
Tobacco Alkaloid Assessment in a DSS-Induced Colitis Mouse Model with a Fully Humanized Immune System

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
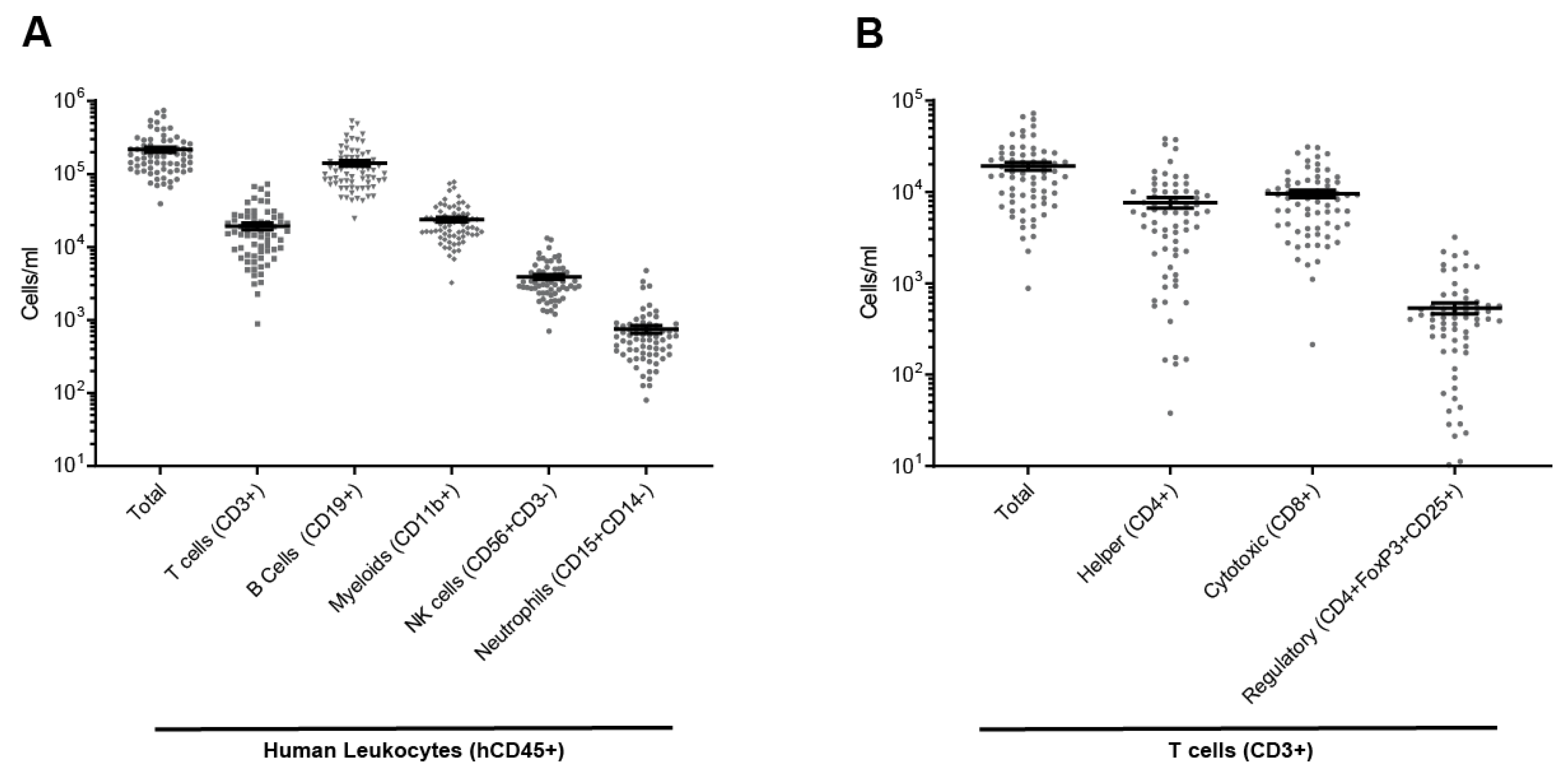
2.1. Full Reconstitution of the Human Immune System in Mouse
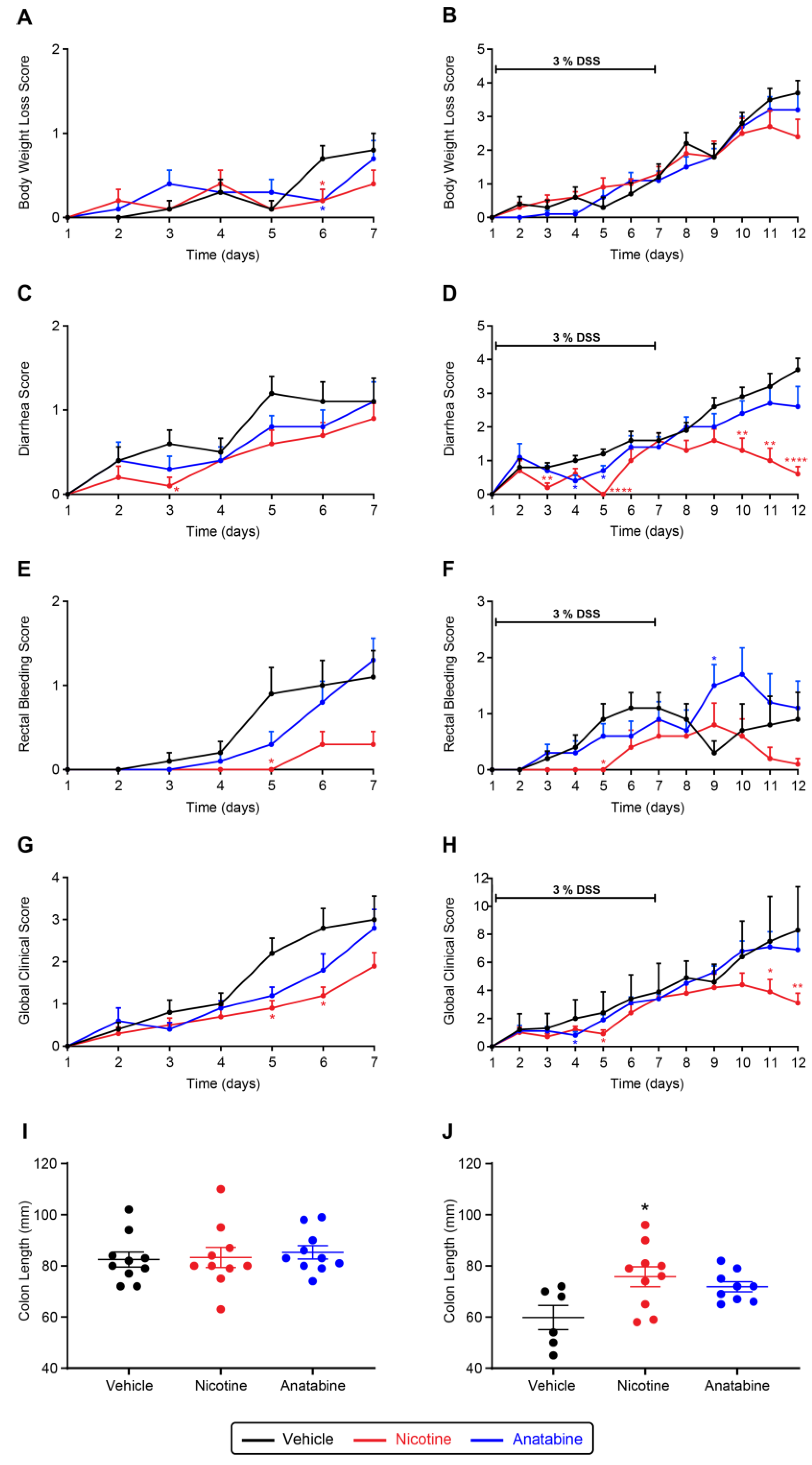
2.2. Nicotine and Anatabine Reduce DSS-Induced UC Symptoms
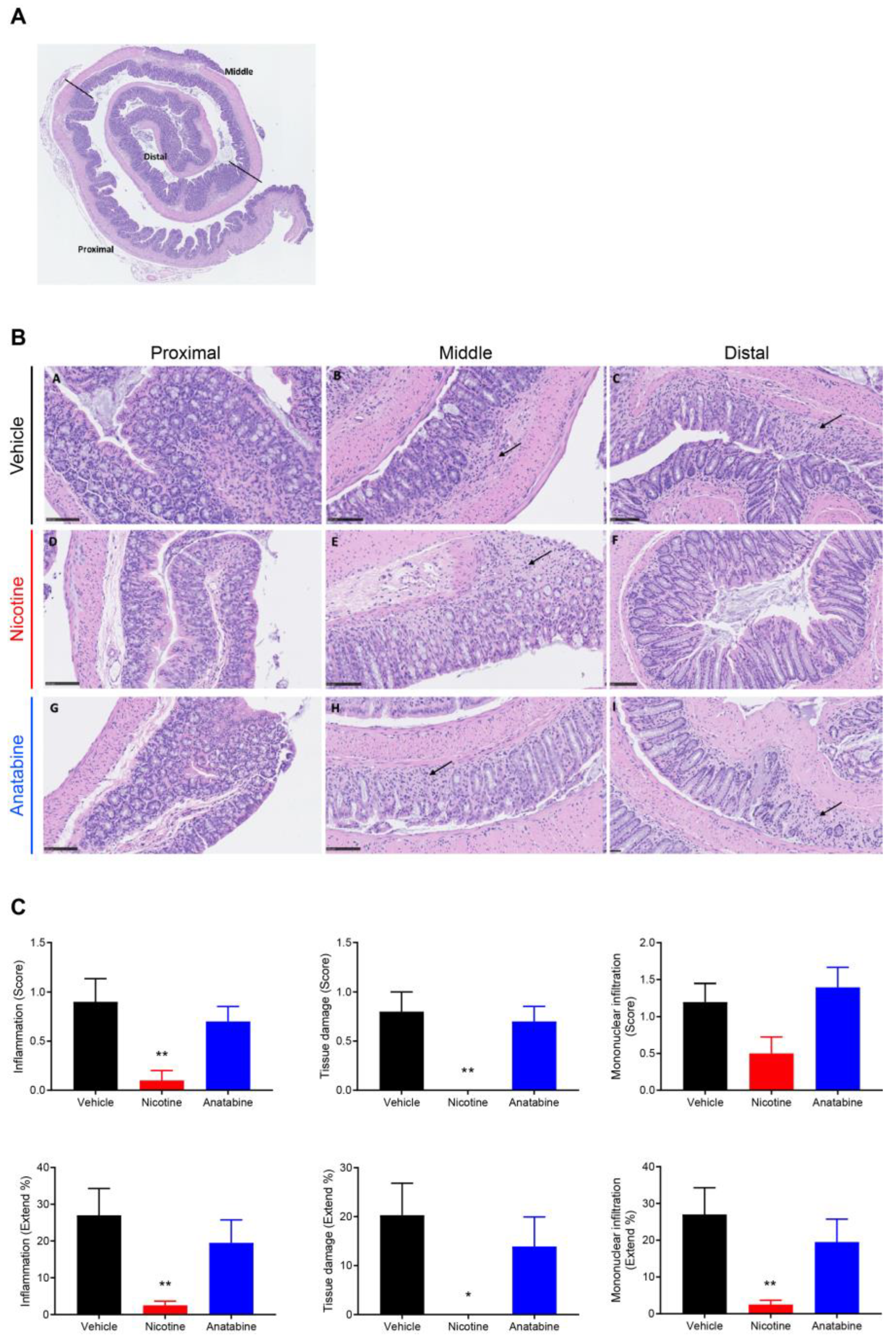
2.3. Nicotine Reduces Tissue Damage and Inflammation
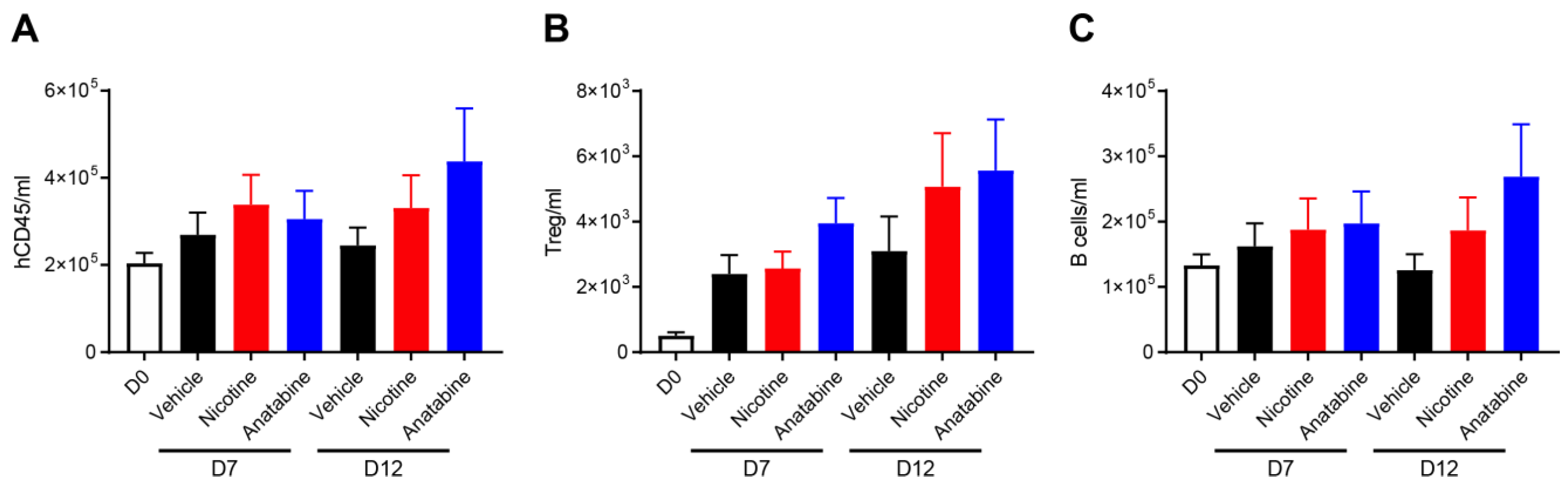
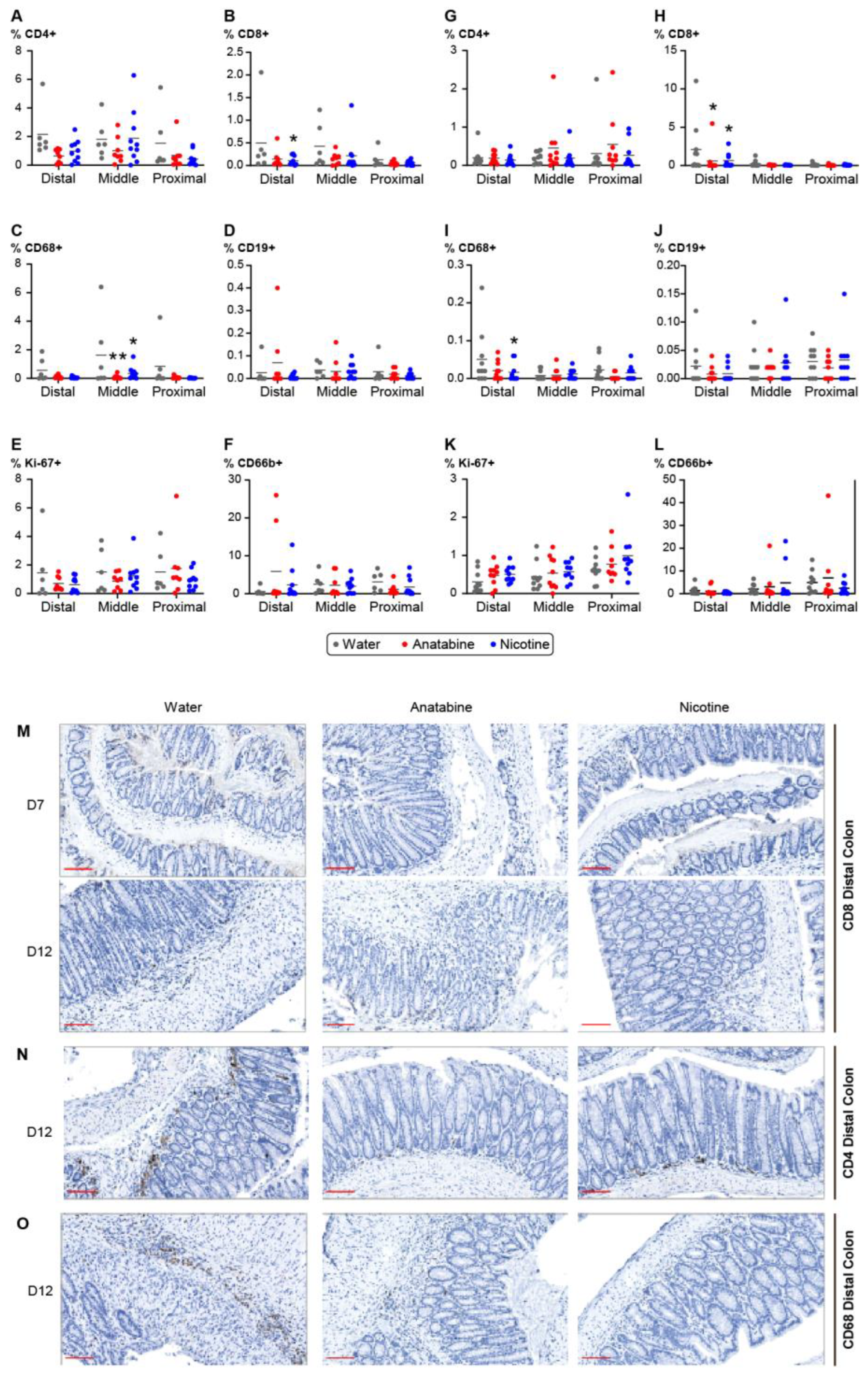
2.4. Effects of Nicotine and Anatabine on Circulating and Intestinal Human Immune Cells
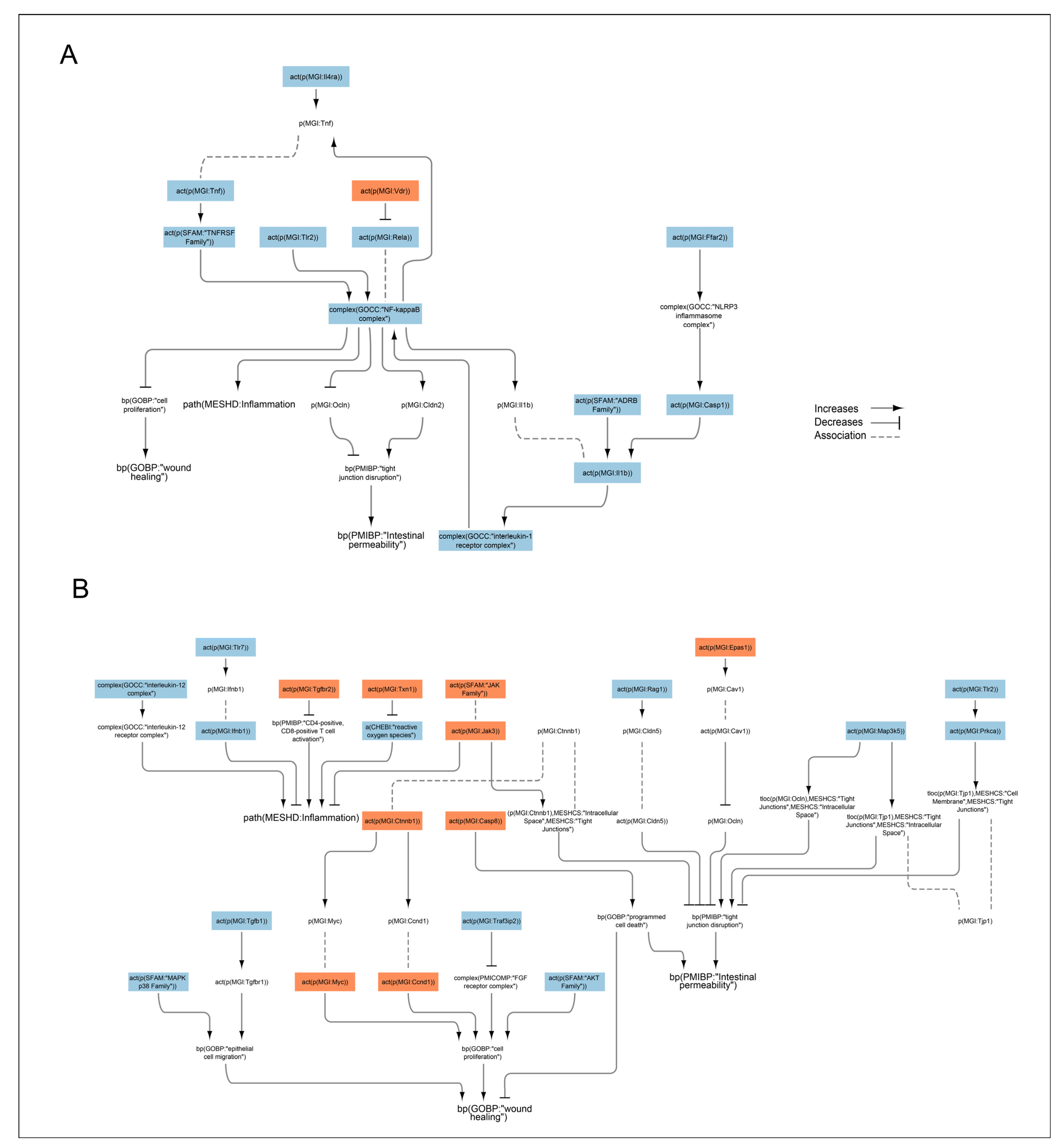
2.5. Transcriptomic Data Analysis of Effects of Nicotine and Anatabine on the Distal Colon of DSS-Treated Mice
3. Discussion
4. Materials and Methods
4.1. Animal Experimentations
4.2. DSS-Induced Acute Colitis
4.3. Treatments
4.4. Flow Cytometry
4.5. Nucleic Acid Extraction
4.6. RNA Sequencing
4.7. Histology and Immunohistochemistry
4.8. Data Analysis
4.9. Transcriptomic Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coward, S.; Clement, F.; Benchimol, E.I.; Bernstein, C.N.; Avina-Zubieta, J.A.; Bitton, A.; Carroll, M.W.; Hazlewood, G.; Jacobson, K.; Jelinski, S.; et al. Past and Future Burden of Inflammatory Bowel Diseases Based on Modeling of Population-Based Data. Gastroenterology 2019, 156, 1345–1353.e1344. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, L.; Schultz, B.M.; Salazar, G.A.; Pardo-Roa, C.; Sebastián, V.P.; Álvarez-Lobos, M.M.; Bueno, S.M. Impact of cigarette smoking on the gastrointestinal tract inflammation: Opposing effects in Crohn’s disease and ulcerative colitis. Front. Immunol. 2018, 9, 74. [Google Scholar] [CrossRef]
- To, N.; Gracie, D.; Ford, A. Systematic review with meta-analysis: The adverse effects of tobacco smoking on the natural history of Crohn’s disease. Aliment. Pharmacol. Ther. 2016, 43, 549–561. [Google Scholar] [CrossRef]
- Mahid, S.S.; Minor, K.S.; Soto, R.E.; Hornung, C.A.; Galandiuk, S. Smoking and inflammatory bowel disease: A meta-analysis. Mayo Clin. Proc. 2006, 81, 1462–1471. [Google Scholar] [CrossRef]
- de Jonge, W.J.; van der Zanden, E.P.; The, F.O.; Bijlsma, M.F.; van Westerloo, D.J.; Bennink, R.J.; Berthoud, H.-R.; Uematsu, S.; Akira, S.; van den Wijngaard, R.M. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 2005, 6, 844–851. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- De Jonge, W.; Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef]
