Abstract
The most common alterations affecting mitochondria, and associated with cardiac pathological conditions, implicate a long list of defects. They include impairments of the mitochondrial electron transport chain activity, which is a crucial element for energy formation, and that determines the depletion of ATP generation and supply to metabolic switches, enhanced ROS generation, inflammation, as well as the dysregulation of the intracellular calcium homeostasis. All these signatures significantly concur in the impairment of cardiac electrical characteristics, loss of myocyte contractility and cardiomyocyte damage found in cardiac diseases. Mitochondrial dynamics, one of the quality control mechanisms at the basis of mitochondrial fitness, also result in being dysregulated, but the use of this knowledge for translational and therapeutic purposes is still in its infancy. In this review we tried to understand why this is, by summarizing methods, current opinions and molecular details underlying mitochondrial dynamics in cardiac diseases.
1. Introduction
Mitochondria were first discovered in 1856 by the physiologist Von Kölliker as sarcosomes while studying human muscle cells; in 1898, they were named mitochondria by Benda from the combination of two Greek words, mitos (thread) and chondros (granule) [1]. Mitochondria possess a double membrane: an outer mitochondrial membrane (OMM) and an inner mitochondrial membrane (IMM), both delimitate the intermembrane space (IMS). Their structures entail phospholipids which play pivotal roles in the overall dynamics and mitochondrial performance; thereby, defects in their compositions can negatively affect the mitochondrial integrity and permeability [2,3]. Additionally, mitochondria are able to interchange their morphology depending on the physiological environment and developmental phase [4]. The accurate functioning of cardiomyocytes needs a steady harmonization of mitochondrial performance, which is intimately linked to a proper balance of cardiac mitochondrial dynamism and quality control (QC) [5]. Although we are witnessing an increased attention among the scientists on the importance of targeting cardiac mitochondria to dissect further potential therapeutic prevention in various cardiovascular diseases (CVDs), a better understanding of the molecular mechanism of mitochondrial dysfunction focusing on mitochondrial dynamics still remains to be fully elaborated. In the present review article, we will mainly provide a comprehensive report on the morphological changes to which mitochondria are subjected in multiple heart pathologies, drawing particular attention to the mitochondrial dynamics to pave the way for further cardioprotective strategies.
2. Most Common Mitochondria-Related Alterations Accompanying Heart Diseases
Mitochondria are the primary energy source for regular cellular function and they are fundamental for the proper physiology of the cardiovascular system. Nevertheless, it is highly recognized that beyond their hallmark role as a powerhouse, mitochondria are the main sites of reactive oxygen species (ROS) generation and lipid synthesis, intimately involved in calcium (Ca2+) homeostasis and various related signaling pathways [6]. It is not surprising that aberrations in cardiac mitochondrial functioning, structure and number are key factors with a central role in the pathogenesis of multiple chronic human disorders, manifoldly implicated in the development of heart diseases [7]. Therefore, despite the divergence in heart pathophysiology, mitochondrial impairment arises to be a common mechanism that affects cardiac functioning and survival.
The most common alterations in mitochondrial function associated with cardiac conditions implicate further defects in the mitochondrial electron transport chain (ETC) activity, which is a crucial element for energy formation. Then, this is followed by the depletion of ATP generation and supply, reverse metabolic substrate, enhanced ROS generation, inflammation, as well as an aberration in the core myocardial activity of Ca2+ homeostasis [8]. These events result in the impairment of cardiac electrical characteristics, loss of myocyte contractility, altered electrical properties and cardiomyocyte damage [9].
Excessive oxidative stress is a common feature in this field, with predominant role in valves disease [10], atherosclerosis progression [11] and coronary artery disease (CAD) [12]. Mitochondria and ROS are closely associated with inflammation. Indeed, mounting pieces of evidence have illustrated the key role of mitochondrial ROS in stimulating the inflammasome NOD-LRR- and pyrin domain-containing protein 3 (NLRP3) and pyroptosis process [13,14]. Mitochondrial dysfunction also results in a dramatic increase in the permeability of the IMM, triggering in turn a profound involvement of ROS formation, mitochondrial bioenergetics impairment and cell death. One of the causes responsible for IMM permeability is the opening of the mitochondrial permeability transition pore (PTP), primarily involved in RI and associated with intracellular Ca2+ overload [15,16,17,18] that leads to apoptosis, necrosis and many other types of cell death.
Moreover, it is well recognized that mitochondria metabolize fatty acids to generate 60–90% of energy through oxidative phosphorylation (OXPHOS), required for the myocardium. Scarcely, the remaining 10–40% of energy is formed by using glucose and carbohydrates to provide sufficient energy for cardiac cells [19,20]. However, this balance in cardiac energy metabolism could be gradually shifted in pathological conditions, resulting in an energy crisis, mitigating cardiac activity, and it is believed to contribute to multiple heart disorders [21,22]. Several lines of evidence point out the switch in metabolism as accompanied by modifications in the expression of several transcriptional factors, including the upregulation of hypoxia-inducible factor-1α (HIF-1α) [23], downregulation of peroxisome proliferator-activated receptor α (PPARα) and PPARγ co-activator-1α (PGC-1α). Collectively, these modifications support glycolysis elevation, a reduction in mitochondrial oxidation, and depletion of ATP production [24], and these events have been described in pressure overload cardiac hypertrophy, ischemic heart diseases and in heart failure (HF) [25,26].
Moreover, some other mitochondrial alterations may also occur in the mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) mutations. Likewise, accumulation of mtDNA causes a drastic increase in mitochondrial dysfunction [27]; mtDNA is a central element for mitochondrial activity, as it encodes 37 genes, implicating in those transfer RNAs (tRNAs) and 2 ribosomal RNAs (rRNAs), and genes encode the subunit enzyme complexes in ETC [28]. The interaction between mtDNA and nDNA is inevitable for regular cardiac mitochondrial functioning. However, mutations in mtDNA are more frequent than in the nuclear genome; this may be highlighted by the fact that mtDNA is more exposed to detrimental agents, unprotected by the histones, minimal DNA repair and that the mtDNA mutations are caused mostly by uncontrolled ROS formation which generates oxidative stress [29]. Many CVDs are reported to be associated to DNA mutagenesis [30,31,32,33,34].
3. Mitochondrial Dynamics in Physiology
Eukaryotic cells continuously need a large amount of energy to perform their functions, from DNA remodeling, DNA replication, cell division to cardiac contraction and nerve impulse transmission; this is allowed by mitochondria and handled by several mechanisms responsible for their QC. Mitochondria are able to change their morphology, number, size and location inside the cell, thanks to the coordination of a complex system of mitochondrial dynamics, which represents one of the QC mechanisms. As a consequence, the mitochondrial network acquires a dynamic organization, important for combining biological information within the population and assuring a correct cellular energy supply through the balanced processes of fusion and fission [35]. The mitochondrial dynamics homeostasis is fundamental for the physiognomy of the network and for cell fate; furthermore, these morphologic transitions respond to intracellular signals which can be represented also by stressors and thus associated to diseased traits [36,37,38].
