2.1. Virtual Screening of Natural Compound Class Flavonoids for MEK2 Inhibition
Figure 1.
Histogram plot of dock-binding affinity of all flavonoids. The docking scores of the selected flavonoids in the left part are highlighted in yellow.
Figure 1.
Histogram plot of dock-binding affinity of all flavonoids. The docking scores of the selected flavonoids in the left part are highlighted in yellow.
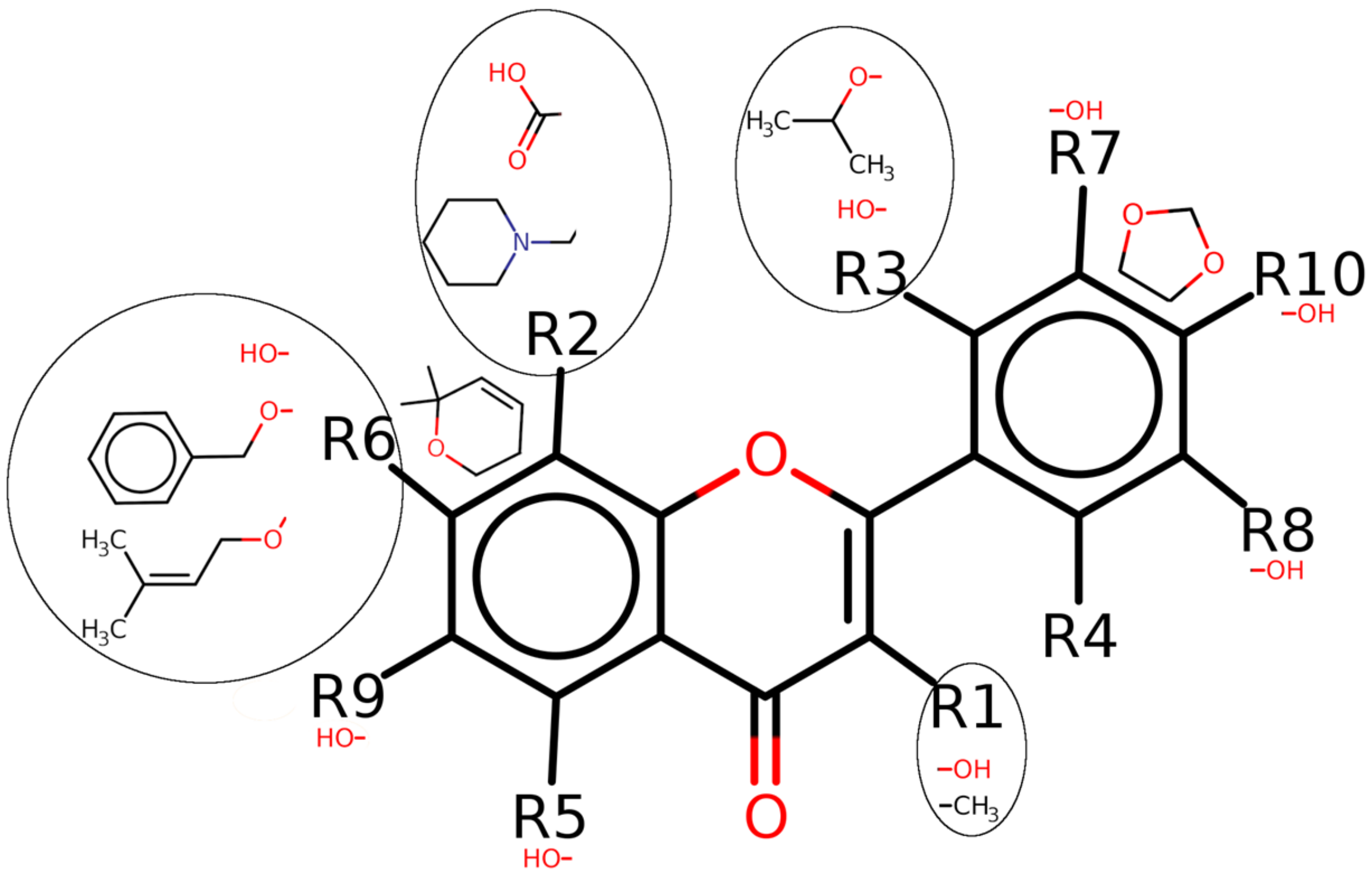
Figure 2.
Two-dimensional sketch of the selected flavonoids for MEK2 inhibition. Oxygen (O), nitrogen (N), and sulfur (S) with their balancing hydrogens are shown as red, blue, and green, respectively.
Figure 2.
