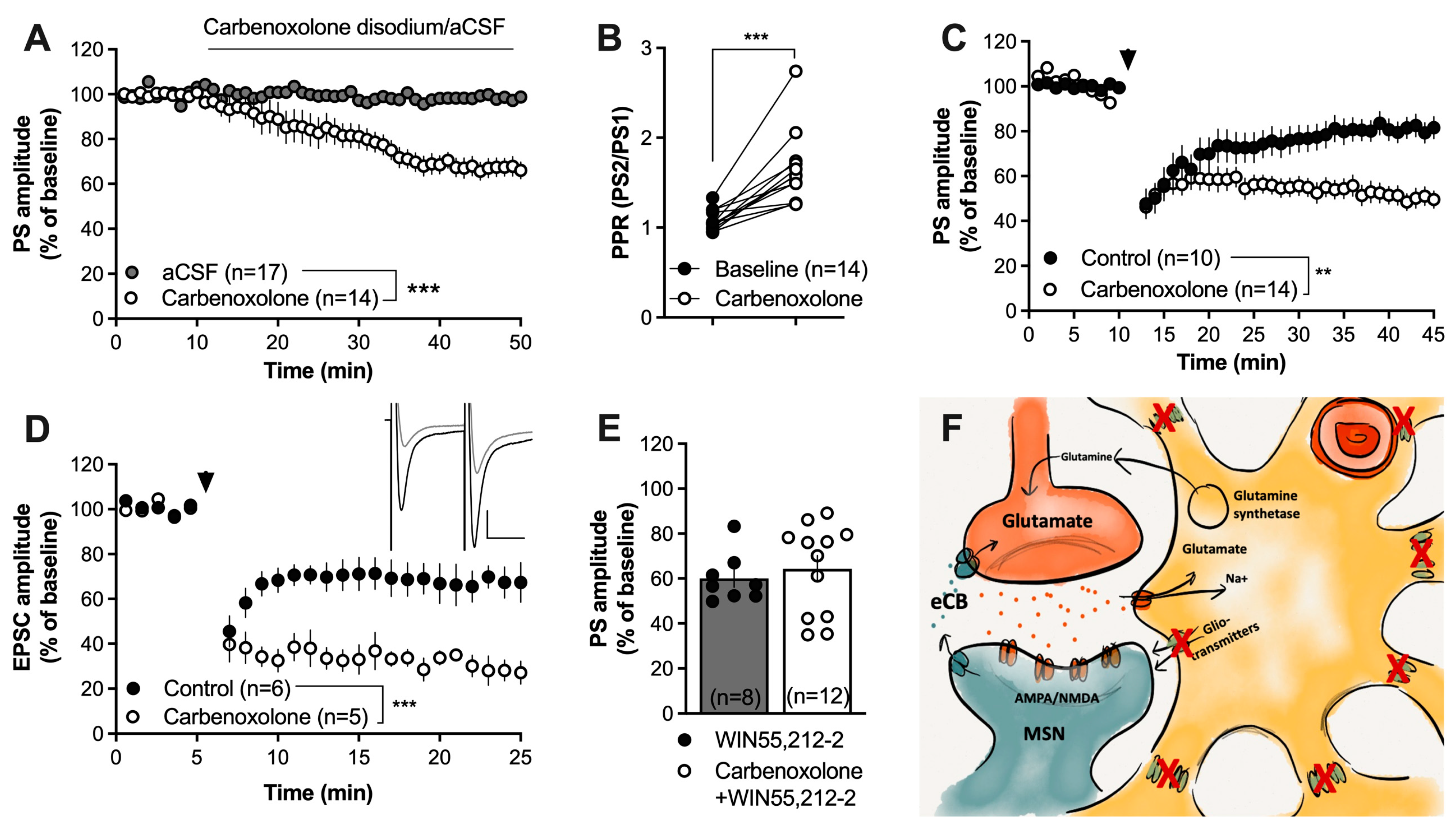
2.3. Impaired Gap-Junction Coupling Enhances HFS-LTD
2.4. Increased Glutamatergic Neurotransmission May Underly the Facilitation of HFS-LTD
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs
4.3. Surgery for Viral Injections
4.4. Ex Vivo Electrophysiology
4.4.1. Brain Slice Preparation
4.4.2. Field-Potential Recordings
4.4.3. Whole-Cell Recordings
4.5. Immunohistochemsitry
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2021, 12, 825816. [Google Scholar] [CrossRef]
- Hansson, E.; Muyderman, H.; Leonova, J.; Allansson, L.; Sinclair, J.; Blomstrand, F.; Thorlin, T.; Nilsson, M.; Ronnback, L. Astroglia and glutamate in physiology and pathology: Aspects on glutamate transport, glutamate-induced cell swelling and gap-junction communication. Neurochem. Int. 2000, 37, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Bergles, D.E.; Jabs, R.; Steinhauser, C. Neuron-glia synapses in the brain. Brain Res. Rev. 2010, 63, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Yang, A.; Boyden, E.S.; Sur, M. Optogenetic astrocyte activation modulates response selectivity of visual cortex neurons in vivo. Nat. Commun. 2014, 5, 3262. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Takano, H.; Dong, J.H.; Haydon, P.G. Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 2007, 27, 6473–6477. [Google Scholar] [CrossRef] [PubMed]
- Pannasch, U.; Vargova, L.; Reingruber, J.; Ezan, P.; Holcman, D.; Giaume, C.; Sykova, E.; Rouach, N. Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 8467–8472. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog. Neurobiol. 2015, 130, 86–120. [Google Scholar] [CrossRef]
- Min, R.; Nevian, T. Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat. Neurosci. 2012, 15, 746–753. [Google Scholar] [CrossRef]
- Lovinger, D.M. Endocannabinoid liberation from neurons in transsynaptic signaling. J. Mol. Neurosci. 2007, 33, 87–93. [Google Scholar] [CrossRef]
- Adermark, L.; Lovinger, D.M. Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. J. Neurosci. 2007, 27, 6781–6787. [Google Scholar] [CrossRef]
- Calabresi, P.; Pisani, A.; Mercuri, N.B.; Bernardi, G. Post-receptor mechanisms underlying striatal long-term depression. J. Neurosci. 1994, 14, 4871–4881. [Google Scholar] [CrossRef] [PubMed]
- Gerdeman, G.L.; Ronesi, J.; Lovinger, D.M. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat. Neurosci. 2002, 5, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Shonesy, B.C.; Winder, D.G.; Patel, S.; Colbran, R.J. The initiation of synaptic 2-AG mobilization requires both an increased supply of diacylglycerol precursor and increased postsynaptic calcium. Neuropharmacology 2015, 91, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Adermark, L.; Lovinger, D.M. Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proc. Natl. Acad. Sci. USA 2007, 104, 20564–20569. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.A.; Zhang, L.; Alger, B.E. Metaplastic control of the endocannabinoid system at inhibitory synapses in hippocampus. Proc. Natl. Acad. Sci. USA 2008, 105, 8142–8147. [Google Scholar] [CrossRef] [PubMed]