When the network becomes fragmented during the fission process, there is an increase in the number of mitochondria, which acquire a smaller and round shape. In physiology, this condition facilitates either the removal of the damaged organelles through mitophagy [39] or the division of the organelles between daughter cells [40]; in contrast, the elongated mitochondria seen in the fused network are fundamental for the maintenance of a healthy mitochondrial status [41]. These two processes, as we will see, need to be in constant balance between them and among the remaining QC mechanisms.
Fission consists in the division of both OMM and IMM with the maintenance of the soluble proteins of the IMS and matrix. When fission is needed for mitochondrial division, at early stages, mtDNA is replicated in order to be distributed to daughter mitochondria, and this event is coupled to the formation of ER-mitochondria contacts, predefined sites of mitochondrial fission [42]. The key protein controlling mitochondrial fission in mammals is dynamin-related protein 1 (DRP1), a large GTPase with cytosolic localization. During fission, DRP1 is recruited to the OMM and through oligomerization induces membrane constriction [43]; GTP hydrolysis is necessary for the conformational changes in DRP1 helices and their constrictive functions [44]. DRP1 binds to the receptors’ mitochondrial fission factor (MFF) [45] and mitochondrial dynamics proteins 49 and 51 (MiD49 and MiD51), also called MIEF 1/2 [46], which help its recruitment and function (Figure 1). However, the immediate translocation of DRP1 to fission sites operated by these receptors is not the unique model to explain its involvement in mitochondrial fission. Indeed, Higgs’ group provided evidence for the formation of a supramolecular complex composed by actin filaments and DRP1, supporting mobility inside cells and then fission. Actin assembly would work as a biochemical signal for DRP1 in stimulating its activity [47]. DRP1 protein is structurally composed by four different domains: the N-terminal with GTPase function, a middle part formed by a central stalk necessary for the assembling of DRP1 at the OMM, a variable domain (B-Insert) and the C-terminal with a GTPase effector domain (GED), fundamental for the formation and the stability of higher order structures at the OMM [48,49].
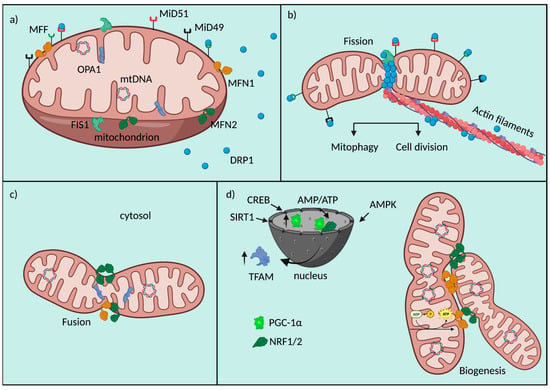
Figure 1.
Fusion/Fission machinery. Schematic representation of mitochondrial fusion and fission machinery and their crosstalk with biogenesis. (a) Mitochondrial players involved in fusion, fission and biogenesis. (b) Mitochondrial fission is orchestrated by a series of molecular events involving OMM receptors (MFF, MiD49, MiD51, FIS1), which help the recruitment of DRP1 to the organelle. DRP1 results to be the main protein effector of mitochondrial fragmentation. (c) Mitochondrial fusion is allowed by the tethering properties of MFNs and the function of OPA1 at the IMM. (d) Mitochondrial biogenesis is at the crosstalk of a nuclear/mitochondrial gene reprogramming involving the upregulation of PGC-1α, NRF1/2, TFAM, their nuclear interactions and fusion proteins.
Another essential OMM protein is FIS1, which is involved in the recruitment of DRP1 at the OMM: this is an important regulator of the assembly of mitochondrial fission complexes [50]. In recent years, a novel role for this protein has been discovered in counteracting the fusion process through the inhibition of the GTPase activity of Mitofusins (MFN) 1, MFN2 and optic atrophy factor 1 (OPA1) [51].
On the other hand, fusion consists in the tethering of two neighboring mitochondria that secondly fuse their OMM and IMM. This process inevitably involves the exchange of biological material of different natures, for example, mtDNA [52]. Usually, material exchange helps to restore mitochondrial homeostasis and to counteract metabolic imbalance, as a defensive mechanism [53]. The core machinery proteins controlling mitochondrial fusion are three GTPase dynamin proteins: MFN1, MFN2 and OPA1. MFNs coordinate the tethering between adjacent mitochondria and consequently OMM fusion; they accumulate at contact areas and can form homo-oligomers (MFN1-MFN1; MFN2-MFN2) and hetero-oligomers (MFN1-MFN2) [54] (Figure 1).
MFNs have been described as composed by a cytosolic N-terminal GTPase domain, a heptad repeat (HR1) domain, two transmembrane anchors and a cytosolic C-terminal HR2 motif. The presentation of this model is based on MFNs yeast topology, which also shows how GTP permits MFNs’ dimerization and how the HR2 domain is able to interact with the N-terminal motif to form a helical bundle, necessary to put membranes in contact and to mediate fusion [55,56]. Recently, Mattie S. and co-workers disputed this model by setting a series of experiments taking advantage of in silico predictive models and in vitro biochemical evidence. Working on animal MFN proteins and highlighting the putative differences between the two topologies, in his model he confirmed a single transmembrane domain with the complete control of oligomerization and consequent fusion by the redox status of two adjacent cysteine residues in the HR2 domain, which is localized exclusively within IMS [57].
OPA1 is a large GTPase responsible for the GTP-dependent fusion of the IMM; its protein structure is mainly localized at the IMS, inserted in the IMM with the N-terminal matrix targeting signal and a transmembrane domain [58]. Mammalian OPA1 gene is transcribed in eight different mRNA variants through alternative splicing of exons 4, 4b and 5b, encoding proteins of 924–1014 amino acids, but the exact function of each isoform is not yet known [59]. Long (L)-OPA1 is the highest molecular weight isoform (~100 kDa) linked to the IMM, that, in case of stress, is cleaved to a soluble short (S)-OPA1 protein (~80 kDa) [60]. L-OPA1 interacts with cardiolipin, a mitochondrial lipid localized in the IMM, with a heterotypic action inducing IMM fusion which is positively regulated by S-OPA1 [61]. In addition to this function, OPA1 acts in the preservation of mitochondrial genome integrity supporting mtDNA replication and distribution [62]; it controls mitochondrial respiratory efficiency and cristae morphology [63].