Two-dimensional sketch of the selected flavonoids for MEK2 inhibition. Oxygen (O), nitrogen (N), and sulfur (S) with their balancing hydrogens are shown as red, blue, and green, respectively.
The first-ranked flavonoid
M2F1 (CID: 129703940) fit well in the MEK2 allosteric site (
Figure 3A). The ligand interacted with ATP and 18 amino acids: Asn-82, Gly-83, Lys-101, Ile-103, Leu-119, Leu-122, Val-131, Gly-132, Phe-133, Ile-145, Met-147, Asp-194, Cys-211, Asp-212, Phe-213, Val-215, Leu-219, and Met-223 (
Figure 4B) (
Table 1). The molecular docking binding affinity was −11.3 Kcal/mol, the binding energy was −10.09 Kcal/mol, and the dissociation constant
, with a value of 7.40, was reasonably high, as needed for the effective inhibition of MEK2 (
Table 2). The interaction (
Appendix A,
Table A1) shows 4 hydrogen bonds and 65 non-bonded contacts (mainly hydrophobic interactions). The hydrogen bond with Asp-194 measures 2.99 Å; two hydrogen bonds with Asp-212 measure 2.75 Å and 2.88 Å, and one hydrogen bond with ATP measures 2.75 Å. The key interacting residues are Ile-145, Asp-212, Phe-213, Leu-219, and Met-223, forming 6, 6, 7, 6, and 6 non-bonding interactions, respectively. The Asp-212 showed the maximum
(48.59 Å
2) upon binding. Comparing to the native inhibitor, of 18 residues, there were ten common residues besides ATP: Asn-82, Lys-101, Leu-122, Ile-145, Asp-194, Asp-212, Phe-213, Val-215, Leu-219, and Met-223 (
Figure 4A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213. The fact that the selected compound shares the same amino acids as the native inhibitor suggests that they may inhibit the protein in the same way.
The second-ranked flavonoid
M2F2 (CID: 56649181) against the MEK2 binding pocket docked well in the allosteric site (
Figure 3A). The ligand interaction with the protein showed 1 hydrogen bond and 56 non-bonded contacts (hydrophobic interactions) through 13 amino acids: Asn-82, Lys-101, Leu-122, Ile-145, Met-147, Arg-193, Asp-194, Cys-211, Asp-212, Phe-213, Gly-214, Val-215, and Met-223 (
Figure 4C) (
Table 1). The docking affinity was −11.1 Kcal/mol, the binding energy was −9.41 Kcal/mol, and the dissociation constant,
, had a value of 6.90, showing as good quality binding as required for adequate inhibition (
Table 2). The hydrogen bond with Lys-101 measured 3.05 Å. The key interacting residues are Asp-194, Asp-212 and Phe-213, forming 11, 17, and 6 non-bonding interactions of each, respectively. The Asp-212 turned out to have the maximum
(48.52 Å
2) upon binding, followed by Asn-82 (38.46 Å
2) and Met-223 with 36.8 Å
2 (
Appendix A,
Table A2). Compared to the native inhibitor, of 13 interacting residues, there were 11 common residues: Asn-82, Lys-101, Leu-122, Ile-145, Arg-193, Asp-194, Asp-212, Phe-213, Val-215, Leu-219, and Met-223 (
Figure 4A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213. The suggested compound holds similar potential as the native inhibitor because it binds to the same group of residues in the allosteric site.
The docking results of the third-ranked flavonoid
M2F3 (CID: 131751372) showed good fitting in the allosteric site of MEK2 (
Figure 3A). The ligand interacted with 15 amino acids: Asn-82, Lys-101, Leu-122, Ile-145, Arg-193, Asp-194, Asp-212, Phe-213, Gly-214, Val-215, Ser-216, Leu-219, Ile-220, and Met-223 (
Figure 4D). The molecular docking affinity was −11.1 Kcal/mol, the binding energy was −10.38 Kcal/mol, and the dissociation constant
, with a value of 7.61, indicates high quality binding for the effective inhibition of MEK2 (
Table 2). The interaction shows 3 hydrogen bonds and 51 non-bonded contacts (mainly hydrophobic interactions). The hydrogen bond with Lys-101 measures 3.01 Å, Val-215 measures 3.11 Å, and Ser-216 measures 2.78 Å. The key interacting residues are Asp-212 and Val-215, forming 8 and 6 non-bonding interactions of each, respectively. Met-223 turned out to show the maximum
(51.44 Å
2) upon binding, followed by Asp-212 (48.86 Å
2) and Asn-82 (38.86 Å
2) (
Appendix A,
Table A3). Compared to the native inhibitor of 15 residues, there are 14 common residues besides ATP: Asn-82, Lys-101, Leu-122, Ile-145, Arg-193, Asp-194, Asp-212, Phe-213, Gly-214, Val-215, Ser-216, Leu-219, Ile-220, and Met-223 (
Figure 4A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213. This compound is a chiral compound and has the stereoisomeric configurations R-R, R-S, S-R, and S-S. The compound’s docking data are shown above in the S-R stereoisomer configuration. Molecular docking was used to examine other stereoisomers’ ability to bind to MEK2. The molecular docking of the R-R configuration of the chiral compound also showed good affinity, with −10.4 Kcal/mol, while the R-S configuration displayed an affinity of −10.7 Kcal/mol, and the S-S configuration had an affinity of −8.1 Kcal/mol. Nevertheless, there is not any literature on this compound’s stereoisomerism. This presents a good opportunity to investigate how the enantiomers of this compound affect its ability to bind to the MEK2 kinase and produce a racemic drug.