- Gerdeman, G.; Lovinger, D.M. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J. Neurophysiol. 2001, 85, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Mathur, B.N.; Tanahira, C.; Tamamaki, N.; Lovinger, D.M. Voltage drives diverse endocannabinoid signals to mediate striatal microcircuit-specific plasticity. Nat. Neurosci. 2013, 16, 1275–1283. [Google Scholar] [CrossRef]
- Ronesi, J.; Gerdeman, G.L.; Lovinger, D.M. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J. Neurosci. 2004, 24, 1673–1679. [Google Scholar] [CrossRef]
- Adermark, L.; Lovinger, D.M. Frequency-dependent inversion of net striatal output by endocannabinoid-dependent plasticity at different synaptic inputs. J. Neurosci. 2009, 29, 1375–1380. [Google Scholar] [CrossRef]
- Adermark, L.; Morud, J.; Lotfi, A.; Ericson, M.; Soderpalm, B. Acute and chronic modulation of striatal endocannabinoid-mediated plasticity by nicotine. Addict. Biol. 2019, 24, 355–363. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Malenka, R.C. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J. Neurosci. 2005, 25, 10537–10545. [Google Scholar] [CrossRef] [PubMed]
- Liput, D.J.; Puhl, H.L.; Dong, A.; He, K.; Li, Y.; Lovinger, D.M. 2-Arachidonoylglycerol mobilization following brief synaptic stimulation in the dorsal lateral striatum requires glutamatergic and cholinergic neurotransmission. Neuropharmacology 2022, 205, 108916. [Google Scholar] [CrossRef] [PubMed]
- Rossi Partridge, J.G.; Apparsundaram, S.; Gerhardt, G.A.; Ronesi, J.; Lovinger, D.M. Nicotinic acetylcholine receptors interact with dopamine in induction of striatal long-term depression. J. Neurosci. 2002, 22, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.H.; Lovinger, D.M. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 8251–8256. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, M.; Stella, N.; Calignano, A.; Lin, S.Y.; Makriyannis, A.; Piomelli, D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 1997, 277, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Rodriguez, A.; Bonilla-Del Rio, I.; Puente, N.; Gomez-Urquijo, S.M.; Fontaine, C.J.; Egana-Huguet, J.; Elezgarai, I.; Ruehle, S.; Lutz, B.; Robin, L.M.; et al. Localization of the cannabinoid type-1 receptor in subcellular astrocyte compartments of mutant mouse hippocampus. Glia 2018, 66, 1417–1431. [Google Scholar] [CrossRef]
- Han, J.; Kesner, P.; Metna-Laurent, M.; Duan, T.; Xu, L.; Georges, F.; Koehl, M.; Abrous, D.N.; Mendizabal-Zubiaga, J.; Grandes, P.; et al. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 2012, 148, 1039–1050. [Google Scholar] [CrossRef]
- Hegyi, Z.; Olah, T.; Koszeghy, A.; Piscitelli, F.; Hollo, K.; Pal, B.; Csernoch, L.; Di Marzo, V.; Antal, M. CB1 receptor activation induces intracellular Ca2+ mobilization and 2-arachidonoylglycerol release in rodent spinal cord astrocytes. Sci. Rep. 2018, 8, 10562. [Google Scholar] [CrossRef]