These processes of fission and fusion are strictly connected to mitochondrial biogenesis [64], another important QC mechanism aimed to preserve cellular homeostasis through the expansion of the mitochondrial network, especially essential in cardiac cells for the generation of an adequate amount of ATP used in muscle contraction. This multistep process includes mtDNA transcription and translation, mitochondrial import of proteins and lipids of nuclear derivation, in order to build new organelles [65] (Figure 1). Given its complexity, biogenesis is controlled by the activation of multiple transcription factors in response to specific stimuli. The main actors working in mitochondrial doubling are two nuclear respiratory factors (NRF1 and NRF2), the estrogen-related receptors (ERR-α, -β, -γ) and PGC-1α [66]. This last one is considered the master regulator of mitochondrial biogenesis, its activity is controlled by an intricate network of signals of different origin (i.e., oxidative stress, exercise or caloric restriction, environmental stimuli) [67]. PGC-1α binds different nuclear transcriptional factors, such as NRF1 and NRF2, which are involved in the control nuclear genes expression linked to mitochondrial respiratory function [68]. Furthermore, PGC-1α can activate transcription factor A (TFAM), a DNA-binding protein responsible for driving the transcription of the mitochondrial genome and of nuclear-encoded proteins with structural properties [69]. Within the major signaling pathways regulating PGC-1α activity, potent inducers of its transcription are members of the transducer of the regulated cAMP response element-binding protein (CREB) family, which are sensors of intracellular Ca2+ and cAMP [70]. In addition, the intracellular AMP/ATP ratio is also important in controlling PGC-1α; when increased, for example, after caloric restriction, it activates AMP-activated kinase (AMPK), inducing the expression of PGC-1α and the increase in mitochondrial biogenesis and fatty acid oxidation rate [71]. AMPK is a metabolic sensor which regulates PGC-1α transcriptional activity also through a direct interaction and phosphorylation, possibly in response to high energy consumption [72] (Figure 1). The regulation of PGC-1α expression is controlled also by Sirtuin 1 (SIRT1), a sensor of redox shifts and a controller of mitochondrial metabolism and inflammation, for example after caloric restriction [73], thanks to post-transcriptional regulation of several targets [74]. Among them, SIRT1 deacetylates PGC-1α leading to its transactivation and inducing the transcriptional activity [75].
A schematic representation of mitochondrial fusion and fission machinery and their crosstalk with biogenesis is as follows. (a) Mitochondrial players are involved in fusion, fission and biogenesis. (b) Mitochondrial fission is orchestrated by a series of molecular events involving OMM receptors (MFF, MiD49, MiD51, FIS1), which help the recruitment of DRP1 to the organelle. DRP1 results to be the main protein effector of mitochondrial fragmentation. (c) Mitochondrial fusion is allowed by the tethering properties of MFNs and the function of OPA1 at the IMM. (d) Mitochondrial biogenesis is at the crosstalk of a nuclear/mitochondrial gene reprogramming involving the upregulation of PGC-1α, NRF1/2, TFAM, their nuclear interactions and fusion proteins.
4. Impairments of Mitochondrial Dynamics in Coronary Artery Diseases
The most common group of cardiovascular pathologies are the so-called CAD, often characterized by a reduction of the blood flow towards the cardiac tissue due to the development of atherosclerotic plaques at the coronary level [76]. CAD includes the phenotypic manifestations of stable and unstable angina, nonfatal myocardial infarction (MI) and HF [77]. The detrimental effect caused by these pathologies is multifactorial: primarily triggered by the inflammatory state that worsens the atherosclerotic process, then promoted to an insufficient balance between the demand and the supply of oxygenated blood to the tissue and to the reperfusion injury (RI), whether the disease is treated at clinical level or not [15,78,79]. Although major mitochondrial alterations in this context have been described (i.e., intense oxidative stress, increased mitochondrial Ca2+ overload, deficiency in ATP production, mitochondrial swelling and cell death [15]), the impairment of QC mechanisms of mitochondria is poorly monitored, but it plays an additional crucial role in CAD and contributes by itself to all those dysregulations (Table 1).

Table 1.
Representative list summarizing alterations in mitochondrial dynamics in different cardiovascular disorders.
4.1. Ischemic Diseases
Overall, fission, fusion and biogenesis are deeply altered after MI, with a significant activation for the first mechanism and the pathological disruption for the last two, as sides of the same coin.
Being the main contributor of mitochondrial fission, DRP1 was the most studied protein in this field. After cardiac stress (i.e., prolonged ischemia, inflammation, mechanical stretch), many authors reported an excessive activation of DRP1 and the consequent mitochondrial fragmentation [80,81,82]. Molecular routes by which DRP1 function is overactivated in ischemia and reperfusion (I/R) are multiple (Figure 2). In 2014, Sharp W. and co-workers reported a significant calcineurin-dependent dephosphorylation of DRP1 at S637, which determined its massive mitochondrial translocation [80]. Another mediator impacting on S637 phosphorylation grade is the suppressor of cytokine signaling 6 (SOCS6); it is usually upregulated during I/R and responsible for the inhibition of S637 phosphorylation and the increased DRP1 translocation to mitochondria with consequent fragmentation [81]. Moreover, it has been shown how DRP1 activity may be indirectly dependent on HIF-1α expression, that is upregulated by the decreased expression of miR-138 during I/R [112] (Figure 2). Experiments of reversion valued these molecular intermediates in the control of fission-dependent mitochondrial impairments; indeed, both the repression of SOCS6 by miR-19b injection [81] and the lowered expression of DRP1 induced by miR-138 administration [112], as well as the direct inhibition of DRP1 through mitochondrial division inhibitor 1 (Mdivi-1) [82,94], successfully controlled mitochondrial fragmentation upon cardiac stress.
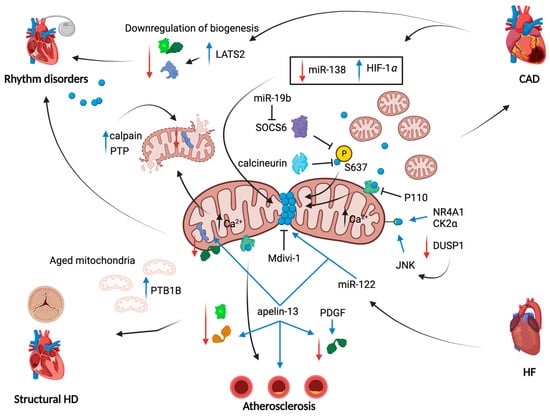
Figure 2.
Mitochondrial dynamics alterations in heart diseases. Schematic overview of molecular pathways through which mitochondrial fission, fusion and biogenesis contribute to pathological aspects of heart diseases. Overall, mitochondrial fragmentation is the main common feature of heart diseases reported in this review. The ways by which mitochondria become fragmented are multiple and are aimed to anchor DRP1 to mitochondria. In CAD, a link with the HIF-1α has been found and involves the downregulation of miR-138; a second miRNA, miR-19b, blocks SOCS6 kinase function to prevent DRP1 phosphorylation on Ser-637. The same effect is obtained by the increase in activity of calcineurin. A concomitant decrease in mitochondrial fusion has been reported: in atherosclerosis development, apelin-13 promotes the downregulation of MFNs and OPA1. Also, mitochondrial biogenesis is downregulated through the loss of PGC-1α, NRF1/2 and TFAM expression, especially in CAD and in rhythm disorders in a Hippo pathway-dependent manner. Red arrows: downregulation of a given protein/pathway; blue arrows: upregulation of a given protein/pathway; black arrows: cause-effect link.