The dock results of the fourth-ranked flavonoid
M2F4 (CID: 44559902) against the MEK2 binding pocket bound to the allosteric site well (
Figure 3B). The ligand interacted with ATP and 12 amino acid residues: Asn-82, Lys-101, Ile-145, Arg-193, Asp-194, Cys-211, Asp-212, Phe-213, Gly-214, Val-215, Met-223, and Met-234 (
Figure 4E) (
Table 1). Molecular docking data show a docking affinity of −10.9 Kcal/mol, a binding energy of −9.26 Kcal/mol, and a dissociation constant
, with a value of 6.79, which was also quite high and was essential for effective MEK2 kinase inhibition (
Table 2). The interaction shows three hydrogen bonds and 57 non-bonded contacts (mainly hydrophobic interactions). The hydrogen bond with Lys-101 measures 2.93 Å, and the two hydrogen bonds with ATP measure: 2.97 Å and 3.01 Å. The key interacting residues are Asp-194, Asp-212, and Phe-213, forming 9, 18, and 9 non-bonding interactions of each, respectively. The Asp-212 turned out to have the maximum
(48.86 Å
2) upon binding, followed by Asn-82 (40.58 Å
2), Asp-194 (38.11 Å
2), and Met-223 (37.38 Å
2) (
Appendix A,
Table A4). Of the 12 interacting residues, 12 residues besides ATP were common among the interacting residues of the native inhibitor: Asn-82, Lys-101, Ile-145, Arg-193, Asp-194, Asp-212, Phe-213, Gly-214, Val-215, and Met-223 (
Figure 4A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213. They may therefore inhibit MEK2 kinase activity in a manner similar to the native inhibitor. This compound is a chiral compound and possesses R-R, R-S, S-R and S-S stereoisomeric configurations. The above docking results of this compound are in the S-S stereoisomer configuration. In order to check the effect of other stereoisomers of this compound on the binding to MEK2, molecular docking was performed. The molecular docking of the R-R configuration of the chiral compound also showed high affinity at −9.1 Kcal/mol, while the R-S configuration displayed an affinity of −9.1 Kcal/mol, and the S-R configuration displayed an affinity of −10.3 Kcal/mol. This compound introduces a good chance to study the effect of enantiomers on the binding to the MEK2 kinase and produce a racemic compound.
The fifth-ranked flavonoid
M2F5 (CID: 154699598) bound well in the MEK2 allosteric site (
Figure 3B). The ligand interacted with ATP and 17 amino acids: Asn-82, Lys-101, Ile-103, Leu-119, Leu-122, Ile-145, Met-147, Asp-194, Cys-211, Asp-212, Phe-213, Gly-214, Val-215, Ser-216, Leu-219, Ile-220, and Met-223 (
Figure 4F) (
Table 1). The molecular docking results show a docking affinity of −10.9 Kcal/mol, a binding energy of −10.02 Kcal/mol, and a dissociation constant
, of 7.34, showing good quality binding as required for the adequate inhibition of MEK2 (
Table 2). The interaction shows 4 hydrogen bonds and 69 non-bonded contacts (mainly hydrophobic interactions). The hydrogen bonds with Asn-82 measure 2.76 Å and 3.20 Å, the hydrogen bond with Lys-101 measures 3.26 Å, and one hydrogen bond with ATP measures 2.78 Å. The key interacting residues are Ile-145, Asp-194, Asp-212 and Phe-213, forming 8, 7, 9 and 9 non-bonding interactions of each, respectively. The Asp-212 turned out to have the maximum
(48.05 Å
2) upon binding, followed by Met-223 (36.98 Å
2) (
Appendix A,
Table A5). Of the 17 interacting residues, 13 residues besides ATP were common among the interacting residues of the native inhibitor: Asn-82, Lys-101, Leu-122, Ile-145, Asp-194, Asp-212, Phe-213, Gly-214, Val-215, Ser-216, Leu-219, Ile-220, and Met-223 (
Figure 4A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213.