- Martin, R.; Bajo-Graneras, R.; Moratalla, R.; Perea, G.; Araque, A. Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 2015, 349, 730–734. [Google Scholar] [CrossRef]
- Walter, L.; Dinh, T.; Stella, N. ATP induces a rapid and pronounced increase in 2-arachidonoylglycerol production by astrocytes, a response limited by monoacylglycerol lipase. J. Neurosci. 2004, 24, 8068–8074. [Google Scholar] [CrossRef]
- Winters, N.D.; Kondev, V.; Loomba, N.; Delpire, E.; Grueter, B.A.; Patel, S. Opposing retrograde and astrocyte-dependent endocannabinoid signaling mechanisms regulate lateral habenula synaptic transmission. Cell Rep. 2023, 42, 112159. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Araque, A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 2010, 68, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Hablitz, L.M.; Gunesch, A.N.; Cravetchi, O.; Moldavan, M.; Allen, C.N. Cannabinoid Signaling Recruits Astrocytes to Modulate Presynaptic Function in the Suprachiasmatic Nucleus. eNeuro 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Secci, M.E.; Mascia, P.; Sagheddu, C.; Beggiato, S.; Melis, M.; Borelli, A.C.; Tomasini, M.C.; Panlilio, L.V.; Schindler, C.W.; Tanda, G.; et al. Astrocytic Mechanisms Involving Kynurenic Acid Control Delta(9)-Tetrahydrocannabinol-Induced Increases in Glutamate Release in Brain Reward-Processing Areas. Mol. Neurobiol. 2019, 56, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.A.; Bekar, L.K.; Nedergaard, M. Astrocytic Endocannabinoids Mediate Hippocampal Transient Heterosynaptic Depression. Neurochem. Res. 2020, 45, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Fonnum, F.; Johnsen, A.; Hassel, B. Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia 1997, 21, 106–113. [Google Scholar] [CrossRef]
- Lian, X.Y.; Stringer, J.L. Energy failure in astrocytes increases the vulnerability of neurons to spreading depression. Eur. J. Neurosci. 2004, 19, 2446–2454. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Suh, S.W.; Swanson, R.A.; Giffard, R.G. Inhibition of mitochondrial function in astrocytes: Implications for neuroprotection. J. Neurochem. 2007, 102, 1383–1394. [Google Scholar] [CrossRef]
- Adermark, L.; Lagstrom, O.; Loften, A.; Licheri, V.; Havenang, A.; Loi, E.A.; Stomberg, R.; Soderpalm, B.; Domi, A.; Ericson, M. Astrocytes modulate extracellular neurotransmitter levels and excitatory neurotransmission in dorsolateral striatum via dopamine D2 receptor signaling. Neuropsychopharmacology 2022, 47, 1493–1502. [Google Scholar] [CrossRef]
- Bonansco, C.; Couve, A.; Perea, G.; Ferradas, C.A.; Roncagliolo, M.; Fuenzalida, M. Glutamate released spontaneously from astrocytes sets the threshold for synaptic plasticity. Eur. J. Neurosci. 2011, 33, 1483–1492. [Google Scholar] [CrossRef]