In some cases, such as the use of Mdivi-1, it was realized that the chronic inhibition of DRP1 cannot be used for therapeutic purposes because it can acquire additional pleiotropic effects in cardiomyocytes. This is the case of the proteolytical cleavage of L-OPA1 and changes in OXPHOS complex proteins’ expression, leading to increased superoxide generation, with impairments of mitochondrial respiration and inhibition of the autophagic process [113]. These results thus suggested that chronic Mdivi-1 treatment could negatively control cardiac tissue function, limiting its potential use in therapy for CAD.
The detrimental effect of enhanced DRP1 expression on cardiac tissue was confirmed also thanks to the use of an additional selective inhibitor of the fission machinery, P110, which acts on the interaction of FIS1/DRP1 fission proteins by inhibiting DRP1 mitochondrial association in several models of I/R. Prevention of excessive mitochondrial fragmentation and the improvement of mitochondrial oxygen consumption, as well as some long-term effects monitored in rats subjected to acute MI, revealed its therapeutic potential [83].
Whatever the regulatory route, the phenotypic effects of the increased DRP1 function result to be major contributors of ROS production, Ca2+ overload, cell death and left ventricular (LV) dysfunction. Direct and indirect inhibition of DRP1 mostly reduces MI size, ROS levels in mouse models subjected to coronary artery occlusion and reperfusion damage [80,82]. In a study carried out on CAD patients and investigating the involvement of rs28362491 polymorphism on the NFKB1 gene in the progression of the pathology, its role in the mitochondrial fission process has been discovered. This gene, linked to CAD, encodes for the nuclear factor kappa B (NF-κB) which has been demonstrated to induce, when mutated, increased sensitivity to oxidative stress-induced apoptosis in endothelial cells. NFKB1 gene mutation significantly increased the expression of DRP1 after stimulation with apoptotic stimuli, causing uncontrolled mitochondrial fission and mitochondrial dysfunction [114].
Despite the evidence furnished on the harmful role of DRP1 upregulation in the ischemic disease the cardiac-specific knockdown of DRP1 in mice has also been classified as lethal. Indeed, several mitochondrial impairments, with an excessive state of elongated mitochondria, caused a significant reduction of the mitophagic process that finally led to LV dysfunction and mice death [84]. These data are not considered to be opposed to the previous ones, but further confirm the crucial role of DRP1 (and fission) and its balance in heart homeostasis during life and disease.
Other proteins involved in mitochondrial fission processes deserve attention in CAD. This is the case of DRP1 receptors at OMM, such as MFF, which help the recruitment of the protein at the mitochondrial level. MFF is demonstrated to be activated during I/R, after a cascade involving the nuclear receptor subfamily 4 group A member 1 (NR4A1) and serine/threonine kinase casein kinase2 α (CK2α), which directly act on MFF. A second molecular axis activates MFF, that involves the I/R-mediated downregulation of Dual-specificity protein phosphatase1 (DUSP1) and the activation of c-Jun N-terminal kinase (JNK) leading to the contribution of the RI by apoptosis [115].
This evidence was deepened thanks to MFF deletion studies carried out both in vitro and in vivo: MFF loss reduced exacerbation of mitochondrial fission in I/R, preserving mitochondrial functionality and tissue rescue from damage. It has been discovered that MFF activation leads also to the dissociation of hexokinase 2 (HK2) from mitochondria, which leads to uncontrolled ROS production, PTP opening, cytochrome c release and cellular apoptosis [85].
If on one hand mitochondrial fragmentation depends on the excessive function of fission proteins, on the other hand it can be easily predicted that also MFN1/2 and OPA1 deregulation may be involved in I/R. However, their exact behavior in these kinds of diseases is poorly understood and results in being controversial. Indeed, most studies established to this aim evaluated the genetic ablation impact of the proteins in mouse or rat model diseases. Although this kind of approach helps to elucidate their role once totally deprived, it is not useful to understand their trend in expression and function during disease.
Among the in vivo experiments that were set up to understand the therapeutic potential of improving mitochondrial fusion, a mitochondrial fusion promoter-M1 has been used on rats subjected to I/R, showing a high grade of MI size reduction and associated mortality when administered before ischemia, and a lower but significant effect at the time of reperfusion [86]. This treatment has gained importance for both the selective way of action in counteracting the decrease of MFN2 and OPA1 (fission processes were not affected) and for the reduction of ROS levels, mitochondrial membrane depolarization and swelling with a specific effect on mitochondrial dynamics [86]. With the same purpose, but using conditional cardiac-specific mouse models of YME1L and OMA1 deletion, research studies have addressed the essential role of the L-OPA1 isoform in preserving cardiac function [88]. YME1L and OMA1 are two mitochondrial proteases involved in OPA1 cleavage from L-OPA1 to S-OPA1 forms; in the absence of YME1L, cardiomyocytes experienced enhanced OPA1 proteolysis with greater mitochondrial fragmentation. This macroscopic feature is linked to increased cell death and a heart phenotype resembling that of dilated cardiomyopathy with metabolic drift from lipid to glucose metabolism [88]. Ex vivo experiments on rat hearts subjected to I/R revealed an impaired balance between L-OPA1 and S-OPA1 forms, due to an increase in OMA1 activation and L-OPA1 cleavage that in turn is associated with cardiac dysfunction [116]. Similar findings were obtained by in vitro experiments on cardiomyocytes, where S-OPA1 expression augmented after I/R and changed localization from the mitochondria to the cytosol [117]. OPA1 can be considered a cardioprotective target for therapies against HF; indeed, enhancing fatty acid utilization in vivo through a high fat diet, OPA1 overexpression improved cardiac function by reducing mitochondrial fragmentation [91].
Moreover, some reports described the mitochondrial Ca2+ uniport (MCU) as upregulated during I/R, with a consequent increase in mitochondrial Ca2+ levels and PTP opening. The consequent calpain expression increase downregulated OPA1-mediated mitochondrial fusion. The final result also in this case is an overactivation of fission, fusion and mitophagy inhibition accompanied by the activation of the apoptotic pathway [87].
Mitochondrial QC mechanisms’ involvement in CAD is not only limited to unbalanced mitochondrial dynamics; in fact, there is evidence that mitochondrial biogenesis is altered, too [118] (Figure 2).