- Lunney, P.C.; Leong, R.W. Review article: Ulcerative colitis, smoking and nicotine therapy. Aliment. Pharmacol. Ther. 2012, 36, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; McDonald, J.W.; Macdonald, J.K. Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database Syst. Rev. 2004, 18, Cd004722. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.; Pereira, D.M.; Valentao, P.; Andrade, P.B. Pyrrolizidine Alkaloids: Chemistry, Pharmacology, Toxicology and Food Safety. Int. J. Mol. Sci. 2018, 19, 1668. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zheng, T.T.; Li, X.; Liang, Y.; Wang, L.J.; Huang, Y.C.; Xiao, H.T. Plant-Derived Alkaloids: The Promising Disease-Modifying Agents for Inflammatory Bowel Disease. Front. Pharmacol. 2019, 10, 351. [Google Scholar] [CrossRef]
- Ruiz Castro, P.A.; Kogel, U.; Lo Sasso, G.; Phillips, B.W.; Sewer, A.; Titz, B.; Garcia, L.; Kondylis, A.; Guedj, E.; Peric, D.; et al. Anatabine ameliorates intestinal inflammation and reduces the production of pro-inflammatory factors in a dextran sulfate sodium mouse model of colitis. J. Inflamm. 2020, 17, 29. [Google Scholar] [CrossRef]
- Thomas, G.A.; Rhodes, J.; Ragunath, K.; Mani, V.; Williams, G.T.; Newcombe, R.G.; Russell, M.A.; Feyerabend, C. Transdermal nicotine compared with oral prednisolone therapy for active ulcerative colitis. Eur. J. Gastroenterol. Hepatol. 1996, 8, 769–776. [Google Scholar]
- Sandborn, W.J.; Tremaine, W.J.; Offord, K.P.; Lawson, G.M.; Petersen, B.T.; Batts, K.P.; Croghan, I.T.; Dale, L.C.; Schroeder, D.R.; Hurt, R.D. Transdermal nicotine for mildly to moderately active ulcerative colitis. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 1997, 126, 364–371. [Google Scholar] [CrossRef]
- Guslandi, M.; Frego, R.; Viale, E.; Testoni, P.A. Distal ulcerative colitis refractory to rectal mesalamine: Role of transdermal nicotine versus oral mesalamine. Can. J. Gastroenterol. Hepatol. 2002, 16, 293–296. [Google Scholar] [CrossRef][Green Version]
- Ingram, J.R.; Thomas, G.A.; Rhodes, J.; Green, J.T.; Hawkes, N.D.; Swift, J.L.; Srivastava, E.D.; Evans, B.K.; Williams, G.T.; Newcombe, R.G. A randomized trial of nicotine enemas for active ulcerative colitis. Clin. Gastroenterol. Hepatol. 2005, 3, 1107–1114. [Google Scholar] [CrossRef]
- Pullan, R.D.; Rhodes, J.; Ganesh, S.; Mani, V.; Morris, J.S.; Williams, G.T.; Newcombe, R.G.; Russell, M.A.; Feyerabend, C.; Thomas, G.A.; et al. Transdermal nicotine for active ulcerative colitis. N. Engl. J. Med. 1994, 330, 811–815. [Google Scholar] [CrossRef]
- Sandborn, W.; Tremaine, W.; Leighton, J.A.; Lawson, G.; Zins, B.; Compton, R.; Mays, D.; Lipsky, J.; Batts, K.; Offord, K. Nicotine tartrate liquid enemas for mildly to moderately active left-sided ulcerative colitis unresponsive to first-line therapy: A pilot study. Aliment. Pharmacol. Ther. 1997, 11, 663–671. [Google Scholar] [CrossRef] [PubMed]