- Largo, C.; Cuevas, P.; Somjen, G.G.; Martin del Rio, R.; Herreras, O. The effect of depressing glial function in rat brain in situ on ion homeostasis, synaptic transmission, and neuron survival. J. Neurosci. 1996, 16, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Padmashri, R.; Suresh, A.; Boska, M.D.; Dunaevsky, A. Motor-Skill Learning Is Dependent on Astrocytic Activity. Neural. Plast. 2015, 2015, 938023. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.E.; Contestabile, A.; Villani, L.; Fonnum, F. The effect of fluorocitrate on transmitter amino acid release from rat striatal slices. Neurochem. Res. 1988, 13, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Westergaard, N.; Waagepetersen, H.S.; Larsson, O.M.; Bakken, I.J.; Sonnewald, U. Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia 1997, 21, 99–105. [Google Scholar] [CrossRef]
- Shen, W.; Chen, S.; Liu, Y.; Han, P.; Ma, T.; Zeng, L.H. Chemogenetic manipulation of astrocytic activity: Is it possible to reveal the roles of astrocytes? Biochem. Pharmacol. 2021, 186, 114457. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Mak, A.; Verheijen, M.H.G. Comparative assessment of the effects of DREADDs and endogenously expressed GPCRs in hippocampal astrocytes on synaptic activity and memory. Front. Cell Neurosci. 2023, 17, 1159756. [Google Scholar] [CrossRef] [PubMed]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte-neuron Communication. Neuroscience 2019, 396, 73–78. [Google Scholar] [CrossRef]
- Gremel, C.M.; Chancey, J.H.; Atwood, B.K.; Luo, G.; Neve, R.; Ramakrishnan, C.; Deisseroth, K.; Lovinger, D.M.; Costa, R.M. Endocannabinoid Modulation of Orbitostriatal Circuits Gates Habit Formation. Neuron 2016, 90, 1312–1324. [Google Scholar] [CrossRef]
- Yin, H.H.; Mulcare, S.P.; Hilario, M.R.; Clouse, E.; Holloway, T.; Davis, M.I.; Hansson, A.C.; Lovinger, D.M.; Costa, R.M. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat. Neurosci. 2009, 12, 333–341. [Google Scholar] [CrossRef]
- Licheri, V.; Lagstrom, O.; Lotfi, A.; Patton, M.H.; Wigstrom, H.; Mathur, B.; Adermark, L. Complex Control of Striatal Neurotransmission by Nicotinic Acetylcholine Receptors via Excitatory Inputs onto Medium Spiny Neurons. J. Neurosci. 2018, 38, 6597–6607. [Google Scholar] [CrossRef]
- Lagstrom, O.; Danielsson, K.; Soderpalm, B.; Ericson, M.; Adermark, L. Voluntary Ethanol Intake Produces Subregion-Specific Neuroadaptations in Striatal and Cortical Areas of Wistar Rats. Alcohol. Clin. Exp. Res. 2019, 43, 803–811. [Google Scholar] [CrossRef]
- Yamamura, S.; Hoshikawa, M.; Dai, K.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. ONO-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharmacol. 2013, 168, 1088–1100. [Google Scholar] [CrossRef]
- Hassel, B.; Paulsen, R.E.; Johnsen, A.; Fonnum, F. Selective inhibition of glial cell metabolism in vivo by fluorocitrate. Brain Res. 1992, 576, 120–124. [Google Scholar] [CrossRef]