Major findings have been obtained from ex vivo studies, in which human samples of failing hearts have been processed for DNA microarrays. In detail, PGC-1α, the key regulatory partner ERR-α and a series of downstream gene targets have been found downregulated [119]. To this, a significant decrease in mtDNA content and expression of mtDNA-encoded proteins [120], the downregulation of NRF1, TFAM and DNA Polymerase Gamma 1 (POLG1) and decreased expression of proteins involved in mtDNA maintenance are observed [118]. In vivo animal models have confirmed this complex pattern, highlighting the decrease in nuclear-encoded mitochondrial gene expression and in PGC-1α, NRF-2α and mtTFA (a nucleus-encoded protein involved in transcription and replication of mtDNA) gene expression [121]. In three pioneer studies, Fabregat-Andres et al. investigated the peripheral blood concentration of PGC-1α (mRNA and protein) in ST-segment elevation acute myocardial infarction (STEMI) patients at admission time. PGC-1α resting levels detected at hospital admission resulted inversely correlated with the extent of tissue damage in patients with MI [122]. However, PGC-1α expression was increased in blood cells at 72 h and this induction was positively correlated with the infarct size and cardiac remodeling [123], so the authors finally suggested that increase of PGC-1α expression can be part of the recovery response of the mitochondrial protection system to MI, and could be an index of cardiac recovery [124].
A recent work by Chen et al. revealed the possibility of the intersection of the Hippo pathway [125] with mitochondrial biogenesis by detecting an increased expression of large tumor suppressor kinase 2 (LATS2) during I/R in cardiomyocytes. When deleted, it supports cardiomyocyte survival and mitochondrial biogenesis; indeed, it induced the increase in the expression of PGC-1α, in association with the rise of mRNA levels of TFAM and NRF1 attenuating RI [126]. Very recently, an innovative approach aimed to reduce RI has been proposed with much interest in the scientific world: mitochondria transplantation. This technique consists of the use of functionally intact autologous mitochondria from healthy cells or tissue that are directly transplanted in the ischemic tissue right before the reperfusion process. The procedure has revealed incredible results in reducing myocardial damage induced by mitochondrial impairments, restoring sufficient energy for cardiac homeostasis [127]. The mechanism of internalization of the autologous mitochondria in a different tissue has been studied and a key role of NRF2 was revealed. This transcription factor controls mitochondrial biogenesis by influencing the expression levels of other critical transcription factors, such as NRF1 and PGC-1α, as well as by improving purine nucleotide biosynthesis [128]. It has been demonstrated that, during mitochondrial transplantation, both NRF2 expression and its downstream targets’ expression are increased and the cardiac injury is reduced due to the stabilization of the mitochondrial fission and fusion process, reduction of apoptosis and improved mitophagy [129]. Modulation of mitochondrial biogenesis might be an efficient technique to restore cellular homeostasis in cardiac disease and pave the way for promising mitochondrial-specific therapies.
4.2. Heart Failure
The last maladaptive stage of most pathologies, not only referring to the cardiac district, is HF. This clinical picture is characterized by several molecular and macroscopic derangements that lead to the absence of an efficient contraction of the cardiac muscle. The first association of mitochondrial fusion and fission proteins in HF dates back to the 2000s, when some groups reported a significant downregulation of OPA1 and MFN2 proteins and FIS1 upregulation as responsible for mitochondrial impairments and apoptosis [89,90]. Of note, the levels of these proteins can vary significantly among the process leading to end-stages of cardiac remodeling, and this fluctuating trend has not been well investigated. In HF, clinicians found the microRNA-122 (miR-122) to be one of the most elevated miRNAs in affected patients [130]. By overexpressing a cardiac-specific version of miR-122, it has been found that increased apoptosis is linked to an increase in DRP1 expression and mitochondrial fission, which contributes to HF [92] (Figure 2). Likewise, analysis on tissue samples from the left ventricular wall of failing canine and human hearts showed an increase in FIS1 and DRP1 proteins and a concomitant reduction of MFN2 and OPA1 expression when compared to control donors [131]. More evidence that matches mitochondrial fission and the onset of HF is given by the establishment of the MFF-KO mouse model. Indeed, MFF-deficient mice died at 13 weeks developing HF, with a cardiac tissue characterized by low mitochondrial density, reduced mitochondrial respiration and augmented mitophagy [132].
A recent study focused on the differences in mitochondrial morphology, dynamics and biogenesis in the two large classes of patients suffering from HF: those with preserved ejection fraction (HFpEF) and those with reduced ejection fraction (HFrEF). HFpEF patients presented mitochondrial fragmentation without changes in protein expression of the fission machinery. HFrEF patients instead showed higher marked mitochondrial structural and morphological changes, with a significant increase in expression of DRP1 and decreased PGC-1α, without differences in the expression of OPA1 and MFN2 [93]. Additionally, in this disease context, PGC-1α played a cardioprotective function in response to cardiac stress like that derived from transverse aortic constriction; in fact, when genetically repressed in mice heart, it leads to an acceleration of the cardiac dysfunction with the final result of HF [133]. A similar role for PGC-1β in controlling cardiac function during HF has been found thanks to the use of PGC-1β-deficient mice, which exhibited an augmented propensity to develop this pathology, due to higher levels of oxidative stress, mitochondrial dysfunctions and a reduction of expression levels of OXPHOS genes [134].
5. Impairments of Mitochondrial Dynamics in Heart Rhythm Disorders
Irregular heartbeat characterizes some cardiac pathologies named as arrhythmias, which make up a large percentage of heart diseases. In arrhythmia, the electrical impulse sequences are inconsistent, either too fast, too slow or non-uniform. The improper beating of the cardiac muscle causes an inefficient pump of blood throughout the other vital organs, which may be ultimately compromised [135]. It has been widely recognized that mitochondrial dysfunctions are prominent in the incidence of cardiac rhythm disorders, mainly due to impaired function of intracellular ion channels, to the insufficient energy generation and excessive ROS production, thus leading to an incorrect action potential propagation and adulterated electrophysiology of cardiac myocytes [8,135,136].
Atrial Fibrillation
Among cardiac arrhythmogenesis disorders, atrial fibrillation (AF) is the most prominent, with an age-related increase in its prevalence [137]. In the last decade, the pathophysiology of AF has been deeply investigated; however, the implications of mitochondrial dysfunctions, in particular the ones which correlate with mitochondrial dynamics, are not fully understood (Table 1).
The first evidence on the involvement of mitochondrial alterations in AF dates back to an early work by Thidemann and Ferrans, in which they found a variation in size (smaller organelles) and number of mitochondria in atrial tissues from patients affected by AF [138]. This finding has been confirmed more recently by Wiersma et al., who assessed that AF is characterized by a shift from tubular mitochondria to a fragmented network, and that this phenotype is associated with cytosolic and mitochondrial Ca2+ overload with compromised bioenergetics [139].
Dong et al. used a rabbit model of rapid atrial pacing to demonstrate for the first time that also mitochondrial biogenesis is impaired and is at the basis of AF etiopathology [140]. In his paper, they found that mtDNA levels were reduced in atrial tissue, and the expression of mitochondrial biogenesis transcription factors (i.e., PGC-1α, NRF-1 and TFAM) were decreased together to the mitochondrial respiratory chain complexes. These data correlate with the fact that AF is also characterized by energy metabolism remodeling.