- AlSharari, S.D.; Akbarali, H.I.; Abdullah, R.A.; Shahab, O.; Auttachoat, W.; Ferreira, G.A.; White, K.L.; Lichtman, A.H.; Cabral, G.A.; Damaj, M.I. Novel insights on the effect of nicotine in a murine colitis model. J. Pharmacol. Exp. Ther. 2013, 344, 207–217. [Google Scholar] [CrossRef]
- Maruta, K.; Watanabe, C.; Hozumi, H.; Kurihara, C.; Furuhashi, H.; Takajo, T.; Okada, Y.; Shirakabe, K.; Higashiyama, M.; Komoto, S.; et al. Nicotine treatment ameliorates DSS-induced colitis by suppressing MAdCAM-1 expression and leukocyte recruitment. J. Leukoc. Biol. 2018, 104, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Wan, J.J.; Sun, Y.; Wu, T.; Wang, P.Y.; Du, P.; Su, D.F.; Yang, Y.; Liu, X. Nicotine protects against DSS colitis through regulating microRNA-124 and STAT3. J. Mol. Med. 2017, 95, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Barreto, G.E.; Iarkov, A.; Moran, V.E. Beneficial effects of nicotine, cotinine and its metabolites as potential agents for Parkinson’s disease. Front. Aging Neurosci. 2014, 6, 340. [Google Scholar] [CrossRef] [PubMed]
- Paris, D.; Beaulieu-Abdelahad, D.; Abdullah, L.; Bachmeier, C.; Ait-Ghezala, G.; Reed, J.; Verma, M.; Crawford, F.; Mullan, M. Anti-inflammatory activity of anatabine via inhibition of STAT3 phosphorylation. Eur. J. Pharmacol. 2013, 698, 145–153. [Google Scholar] [CrossRef]
- Paris, D.; Beaulieu-Abdelahad, D.; Mullan, M.; Ait-Ghezala, G.; Mathura, V.; Bachmeier, C.; Crawford, F.; Mullan, M.J. Amelioration of experimental autoimmune encephalomyelitis by anatabine. PLoS ONE 2013, 8, e55392. [Google Scholar] [CrossRef] [PubMed]
- Dewey, R.E.; Xie, J. Molecular genetics of alkaloid biosynthesis in Nicotiana tabacum. Phytochemistry 2013, 94, 10–27. [Google Scholar] [CrossRef]
- Paris, D.; Beaulieu-Abdelahad, D.; Bachmeier, C.; Reed, J.; Ait-Ghezala, G.; Bishop, A.; Chao, J.; Mathura, V.; Crawford, F.; Mullan, M. Anatabine lowers Alzheimer's Aβ production in vitro and in vivo. Eur. J. Pharmacol. 2011, 670, 384–391. [Google Scholar] [CrossRef]
- Verma, M.; Beaulieu-Abdelahad, D.; Ait-Ghezala, G.; Li, R.; Crawford, F.; Mullan, M.; Paris, D. Chronic Anatabine Treatment Reduces Alzheimer’s Disease (AD)-Like Pathology and Improves Socio-Behavioral Deficits in a Transgenic Mouse Model of AD. PLoS ONE 2015, 10, e0128224. [Google Scholar] [CrossRef]
- Marescotti, D.; Lo Sasso, G.; Guerrera, D.; Renggli, K.; Ruiz Castro, P.A.; Piault, R.; Jaquet, V.; Moine, F.; Luettich, K.; Frentzel, S.; et al. Development of an Advanced Multicellular Intestinal Model for Assessing Immunomodulatory Properties of Anti-Inflammatory Compounds. Front. Pharmacol. 2021, 12, 639716. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Kolli, A.R.; Koshibu, K.; Martin, F.; Kondylis, A.; Kuczaj, A.; Tan, W.T.; Yeo, Y.S.; Tan, G.; Teng, C. In Vivo Profiling of a Natural Alkaloid, Anatabine, in Rodents: Pharmacokinetics and Anti-Inflammatory Efficacy. J. Nat. Prod. 2021, 84, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Messinis, D.E.; Poussin, C.; Latino, D.A.R.S.; Eb-Levadoux, Y.; Dulize, R.; Peric, D.; Guedj, E.; Titz, B.; Ivanov, N.V.; Peitsch, M.C.; et al. Systems biology reveals anatabine to be an NRF2 activator. Front. Pharmacol. 2022, 13, 1011184. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Castro, P.A.; Yepiskoposyan, H.; Gubian, S.; Calvino-Martin, F.; Kogel, U.; Renggli, K.; Peitsch, M.C.; Hoeng, J.; Talikka, M. Systems biology approach highlights mechanistic differences between Crohn’s disease and ulcerative colitis. Sci. Rep. 2021, 11, 11519. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Thomson, T.M.; Sewer, A.; Drubin, D.A.; Mathis, C.; Weisensee, D.; Pratt, D.; Hoeng, J.; Peitsch, M.C. Assessment of network perturbation amplitudes by applying high-throughput data to causal biological networks. BMC Syst. Biol. 2012, 6, 54. [Google Scholar] [CrossRef]
- Yepiskoposyan, H.; Peitsch, M.C.; Talikka, M. Causal Biological Network Model for Inflammasome Signaling Applied for Interpreting Transcriptomic Changes in Various Inflammatory States. Int. J. Inflamm. 2022, 2022, 4071472. [Google Scholar] [CrossRef]
- Boué, S.; Talikka, M.; Westra, J.W.; Hayes, W.; Di Fabio, A.; Park, J.; Schlage, W.K.; Sewer, A.; Fields, B.; Ansari, S.; et al. Causal biological network database: A comprehensive platform of causal biological network models focused on the pulmonary and vascular systems. Database (Oxford). 2015, 2015, bav030. [Google Scholar] [CrossRef][Green Version]
- Talikka, M.; Boue, S.; Schlage, W.K. Causal biological network database: A comprehensive platform of causal biological network models focused on the pulmonary and vascular systems. In Computational Systems Toxicology. Methods in Pharmacology and Toxicology; Hoeng, J., Peitsch, M., Eds.; Humana Press: New York, NY, USA, 2015. [Google Scholar] [CrossRef]
- Zabrodskii, P.F.; Gromov, M.S.; Maslyakov, V.V. Effect of α7n-Acetylcholine Receptor Activation and Antibodies to TNF-α on Mortality of Mice and Concentration of Proinflammatory Cytokines During Early Stage of Sepsis. Bull. Exp. Biol. Med. 2015, 159, 740–742. [Google Scholar] [CrossRef]
- Bai, A.; Guo, Y.; Lu, N. The effect of the cholinergic anti-inflammatory pathway on experimental colitis. Scand. J. Immunol. 2007, 66, 538–545. [Google Scholar] [CrossRef]
- Melgar, S.; Karlsson, L.; Rehnström, E.; Karlsson, A.; Utkovic, H.; Jansson, L.; Michaëlsson, E. Validation of murine dextran sulfate sodium-induced colitis using four therapeutic agents for human inflammatory bowel disease. Int. Immunopharmacol. 2008, 8, 836–844. [Google Scholar] [CrossRef]