- Willoughby, J.O.; Mackenzie, L.; Broberg, M.; Thoren, A.E.; Medvedev, A.; Sims, N.R.; Nilsson, M. Fluorocitrate-mediated astroglial dysfunction causes seizures. J. Neurosci. Res. 2003, 74, 160–166. [Google Scholar] [CrossRef]
- Xia, M.; Anderson, T.L.; Prantzalos, E.R.; Hawkinson, T.R.; Clarke, H.A.; Keohane, S.B.; Sun, R.C.; Turner, J.R.; Ortinski, P.I. Voltage-gated potassium channels control extended access cocaine seeking: A role for nucleus accumbens astrocytes. Neuropsychopharmacology, 2023; online ahead of print. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Y.; Yang, H.; Cao, S.; Luo, Y.; Yu, T. Astrocytes Are Involved in the Effects of Ketamine on Synaptic Transmission in Rat Primary Somatosensory Cortex. J. Integr. Neurosci. 2023, 22, 116. [Google Scholar] [CrossRef]
- Morioka, N.; Sugimoto, T.; Tokuhara, M.; Nakamura, Y.; Abe, H.; Hisaoka, K.; Dohi, T.; Nakata, Y. Spinal astrocytes contribute to the circadian oscillation of glutamine synthase, cyclooxygenase-1 and clock genes in the lumbar spinal cord of mice. Neurochem. Int. 2012, 60, 817–826. [Google Scholar] [CrossRef]
- Hassel, B.; Westergaard, N.; Schousboe, A.; Fonnum, F. Metabolic differences between primary cultures of astrocytes and neurons from cerebellum and cerebral cortex. Effects of fluorocitrate. Neurochem. Res. 1995, 20, 413–420. [Google Scholar] [CrossRef]
- Hassel, B.; Sonnewald, U.; Unsgard, G.; Fonnum, F. NMR spectroscopy of cultured astrocytes: Effects of glutamine and the gliotoxin fluorocitrate. J. Neurochem. 1994, 62, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Adermark, L.; Lovinger, D.M. Electrophysiological properties and gap junction coupling of striatal astrocytes. Neurochem. Int. 2008, 52, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Wolosker, H.; Balu, D.T.; Coyle, J.T. The Rise and Fall of the d-Serine-Mediated Gliotransmission Hypothesis. Trends Neurosci. 2016, 39, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Cavaccini, A.; Durkee, C.; Kofuji, P.; Tonini, R.; Araque, A. Astrocyte Signaling Gates Long-Term Depression at Corticostriatal Synapses of the Direct Pathway. J. Neurosci. 2020, 40, 5757–5768. [Google Scholar] [CrossRef] [PubMed]
- Eraso-Pichot, A.; Pouvreau, S.; Olivera-Pinto, A.; Gomez-Sotres, P.; Skupio, U.; Marsicano, G. Endocannabinoid signaling in astrocytes. Glia 2023, 71, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Durkee, C.A.; Covelo, A.; Lines, J.; Kofuji, P.; Aguilar, J.; Araque, A. G(i/o) protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 2019, 67, 1076–1093. [Google Scholar] [CrossRef]
- Mariotti, L.; Losi, G.; Sessolo, M.; Marcon, I.; Carmignoto, G. The inhibitory neurotransmitter GABA evokes long-lasting Ca2+ oscillations in cortical astrocytes. Glia 2016, 64, 363–373. [Google Scholar] [CrossRef]
- Vaidyanathan, T.V.; Collard, M.; Yokoyama, S.; Reitman, M.E.; Poskanzer, K.E. Cortical astrocytes independently regulate sleep depth and duration via separate GPCR pathways. eLife 2021, 10, e63329. [Google Scholar] [CrossRef]