It is important to mention that several risk factors promote AF; however, type 2 diabetes mellitus (T2DM) is the most prevalent [141]. In the study by Shao and co-workers, the T2DM animal group exhibited higher incidence of AF; this event was correlated to a significant reduction in protein expression of PGC-1α, NRF-1 and TFAM, as well as the concomitant downregulation of DRP1, MFN1 and OPA1 [95]. These data suggest impairment of both mitochondrial biogenesis and the fusion-fission process in cardiac tissue. Similar results were obtained by another independent group with the use of a rabbit model of T2DM: their data confirmed a correlation between T2DM and a higher incidence of AF, which is associated with downregulation of mitochondrial biogenesis and fusion and the upregulation of fission-related proteins [142]. Accordingly, Montaigne and co-workers confirmed the presence of fragmented mitochondria directly in patients, as a consequence of MFN1 downregulation in the atrial tissue [96]. Interestingly, patients affected by post-operative AF also exhibited an impaired level of PGC-1α [143], suggesting that mitochondrial biogenesis could be a target platform for AF treatments.
Cardiac arrhythmogenesis involves not only atria but can also occur at the ventricular level. Ventricular fibrillation (VF) is the main cause of sudden cardiac death and is one of the major causes of mortality worldwide [144]. New evidence shows the involvement of proteins which regulate mitochondrial dynamics in VF. Ventricular tissues from rabbit models displayed increased levels of DRP1 and no variation in MFNs [145]; a different result was obtained in an in vivo model of catecholaminergic polymorphic ventricular tachycardia (CPVT) aggravated by the mutation in the trans-2, 3-enoyl-CoA reductase-like (TECRL) gene which encodes for an endoplasmic reticulum protein, published to be associated to inherited arrhythmia [146,147]. The authors showed how TECRL mutation or deficiency triggers significant mitochondrial impairments with decreased mitochondrial biogenesis and fusion protein MFN2 [147]. Interestingly, both AF and VF have been recently seen to be associated with degenerative cardiac PGC-1β dysfunction. It has been demonstrated that PGC-1β null mice exhibit impaired action potential conduction, together with the presence of abnormal mitochondria and altered metabolism [148,149,150].
6. Impairments of Mitochondrial Dynamics in Structural Heart Diseases
Structural heart diseases include all those categories of cardiovascular disorders in which an abnormal structure of the heart muscle (valves, walls and chambers) is associated with a pathological function. Any problem with one of the four heart valves can lead to obstructive blood flow. It is very common among the population and it includes a heterogeneous group of valvular impairments with different incidence; they can be congenital [151,152] or develop with age [153,154]. Sometimes, they are associated with other heart diseases, and if not treated in a timely fashion, they might lead to HF. Otherwise, when the structural problems involve the muscle, we can speak about cardiomyopathies (CM). Their classification has evolved along the years [155]. Hypertrophic cardiomyopathy (HCM) [156,157] and dilated cardiomyopathy (DCM) [158,159] are the most prevalent worldwide.
6.1. Valvular Heart Disease
In the last decade, mitochondrial dysfunctions have been much more investigated in valvular diseases. In 2017, Rogers and colleagues demonstrated for the first time that calcific regions of human carotid arteries and calcified human aortic valves (CAVS) express very high levels of DRP1 [97]. Microscopic analysis investigated mitochondrial morphology and reported increased mitochondrial fragmentation. This altered morphology was independent from DRP1, as Mdivi-1-mediated inhibition fully restored morphology [97]. Moreover, inhibition of DRP1 reduced the calcification process both in primary human smooth muscle cells and valve interstitial cells via the increase in SRY-box transcription factor 9 expression, suggesting the role of DRP1 in the promotion of osteogenic differentiation [97]. Four years later, in the paper by Morciano et al., an additional puzzle was put down regarding the investigation of several mitochondrial QC mechanisms [98]. Besides the contribution of mitophagy, mitochondrial biogenesis has been evaluated in human calcific aortic valves, showing a significant downregulation of PGC-1α which correlated with aged mitochondria and insufficient mitochondrial turnover [98,160].
In 2022, it was demonstrated that a protein located at the endoplasmic reticulum (ER), the protein tyrosine phosphatase 1B (PTP1B), is expressed in aortic valve stenosis. Immunohistochemistry of a human sample of CAVS displayed increased expression of PTP1B compared with healthy valves. The pharmacological inhibition and the downregulation of PTP1B limited osteogenic differentiation of interstitial valvular cells through the regulation of OPA1 homeostasis [161] (Figure 2). Moreover, drug-induced PTP1B inhibition promoted mitochondrial biogenesis [161], which was demonstrated from our group to be downregulated in CAVS [98].
Bicuspid aortic valve (BAV) is one the most common congenital heart diseases, characterized by the presence of only two cups, which affect almost 1.4% of the population [162]. It has been reported that there is a strong association between BAV and thoracic aortic aneurism (TAA) [162]. A recent study highlighted unbalanced mitochondrial dynamics and mitochondrial bioenergetics in BAV-TAA tissues accompanied with reduced expression of NOTCH1 [99]. The authors used an innovative model, aorta smooth muscle-on-a-chip, to assess the correlation between NOTCH1 downregulation and mitochondrial dynamics. They demonstrated that MFN1 and MFN2 expression was reduced in the BAV-TAA group; on the contrary, DRP1 and MFF levels were increased even if not correlating significantly with the downregulation of NOTCH1 [99]. Although this study has many limitations, it provides the basis for future deeper investigation of the connection between BAV and mitochondrial dynamics dysfunctions, which is still very lacking.
6.2. Cardiomyopathies
There are multiple works that demonstrate mitochondrial dynamic impairments in the pathogenesis of DCM [158] (Table 1). A notable example derives from the studies by Ashrafian H., who identified a mouse model, called “Python mouse”, with a missense mutation in the middle domain of the Dnm1l gene that spontaneously developed DCM in a fully penetrant way [163]. C452F mutation in that gene confers to mitochondria an unexpected elongated morphology with the disruption of the fission process; additionally, energetic defects have been found, resembling the DCM phenotype. The chronic elongated mitochondrial phenotype has deleterious consequences, including loss of mitophagy induction, depolarization and impaired ATP production with a progressive decline in OXPHOS [100].