- Gonzalez, L.; Strbo, N.; Podack, E.R. Humanized mice: Novel model for studying mechanisms of human immune-based therapies. Immunol. Res. 2013, 57, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.A.; Cuthbert, A.; Yang, C.; Miller, D.M.; DiIorio, P.; Laning, J.; Burzenski, L.; Gott, B.; Foreman, O.; Kavirayani, A.; et al. Parameters for establishing humanized mouse models to study human immunity: Analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rgamma(null) mutation. Clin. Immunol. 2010, 135, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Lepus, C.M.; Gibson, T.F.; Gerber, S.A.; Kawikova, I.; Szczepanik, M.; Hossain, J.; Ablamunits, V.; Kirkiles-Smith, N.; Herold, K.C.; Donis, R.O.; et al. Comparison of human fetal liver, umbilical cord blood, and adult blood hematopoietic stem cell engraftment in NOD-scid/gammac-/-, Balb/c-Rag1-/-gammac-/-, and C.B-17-scid/bg immunodeficient mice. Hum. Immunol. 2009, 70, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Karmele, E.P.; Pasricha, T.S.; Ramalingam, T.R.; Thompson, R.W.; Gieseck, R.L., 3rd; Knilans, K.J.; Hegen, M.; Farmer, M.; Jin, F.; Kleinman, A.; et al. Anti-IL-13Rα2 therapy promotes recovery in a murine model of inflammatory bowel disease. Mucosal Immunol. 2019, 12, 1174–1186. [Google Scholar] [CrossRef]
- Kugathasan, S.; Saubermann, L.J.; Smith, L.; Kou, D.; Itoh, J.; Binion, D.G.; Levine, A.D.; Blumberg, R.S.; Fiocchi, C. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut 2007, 56, 1696–1705. [Google Scholar] [CrossRef]
- Hayashi, S.; Hamada, T.; Zaidi, S.F.; Oshiro, M.; Lee, J.; Yamamoto, T.; Ishii, Y.; Sasahara, M.; Kadowaki, M. Nicotine suppresses acute colitis and colonic tumorigenesis associated with chronic colitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G968–G978. [Google Scholar] [CrossRef]
- Galitovskiy, V.; Qian, J.; Chernyavsky, A.I.; Marchenko, S.; Gindi, V.; Edwards, R.A.; Grando, S.A. Cytokine-induced alterations of α7 nicotinic receptor in colonic CD4 T cells mediate dichotomous response to nicotine in murine models of Th1/Th17- versus Th2-mediated colitis. J. Immunol. 2011, 187, 2677–2687. [Google Scholar] [CrossRef]
- Serafini, M.A.; Paz, A.H.; Nunes, N.S. Cholinergic immunomodulation in inflammatory bowel diseases. Brain Behav. Immun. Health 2022, 19, 100401. [Google Scholar] [CrossRef]