- Yu, G.; Cao, F.; Hou, T.; Cheng, Y.; Jia, B.; Yu, L.; Chen, W.; Xu, Y.; Chen, M.; Wang, Y. Astrocyte reactivation in medial prefrontal cortex contributes to obesity-promoted depressive-like behaviors. J. Neuroinflamm. 2022, 19, 166. [Google Scholar] [CrossRef]
- PereaBlomstrand, F.; Aberg, N.D.; Eriksson, P.S.; Hansson, E.; Ronnback, L. Extent of intercellular calcium wave propagation is related to gap junction permeability and level of connexin-43 expression in astrocytes in primary cultures from four brain regions. Neuroscience 1999, 92, 255–265. [Google Scholar] [CrossRef]
- Giaume, C.; Venance, L. Intercellular calcium signaling and gap junctional communication in astrocytes. Glia 1998, 24, 50–64. [Google Scholar] [CrossRef]
- Labra, V.C.; Santibanez, C.A.; Gajardo-Gomez, R.; Diaz, E.F.; Gomez, G.I.; Orellana, J.A. The Neuroglial Dialog between Cannabinoids and Hemichannels. Front. Mol. Neurosci. 2018, 11, 79. [Google Scholar] [CrossRef]
- Stehberg, J.; Moraga-Amaro, R.; Salazar, C.; Becerra, A.; Echeverria, C.; Orellana, J.A.; Bultynck, G.; Ponsaerts, R.; Leybaert, L.; Simon, F.; et al. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 2012, 26, 3649–3657. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Royal, C.; Johnston, A.D.; Boyce AK, J.; Diaz-Castro, B.; Institoris, A.; Peringod, G.; Zhang, O.; Stout, R.F.; Spray, D.C.; Thompson, R.J.; et al. Stress gates an astrocytic energy reservoir to impair synaptic plasticity. Nat. Commun. 2020, 11, 2014. [Google Scholar] [CrossRef] [PubMed]
- Rouach, N.; Koulakoff, A.; Abudara, V.; Willecke, K.; Giaume, C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008, 322, 1551–1555. [Google Scholar] [CrossRef]
- Onodera, M.; Meyer, J.; Furukawa, K.; Hiraoka, Y.; Aida, T.; Tanaka, K.; Tanaka, K.F.; Rose, C.R.; Matsui, K. Exacerbation of Epilepsy by Astrocyte Alkalization and Gap Junction Uncoupling. J. Neurosci. 2021, 41, 2106–2118. [Google Scholar] [CrossRef]
- McKeon, P.N.; Bunce, G.W.; Patton, M.H.; Chen, R.; Mathur, B.N. Cortical control of striatal fast-spiking interneuron synchrony. J. Physiol. 2022, 600, 2189–2202. [Google Scholar] [CrossRef]
- Buckley, C.; Zhang, X.; Wilson, C.; McCarron, J.G. Carbenoxolone and 18beta-glycyrrhetinic acid inhibit inositol 1,4,5-trisphosphate-mediated endothelial cell calcium signalling and depolarise mitochondria. Br. J. Pharmacol. 2021, 178, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Vessey, J.P.; Lalonde, M.R.; Mizan, H.A.; Welch, N.C.; Kelly, M.E.; Barnes, S. Carbenoxolone inhibition of voltage-gated Ca channels and synaptic transmission in the retina. J. Neurophysiol. 2004, 92, 1252–1256. [Google Scholar] [CrossRef]
- Karlsson, R.M.; Adermark, L.; Molander, A.; Perreau-Lenz, S.; Singley, E.; Solomon, M.; Holmes, A.; Tanaka, K.; Lovinger, D.M.; Spanagel, R.; et al. Reduced alcohol intake and reward associated with impaired endocannabinoid signaling in mice with a deletion of the glutamate transporter GLAST. Neuropharmacology 2012, 63, 181–189. [Google Scholar] [CrossRef]