Overall, literature teaches us that almost all animal models characterized by heart-specific KO of a given mitochondrial dynamics-related protein show profound alterations, that all together concur with age to DCM onset. DRP1 KO mice exhibited hypertrophy, LV dysfunction [84] and lethal DCM [101]; when conditionally turned off, they also displayed a decrease in both mitochondrial fission (MFF and FIS1) and fusion proteins (MFN1/2 and L-OPA1), confirming an impaired balance in the mitochondrial dynamics machinery of these hearts [101]. To this regard, Wai et al. demonstrated that cardiac-specific deletion of YME1L triggers the onset of DCM through the activation of OMA1 and induced proteolysis of OPA1 [88], subsequently showing fragmented mitochondria in cardiomyocytes isolated from these mice hearts. These data were confirmed by an independent study, in which heterozygote OPA+/− mice had a concomitantly slow development of DCM and exhibited a reduction in the mitochondrial fusion process [164]. Additionally, the deletion of MFF, as investigated by Chen et al., triggered the death of mice at 13 weeks, due to severe DCM [132]. Interestingly, the same study showed that the simultaneous deletion of MFF and MFN1 prevented cardiac defects, probably balancing mitochondrial fission and fusion mechanisms. However, with the cardiac-specific deletion of both MFN1 and MFN2, DCM is lethal to mice, with a severe mitochondrial fragmentation, reduced mtDNA content and alterations in the biogenesis process [38,102,103]. Recently, these results have started to be partially confirmed also in humans, who are affected by DCM and low levels of MFN1 and mitochondrial fragmentation in cardiomyocytes [165]. In the same diseased patients, but from a study of an independent research group, mitochondrial biogenesis was found as a compensatory mechanism, extremely upregulated, sustained by the upregulation of PGC-1α, NRF1/2 and TFAM [166]. Abnormal biogenesis in DCM human samples correlated with morphological and respiratory dysfunctions [167].
7. Insights on the Role of Mitochondrial Dynamics in Atherosclerosis
Atherosclerosis is a multifactorial degenerative disease characterized by alterations of the artery wall, which loses elasticity due to the accumulation of Ca2+, cholesterol, inflammatory cells and fibrotic material.
Compelling evidence highlighted a key role for mitochondrial dynamics in endothelial dysfunction, a hallmark of early atherosclerosis (Table 1). Atherosclerosis is an increasing complication in patients with diabetes; therefore, many studies have focused on the role of endothelial dysfunction in diabetes-induced CVD. Accordingly, it was found that mitochondrial fragmentations and FIS1 expression were increased in venous endothelial cells isolated from patients with diabetes mellitus [104]. A similar trend was also observed in cultured human aortic endothelial cells (HAEC), which showed a loss of the mitochondrial network and an increase in FIS1 and DRP1 expression upon glucose [104] or adipokine RBP4 [168] treatment. This phenotype was rescued by the inhibition of mitochondrial fission [104]. Moreover, DRP1 inhibition by Metformin was remarked to improve the endothelial activity and to prevent the atherosclerotic lesions in apolipoprotein E-deficient mice [105]. Metformin does not directly act on DRP1, but it triggers AMPK activation that in turn reduces the levels of the fission protein; additionally, AMPK promotes DRP1 Ser-637 phosphorylation to limit its translocation to mitochondria. Of note, only the AMPK-α2 isoform blocks the effect of Metformin on mitochondrial dynamics when depleted. All these studies confirm that alterations of mitochondrial fission contribute to the development of atherosclerosis in diabetes.
Chehaitly A. and co-workers recently investigated the role of fusion in endothelial cells’ responses to flow since shear stress-mediated dilation is another risk factor for atherosclerosis. They found that the disturbed flow decreased OPA1 expression and mitochondrial length in vitro. In addition, shear stress-mediated dilation was reduced in the arteries of OPA1+/− mice. The absence of OPA1 did not influence the acetylcholine-mediated dilation, thus OPA1 deficiency may selectively affect the endothelial response to flow [106]. Beside the endothelial component, vascular smooth muscle cells (VSMC) also play a central role in the formation of atherosclerotic plaque. Abnormal proliferation and migration of VSMCs promotes plaque formation and they are closely related to plaque vulnerability. The activation of VSMC by platelet-derived growth factor (PDGF) was found to induce a loss of the mitochondrial network associated with a significant decrease in MFN2 levels, without affecting the number of mitochondria. Inhibition of mitochondrial fragmentation avoided the hyperproliferation response of VSMCs to PDGF, demonstrating that the conversion of VSMCs in the proliferating phenotype was associated with mitochondrial morphology [108] (Figure 2). Along this line, another study observed that MFN2 overexpression significantly reduced VSMCs’ proliferation and intima thickening in vivo by suppressing AKT-mediated survival pathways [109,110]. These results are consistent with studies demonstrating that mitochondrial fission also plays a role in VSMCs’ hyperproliferation. Interestingly, apelin-13, a stimulator of VSMCs’ proliferative activity, reduced not only the expression of MFN2, MFN1 and OPA1, but it also increased the expression levels of DRP1 in human aortic cells [111]. The same group found that apelin 13 accelerated atherosclerosis in the aorta, while the pharmacological inhibition of DRP1 restored the physiological mechanism of fission and attenuated the proliferating activity of VSMCs in APOE−/− mice [111]. Marsboom et al. also confirmed that the inhibition of DRP1 by Mdivi-1 restored the mitochondrial network and slowed the proliferation of pulmonary artery smooth muscle cells [107].
Arterial calcification is another hallmark of atherosclerosis and DRP1 seems to play an important role in this phenotype. In fact, DRP1 promotes human cardiovascular calcification via regulating osteogenic differentiation [97]. DRP1 inhibition attenuates matrix mineralization, cytoskeletal rearrangement, leading to the lowering of VSMC calcification. Taken together, these findings strongly suggest that both the enhancement of fission and the reduction of fusion have a relation with atherosclerosis pathophysiology.
Among the mitochondrial quality control mechanisms, the role of mitochondrial biogenesis in the onset of atherosclerosis has drawn the attention of the scientific community. The overexpression of PGC-1α in VSMCs significantly ameliorated HCD-induced atherosclerotic plaque formation in rabbit models. High levels of PGC-1α in VSMCs inhibited the switch of VSMCs in the proliferating status [169]. In contrast, Steins’ paper reported that the mouse model PGC-1α−/−/APOE−/− exhibited a reduction of total body and visceral fat weight, but it did not show differences in the atherosclerotic burden compared to PGC1α+/+/APOE−/−. The decreased inflammation as a consequence of reduced visceral fat in PGC-1α KO animals could explain why they did not develop atherosclerosis [170]. Moreover, it was reported that PGC-1α polymorphisms correlated to the atherosclerosis onset and its complications [171,172,173]. Although the role of mitochondrial biogenesis in atherosclerosis is not well established, collectively, these data make it clear that modulation of mitochondrial dynamics could be a possible therapeutic strategy to slow atherosclerosis progression and plaque formation.