- Carlson, N.G.; Bacchi, A.; Rogers, S.W.; Gahring, L.C. Nicotine blocks TNF-alpha-mediated neuroprotection to NMDA by an alpha-bungarotoxin-sensitive pathway. J. Neurobiol. 1998, 35, 29–36. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, X.D.; Kolosov, V.P.; Perelman, J.M. Nicotine reduces TNF-alpha expression through a alpha7 nAChR/MyD88/NF-kB pathway in HBE16 airway epithelial cells. Cell Physiol. Biochem. 2011, 27, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Sugano, N.; Shimada, K.; Ito, K.; Murai, S. Nicotine inhibits the production of inflammatory mediators in U937 cells through modulation of nuclear factor-kappaB activation. Biochem. Biophys. Res. Commun. 1998, 252, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Kwan, K.; Levine, Y.A.; Olofsson, P.S.; Yang, H.; Li, J.; Joshi, S.; Wang, H.; Andersson, U.; Chavan, S.S. α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 2014, 20, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.P.S.; Yu, L.; Lam, E.K.Y.; Tai, E.K.K.; Wu, W.K.K.; Cho, C.-H. Nicotine promotes colon tumor growth and angiogenesis through β-adrenergic activation. Toxicol. Sci. 2007, 97, 279–287. [Google Scholar] [CrossRef]
- El-Gowilly, S.M.; Ghazal, A.R.; Gohar, E.Y.; El-Mas, M.M. Exacerbation by nicotine of the cyclosporine A-induced impairment of beta-adrenoceptor-mediated renal vasodilation in rats. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1164–1171. [Google Scholar] [CrossRef]
- Kishibe, M.; Griffin, T.M.; Goslawski, M.; Sinacore, J.; Kristian, S.A.; Radek, K.A. Topical nicotinic receptor activation improves wound bacterial infection outcomes and TLR2-mediated inflammation in diabetic mouse wounds. Wound Repair Regen. 2018, 26, 403–412. [Google Scholar] [CrossRef]
- Kishibe, M.; Griffin, T.M.; Radek, K.A. Keratinocyte nicotinic acetylcholine receptor activation modulates early TLR2-mediated wound healing responses. Int. Immunopharmacol. 2015, 29, 63–70. [Google Scholar] [CrossRef]
- Diebold, S.S. Recognition of viral single-stranded RNA by Toll-like receptors. Adv. Drug Deliv. Rev. 2008, 60, 813–823. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Kim, M.-S.; Kim, E.; Cheon, J.H.; Lee, Y.-S.; Kim, Y.; Lee, S.-H.; Seo, S.-U.; Shin, S.-H.; Choi, S.S. Enteric viruses ameliorate gut inflammation via toll-like receptor 3 and toll-like receptor 7-mediated interferon-β production. Immunity 2016, 44, 889–900. [Google Scholar] [CrossRef]
- Grasa, L.; Abecia, L.; Forcén, R.; Castro, M.; de Jalón, J.A.G.; Latorre, E.; Alcalde, A.I.; Murillo, M.D. Antibiotic-induced depletion of murine microbiota induces mild inflammation and changes in toll-like receptor patterns and intestinal motility. Microb. Ecol. 2015, 70, 835–848. [Google Scholar] [CrossRef]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Conrad, C.; Geiges, M.; Cozzio, A.; Thürlimann, W.; Burg, G.; Nestle, F.O.; Dummer, R. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch. Dermatol. 2004, 140, 1490–1495. [Google Scholar] [CrossRef]