- Sergeeva, O.A.; Doreulee, N.; Chepkova, A.N.; Kazmierczak, T.; Haas, H.L. Long-term depression of cortico-striatal synaptic transmission by DHPG depends on endocannabinoid release and nitric oxide synthesis. Eur. J. Neurosci. 2007, 26, 1889–1894. [Google Scholar] [CrossRef]
- Adermark, L.; Bowers, M.S. Disentangling the Role of Astrocytes in Alcohol Use Disorder. Alcohol. Clin. Exp. Res. 2016, 40, 1802–1816. [Google Scholar] [CrossRef]
- Mok, M.H.; Fricker, A.C.; Weil, A.; Kew, J.N. Electrophysiological characterisation of the actions of kynurenic acid at ligand-gated ion channels. Neuropharmacology 2009, 57, 242–249. [Google Scholar] [CrossRef]
- Dang, M.T.; Yokoi, F.; Yin, H.H.; Lovinger, D.M.; Wang, Y.; Li, Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc. Natl. Acad. Sci. USA 2006, 103, 15254–15259. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, F.B.; Min, R.; Nevian, T. Presynaptic NMDA Receptors Influence Ca2+ Dynamics by Interacting with Voltage-Dependent Calcium Channels during the Induction of Long-Term Depression. Neural. Plast. 2022, 2022, 2900875. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Umetani, Y.; Amitani, M.; Hosoi, R.; Momosaki, S.; Hatazawa, J.; Gee, A.; Inoue, O. Role of NMDA receptors in the increase of glucose metabolism in the rat brain induced by fluorocitrate. Neurosci. Lett. 2007, 415, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Adermark, L.; Gutierrez, S.; Lagstrom, O.; Hammarlund, M.; Licheri, V.; Johansson, M.E. Weight gain and neuroadaptations elicited by high fat diet depend on fatty acid composition. Psychoneuroendocrinology 2021, 126, 105143. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, B.; McDaid, L.; Wade, J.J.; Toman, M.; Wong-Lin, K.; Harkin, J. A Computational Study of Astrocytic GABA Release at the Glutamatergic Synapse: EAAT-2 and GAT-3 Coupled Dynamics. Front. Cell Neurosci. 2021, 15, 682460. [Google Scholar] [CrossRef] [PubMed]
- Heja, L.; Barabas, P.; Nyitrai, G.; Kekesi, K.A.; Lasztoczi, B.; Toke, O.; Tarkanyi, G.; Madsen, K.; Schousboe, A.; Dobolyi, A.; et al. Glutamate uptake triggers transporter-mediated GABA release from astrocytes. PLoS ONE 2009, 4, e7153. [Google Scholar] [CrossRef]
- Hertz, L.; Wu, P.H.; Schousboe, A. Evidence for net uptake of GABA into mouse astrocytes in primary cultures—its sodium dependence and potassium independence. Neurochem. Res. 1978, 3, 313–323. [Google Scholar] [CrossRef]
- Kilb, W.; Kirischuk, S. GABA Release from Astrocytes in Health and Disease. Int. J. Mol. Sci. 2022, 23, 15859. [Google Scholar] [CrossRef]
- Paulsen, R.E.; Fonnum, F. Role of glial cells for the basal and Ca2+-dependent K+-evoked release of transmitter amino acids investigated by microdialysis. J. Neurochem. 1989, 52, 1823–1829. [Google Scholar] [CrossRef]