The sixth-ranked flavonoid
M2F6 (CID: 100986463) docked well in the MEK2 allosteric site (
Figure 3B). The protein-ligand interaction showed 1 hydrogen bond and 72 non-bonded contacts (mainly hydrophobic interactions) through 19 amino acids: Gly-83, Lys-101, Ile-103, Leu-119, Leu-122, Val-131, Gly-132, Phe-133, Ile-145, Met-147, Arg-193, Asp-194, Cys-211, Asp-212, Phe-213, Val-215, Leu-219, Ile-220, and Met-223 (
Figure 5B) (
Table 1). The docking affinity was −10.6 Kcal/mol, while the binding energy was −10.40 Kcal/mol, and the dissociation constant
, was 7.62, which demonstrated a good binding affinity, as needed for the effective inhibition of MEK2 (
Table 2). The hydrogen bond with Lys-101 measures 3.12 Å. The key interacting residues are Lys-101, Ile-145, Asp-212, Phe-213 and Leu-219, forming 7, 6, 9, 6, and 6 non-bonding interactions of each, respectively. Asp-212 proved to have the maximum
with 49.47 Å
2 upon binding, followed by Met-223 (36.55 Å
2) (
Appendix A,
Table A6). Of the 19 interacting residues, 11 residues besides ATP were common among the interacting residues of the native inhibitor: Lys-101, Leu-122, Ile-145, Arg-193, Asp-194, Asp-212, Phe-213, Val-215, Leu-219, Ile-220, and Met-223 (
Figure 5A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213. This suggests that the proposed compound was blocking the same group of residues as the native inhibitor, and hence, it may inhibit the activity of the protein in the same manner.
The dock results of the seventh-ranked flavonoid
M2F7 (CID: 102577930) against the MEK2 binding pocket fitted well in the allosteric site (
Figure 3C). The ligand interacted with 17 amino acids: Asn-82, Gly-83, Lys-101, Leu-119, Leu-122, Val-131, Gly-132, Phe-133, Ile-145, Met-147, Asp-194, Cys-211, Asp-212, Phe-213, Val-215, Leu-219, and Met-223 (
Figure 5C) (
Table 1). The molecular docking results show a docking affinity of −10.5 Kcal/mol, a binding energy of −9.99 Kcal/mol, and a dissociation constant
, of 7.33, showing good quality binding, as required for the adequate inhibition of MEK2 (
Table 2). The interaction shows 2 hydrogen bonds and 56 non-bonded contacts (mainly hydrophobic interactions). The hydrogen bonds with Val-131 measure 2.92 Å, and the hydrogen bonds with Gly-132 measure 2.70 Å. The key interacting residues are Asp-212, Phe-213, and Leu-219, forming 9, 6, and 6 non-bonding interactions of each, respectively. Asp-212 turned out to have the maximum
(49.6 Å
2) upon binding, followed by Met-223 (41.51 Å
2) (
Appendix A,
Table A7). Of the 17 interacting residues, 11 residues besides ATP were common among the interacting residues of the native inhibitor: Asn-82, Lys-101, Leu-122, Ile-145, Asp-194, Asp-212, Phe-213, Val-215, Leu-219, and Met-223 (
Figure 5A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213.
The eighth-ranked flavonoid
M2F8 (CID: 129696793) docked to the MEK2 binding pocket and bound well to the allosteric site (
Figure 3C). The ligand interacted with 17 residues: Asn-82, Lys-101, Ile-103, Leu-122, Val-131, Gly-132, Phe-133, Ile-145, Arg-193, Asp-194, Cys-211, Asp-212, Phe-213, Gly-214, Leu-219, Ile-220, and Met-223 (
Figure 5D) (
Table 1). The molecular docking data show a docking affinity of −10.4 Kcal/mol, a binding energy of −9.71 Kcal/mol, and a dissociation constant
, of 7.12, which was reasonably high, as required for the effective inhibition of MEK2 (
Table 2). The interaction (
Appendix A,
Table A8) shows 2 hydrogen bonds and 57 non-bonded contacts (mainly hydrophobic interactions). The hydrogen bonds with Val-131 measure 2.62 Å, and the hydrogen bonds with Gly-132 measure 2.95 Å. The key interacting residues are Ile-145, Asp-194, and Asp-212, forming 7, 9, and 14 non-bonding interactions of each, respectively. Asp-212 turned out to have the maximum
(48.94 Å
2) upon binding, followed by Met-223 (37.29 Å
2), Asp-194 (34.13 Å
2), and Asn-82 (33.47 Å
2). Of the 17 interacting residues, 12 residues were common among the interacting residues of the native inhibitor: Asn-82, Lys-101, Leu-122, Val-131, Gly-132, Ile-145, Arg-193, Asp-194, Asp-212, Phe-213, Gly-214, Leu-219, Ile-220, and Met-223 (
Figure 5A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213. They may therefore inhibit MEK2 kinase activity in a manner similar to the native inhibitor.