- Lo, S.-M.; Hwang, Y.-S.; Liu, C.-L.; Shen, C.-N.; Hong, W.-H.; Yang, W.-C.; Lee, M.-H.; Shen, C.-R. Inhibiting TLR7 Expression in the Retinal Pigment Epithelium Suppresses Experimental Autoimmune Uveitis. Front. Immunol. 2022, 12, 736261. [Google Scholar] [CrossRef]
- Eigenbrod, T.; Dalpke, A.H. TLR7 inhibition: A novel strategy for pancreatic cancer treatment? JAKSTAT 2013, 2, e23011. [Google Scholar] [CrossRef]
- Waetzig, G.H.; Seegert, D.; Rosenstiel, P.; Nikolaus, S.; Schreiber, S. p38 mitogen-activated protein kinase is activated and linked to TNF-α signaling in inflammatory bowel disease. J. Immunol. 2002, 168, 5342–5351. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, E.; Neumann, M.; Vieth, M.; Roessner, A.; Malfertheiner, P.; Naumann, M. Inhibition of p38 MAP kinase-and RICK/NF-κB-signaling suppresses inflammatory bowel disease. FASEB J. 2004, 18, 1550–1552. [Google Scholar] [CrossRef]
- Costantini, T.W.; Krzyzaniak, M.; Cheadle, G.A.; Putnam, J.G.; Hageny, A.-M.; Lopez, N.; Eliceiri, B.P.; Bansal, V.; Coimbra, R. Targeting α-7 nicotinic acetylcholine receptor in the enteric nervous system: A cholinergic agonist prevents gut barrier failure after severe burn injury. Am. J. Pathol. 2012, 181, 478–486. [Google Scholar] [CrossRef]
- Zhou, H.; Liang, H.; Li, Z.-F.; Xiang, H.; Liu, W.; Li, J.-G. Vagus nerve stimulation attenuates intestinal epithelial tight junctions disruption in endotoxemic mice through α7 nicotinic acetylcholine receptors. Shock 2013, 40, 144–151. [Google Scholar] [CrossRef]
- Du, J.; Chen, Y.; Shi, Y.; Liu, T.; Cao, Y.; Tang, Y.; Ge, X.; Nie, H.; Zheng, C.; Li, Y.C. 1,25-Dihydroxyvitamin D protects intestinal epithelial barrier by regulating the myosin light chain kinase signaling pathway. Inflamm. Bowel Dis. 2015, 21, 2495–2506. [Google Scholar] [CrossRef]
- Palmer, M.T.; Weaver, C.T. Linking vitamin d deficiency to inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, H.; Wu, H.; Li, H.; Liu, L.; Guo, J.; Li, C.; Shih, D.Q.; Zhang, X. Protective role of 1, 25 (OH) 2vitamin D3 in the mucosal injury and epithelial barrier disruption in DSS-induced acute colitis in mice. BMC Gastroenterol. 2012, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Sewer, A.; Talikka, M.; Xiang, Y.; Hoeng, J.; Peitsch, M.C. Quantification of biological network perturbations for mechanistic insight and diagnostics using two-layer causal models. BMC Bioinform. 2014, 15, 238. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Adobe Inc. Adobe Illustrator. Available online: https://adobe.com/products/illustrator (accessed on 29 November 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score | Inflammation Score | Inflammation Extent | Mucosal Damage Score | Mucosal Damage Extent | Density Score of Mononuclear Infiltration |