- Roberts, B.M.; Doig, N.M.; Brimblecombe, K.R.; Lopes, E.F.; Siddorn, R.E.; Threlfell, S.; Connor-Robson, N.; Bengoa-Vergniory, N.; Pasternack, N.; Wade-Martins, R.; et al. GABA uptake transporters support dopamine release in dorsal striatum with maladaptive downregulation in a parkinsonism model. Nat. Commun. 2020, 11, 4958. [Google Scholar] [CrossRef] [PubMed]
- Adermark, L.; Lovinger, D.M. Ethanol effects on electrophysiological properties of astrocytes in striatal brain slices. Neuropharmacology 2006, 51, 1099–1108. [Google Scholar] [CrossRef]
- Mennerick, S.; Dhond, R.P.; Benz, A.; Xu, W.; Rothstein, J.D.; Danbolt, N.C.; Isenberg, K.E.; Zorumski, C.F. Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J. Neurosci. 1998, 18, 4490–4499. [Google Scholar] [CrossRef] [PubMed]
- Augustin, S.M.; Gracias, A.L.; Luo, G.; Anumola, R.C.; Lovinger, D.M. Striatonigral direct pathway 2-arachidonoylglycerol contributes to ethanol effects on synaptic transmission and behavior. Neuropsychopharmacology 2023, 48, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Hilario, M.R.; Clouse, E.; Yin, H.H.; Costa, R.M. Endocannabinoid signaling is critical for habit formation. Front. Integr. Neurosci. 2007, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Orozco, P.E.; Montes-Rodriguez, C.J.; Soria-Gomez, E.; Mendez-Diaz, M.; Prospero-Garcia, O. Impairment of endocannabinoids activity in the dorsolateral striatum delays extinction of behavior in a procedural memory task in rats. Neuropharmacology 2008, 55, 55–62. [Google Scholar] [CrossRef]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A glycine site associated with N-methyl-D-aspartic acid receptors: Characterization and identification of a new class of antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef]
- Hassel, B.; Bachelard, H.; Jones, P.; Fonnum, F.; Sonnewald, U. Trafficking of amino acids between neurons and glia in vivo. Effects of inhibition of glial metabolism by fluoroacetate. J. Cereb. Blood Flow Metab. 1997, 17, 1230–1238. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adermark, L.; Stomberg, R.; Söderpalm, B.; Ericson, M. Astrocytic Regulation of Endocannabinoid-Dependent Synaptic Plasticity in the Dorsolateral Striatum. Int. J. Mol. Sci. 2024, 25, 581. https://doi.org/10.3390/ijms25010581
Adermark L, Stomberg R, Söderpalm B, Ericson M. Astrocytic Regulation of Endocannabinoid-Dependent Synaptic Plasticity in the Dorsolateral Striatum. International Journal of Molecular Sciences. 2024; 25(1):581. https://doi.org/10.3390/ijms25010581
Chicago/Turabian StyleAdermark, Louise, Rosita Stomberg, Bo Söderpalm, and Mia Ericson. 2024. "Astrocytic Regulation of Endocannabinoid-Dependent Synaptic Plasticity in the Dorsolateral Striatum" International Journal of Molecular Sciences 25, no. 1: 581. https://doi.org/10.3390/ijms25010581
APA StyleAdermark, L., Stomberg, R., Söderpalm, B., & Ericson, M. (2024). Astrocytic Regulation of Endocannabinoid-Dependent Synaptic Plasticity in the Dorsolateral Striatum. International Journal of Molecular Sciences, 25(1), 581. https://doi.org/10.3390/ijms25010581