The ninth-ranked flavonoid
M2F9 (CID: 44382144) against the MEK2 binding pocket fitted well in the allosteric site (
Figure 3C). The protein-ligand interaction showed 1 hydrogen bond and 52 non-bonded contacts (mainly hydrophobic interactions) through 14 amino acids: Asn-82, Lys-101, Leu-119, Leu-122, Gly-132, Phe-133, Ile-145, Asp-194, Cys-211, Asp-212, Phe-213, Val-215, Leu-219, and Met-223 (
Figure 5E) (
Table 1). Molecular docking results demonstrated an affinity of −10.4 Kcal/mol, a binding energy of −10.16 Kcal/mol, and a dissociation constant
, of 7.45, showing high-quality binding for the effective inhibition of MEK2 (
Table 2). The hydrogen bond with Lys-101 measures 2.80 Å. The key interacting residues are Asp-212, Phe-213, and Leu-219, forming 12, 6, and 6 non-bonding interactions of each, respectively. The Asp-212 turned out to have the maximum
(50.02 Å
2) upon binding, then Met-223 (40.9 Å
2) (
Appendix A,
Table A9). Of the 14 interacting residues, 10 residues besides ATP were common among the interacting residues of the native inhibitor: Asn-82, Lys-101, Leu-122, Ile-145, Asp-194, Asp-212, Phe-213, Val-215, Leu-219, and Met-223 (
Figure 5A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213.
The tenth-ranked flavonoid
M2F10 (CID: 14626316) bound in MEK2 binding pocket and fitted well in the allosteric site (
Figure 3C). The protein-ligand complex shows 7 hydrogen bonds and 74 non-bonded contacts (mainly hydrophobic interactions) through ATP and 21 residues: Gly-81, Asn-82, Gly-83, Lys-101, Ile-103, Leu-119, Leu-122, Val-131, Gly-132, Phe-133, Ile-145, Met-147, Asp-194, Cys-211, Asp-212, Phe-213, Val-215, Leu-219, Ile-220, and Met-223 (
Figure 5F) (
Table 1). The interaction shows a docking affinity of −10.4 Kcal/mol, a binding energy of −10.35 Kcal/mol, and a dissociation constant
, of 7.59, which is considered good quality binding, as required for the adequate inhibition of MEK2 (
Table 2). The hydrogen bonds with Gly-81 measure 3.04 Å, while the bonds with Asn-82 measure 2.81 Å, the bonds with Gly-83 measure 3.21 Å, the bonds with Lys-101 measure 3.30 Å, the bonds with Asp-194 measure 3.06 Å, and two hydrogen bonds with ATP measure 2.76 Å and 3.14 Å. The key interacting residues are Lys-101, Ile-145, Asp-212, and Phe-213, with 6 non-bonding interactions of each and with Asp-194 demonstrated 7 non-bonding interactions. Asp-212 turned out to have the maximum
(48.8 Å
2) upon binding, followed by Met-223 (37.61 Å
2) (
Appendix A,
Table A10). Of the 21 interacting residues, 12 residues besides ATP were common among the interacting residues of the native inhibitor: Asn-82, Lys-101, Leu-122, Ile-145, Arg-193, Asp-194, Asp-212, Phe-213, Val-215, Leu-219, Ile-220, and Met-223 (
Figure 5A) (
Table 1), and they share the same key residues, Asp-212 and Phe-213. They may therefore inhibit MEK2 kinase activity in a manner similar to the native inhibitor.
All the ten selected flavonoids showed a high docking affinity, binding energy, and dissociation constant score and are proposed as potential inhibitors of MEK2 kinase. Asp-212 was observed as a common key interacting residue, playing a role in the binding of the ten proposed flavonoids as it showed a maximum due to binding (except for M2F3, where it was the second maximum). Additionally, another key residue, Lys-101, was found to form a hydrogen bond in seven cases out of ten. In the remaining three cases, where it did not form a hydrogen bond but appeared as an important interacting residue.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