|---|---|---|---|---|---|
| 0 | None or residual | % surface | None | % surface | None |
| 1 | Minimal (small scattered foci of inflammation) | Focal crypt lesion, minimal erosion but not ulceration | Minimal | ||
| 2 | Slight (slightly larger and/or more foci) | Extension in the mucosa (large erosion and/or ulceration) involving at most half of mucosal thickness | Slight (several clusters and aggregate or minimal but diffuse) | ||
| 3 | Moderate (relatively marked but multifocal in mucosa) | Extensive damage affecting more than half of the mucosa (50/75%); loss of epithelium | Moderate density (numerous clusters of slight and diffuse) | ||
| 4 | Marked transmural | Extensive damage to the entire mucosa or almost | Marked density |
| Antibodies | Dilution | Reference | Company a |
|---|---|---|---|
| Anti-CD4 (EPR6855) | 1/500 | ab133616 | Abcam |
| Anti-CD8 (SP16) | 1/500 | 108R-14 | Invitrogen |
| Recombinant anti-CD68 (EPR20545) | 1/6500 | ab213363 | Abcam |
| Anti-CD19 antibody | 1/300 | ab134114 | Abcam |
| Ki-67 monoclonal antibody (SP6) | 1/1200 | MA5-14520 | Invitrogen |
| Anti-CD66b | 1/300 | ab214175 | Abcam |
| Anti-rabbit IgG | 1/2000 | ab205718 | Abcam |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verhaeghe, C.; Talikka, M.; Sewer, A.; Sierro, N.; Auberson, M.; Peric, D.; Bornand, D.; Dulize, R.; Guedj, E.; Nef, P.; et al. Tobacco Alkaloid Assessment in a DSS-Induced Colitis Mouse Model with a Fully Humanized Immune System. Int. J. Mol. Sci. 2023, 24, 6419. https://doi.org/10.3390/ijms24076419
Verhaeghe C, Talikka M, Sewer A, Sierro N, Auberson M, Peric D, Bornand D, Dulize R, Guedj E, Nef P, et al. Tobacco Alkaloid Assessment in a DSS-Induced Colitis Mouse Model with a Fully Humanized Immune System. International Journal of Molecular Sciences. 2023; 24(7):6419. https://doi.org/10.3390/ijms24076419
Chicago/Turabian StyleVerhaeghe, Catherine, Marja Talikka, Alain Sewer, Nicolas Sierro, Mehdi Auberson, Dariusz Peric, David Bornand, Remi Dulize, Emmanuel Guedj, Patrick Nef, and et al. 2023. "Tobacco Alkaloid Assessment in a DSS-Induced Colitis Mouse Model with a Fully Humanized Immune System" International Journal of Molecular Sciences 24, no. 7: 6419. https://doi.org/10.3390/ijms24076419
APA StyleVerhaeghe, C., Talikka, M., Sewer, A., Sierro, N., Auberson, M., Peric, D., Bornand, D., Dulize, R., Guedj, E., Nef, P., Tabruyn, S. P., Hoeng, J., Peitsch, M. C., & Lo Sasso, G. (2023). Tobacco Alkaloid Assessment in a DSS-Induced Colitis Mouse Model with a Fully Humanized Immune System. International Journal of Molecular Sciences, 24(7), 6419. https://doi.org/10.3390/ijms24076419