A schematic overview of molecular pathways through which mitochondrial fission, fusion and biogenesis contribute to pathological aspects of heart diseases is as follows. Overall, mitochondrial fragmentation is the main common feature of heart diseases reported in this review. The ways by which mitochondria become fragmented are multiple and are aimed to anchor DRP1 to mitochondria. In CAD, a link with the HIF-1α has been found and involves the downregulation of miR-138; a second miRNA, miR-19b, blocks SOCS6 kinase function to prevent DRP1 phosphorylation on Ser-637. The same effect is obtained by the increase in the activity of calcineurin. A concomitant decrease in mitochondrial fusion has been reported: in atherosclerosis development, apelin-13 promotes the downregulation of MFNs and OPA1. Additionally, mitochondrial biogenesis is downregulated through the loss of PGC-1α, NRF1/2 and TFAM expression, especially in CAD and in rhythm disorders in a Hippo pathway-dependent manner. Red arrows: downregulation of a given protein/pathway; blue arrows: upregulation of a given protein/pathway; and black arrows: cause-effect link.
8. Still Little Data from Clinical Studies
From data reported so far, the accumulation of fragmented and damaged mitochondria results to be closely related to the development of CVD. Despite the therapeutic potential of fission/fusion modulation in CVD, there are currently few active clinical trials. Much preclinical evidence supports the potential effectiveness of molecules targeting mitochondrial dynamics and biogenesis to treat CVD. Future challenges will be to turn these molecules into treatments, but clinical studies are still needed to validate their efficacy and safety.
In 2013, the clinical trial of Metformin in patients with peripheral artery disease (PAD) was started to evaluate its beneficial effects on skeletal muscles (NTC01901224). Unfortunately, the results of this clinical study have not been published because it was terminated prematurely due to lack of funds. Based on preliminary data showing that DRP1 inhibition reduces atherosclerotic plaque formation in ApoE KO models [105], another trial aimed to investigate the role of DRP1 in human macrophages and to identify new therapeutic targets has been established (NCT03980548). The study is currently ongoing. A non-randomized interventional study that began this year aims to compare mitochondrial dynamics between patients operated on for ascending aortic aneurysm, type A aortic dissection and a control group (NCT05434481).
Many clinical trials have investigated sodium-dependent glucose transporter 2 (SGLT2) inhibitors in treating diabetic-induced cardiac diseases. Particularly, a phase III double-blind randomized study (NCT01131676) evaluated the effects of Empagliflozin, as a SGLT2 inhibitor, in patients with T2DM and high cardiovascular risk. The inhibitor’s administration reduced cardiovascular mortality and hospitalization [174]. Experimental studies unveiled the positive effects of SGLT2 inhibitors on mitochondrial biogenesis by PGC-1α and SIRT1 activation [175]. SGLT2 inhibition reduces FIS1 and enhances expression levels of MFN2 and OPA1 both in vitro and in vivo [176]. Although the effects on mitochondrial dynamics in humans are still to be confirmed, SGLT2 represents a promising target against cardiac disease. Melatonin was also found to be a cardioprotective molecule against different cardiovascular pathologies. Preclinical studies recently investigated the role of melatonin on vascular calcification and demonstrated that melatonin promoted OPA1-mediated mitochondrial fusion and decreased VSMCs’ calcification [177,178]. A meta-analysis of seven randomized clinical trials revealed that melatonin administration attenuated LV ejection fraction and heart dysfunctions [179]. The effects of melatonin on mitochondrial dynamics have not yet been evaluated in humans, and further trials are needed to confirm the data obtained in the experimental studies. Taking all of this into consideration, the lack of clinical studies has not yet allowed for the validation of the effectiveness of preclinical strategies in improving mitochondrial morphology and metabolism in cardiovascular disease.
9. Mitophagy and Mitochondrial Unfolded Protein Response
Mitophagy and mitochondrial unfolded protein response (mtUPR) are considered additional mechanisms of QC, because they are aimed to restore a condition of mitochondrial integrity following stress. The first one is expected to remove damaged organelles through a localized and selective process of lysosomal digestion [180], the second one is a cellular protective response which follows the accumulation of unfolded proteins in the mitochondrial matrix [181].
Balanced mitophagy counteracts several mitochondria-mediated pathological processes, especially those linked to mtROS overproduction and the activation of the chronic inflammatory response. An obvious example is represented by atherosclerosis, in which the activation of the canonical pathway attenuates NLRP3 inflammasome activity, IL-1β maturation and mtROS levels; this helps to stabilize atherosclerotic plaques and to downregulate the inflammatory response [11,182]. Mitophagy is also induced after I/R events with cardioprotective properties which allow cellular ATP preservation [183] and cell survival [184]. These notions make us understand how essential this process is. Inefficient mitophagy has been found to be linked to HF [185,186], vascular calcification [98] and congenital heart disease [187]; here, the targeting of mitophagy has improved the disease traits.
Otherwise, the causes and the effects of the activation of mtUPR in heart diseases are still little known; nevertheless, the induction of mtUPR is considered to be a pro-survival pathway [188] to the same extent as mitophagy. Some in vitro stressors (i.e., complex I inhibition, Hsp90 loss of function, unfolded proteins) and in vivo changes in hemodynamic parameters (i.e., pressure overload in HF) trigger the transient activation of mtUPR in the heart. Through a linked nuclear reprogramming, which is induced by mtUPR and largely independent from ATF5, an upregulation of an adaptive gene response is able to restore mitochondrial dysfunctions and cardiac contractility [189,190].
10. Conclusions
From all these notions, in a controversial appearance, we can exclude the possibility of clearly defining a disease trait as characterized by a well-defined pattern of proteins’ expression and function related to mitochondrial dynamics and biogenesis impairments. Although mitochondrial fragmentation and disruption of mitochondrial biogenesis are recognized as predominant mechanisms in heart diseases, the protein pool involved in these routes is not equally described among reports. As a consequence, selective therapeutic approaches against CVDs with the use of inhibitors of mitochondrial fission or promoters of mitochondrial fusion and biogenesis are still in their infancy. One thing stands out to be clear and homogeneous among the studies analyzed: the need for balance among all QC mechanisms, including mitochondrial dynamics, mitophagy and biogenesis, at each stage of heart development. This is a delicate balance that is difficult to measure and to be targeted, but once dysregulated, leads to pathological effects.
Author Contributions
Conceptualization, G.M.; validation, G.M., P.P. and E.T., writing—original draft preparation, G.M., C.B., G.P., D.R. and E.B.; writing—review and editing, G.M. and C.G.; figures, G.M. All authors have read and agreed to the published version of the manuscript.
Funding
The Signal Transduction Laboratory is supported by the Italian Association for Cancer Research grants IG-23670 (to P.P.) and IG-19803 (to C.G.), A-ROSE (Associazione Ricerca Oncologica Sperimentale Estense); Progetti di Rilevante Interesse Nazionale grants PRIN2017E5L5P3 (to P.P.) and PRIN20177E9EPY (to C.G.); Italian Ministry of Health grants GR-2018-12367114 (to G.M.) and GR-2019-12369862 (to G.M.); European Research Council grant 853057-InflaPML (to C.G.); local funds from the University of Ferrara (to P.P. and C.G.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
P.P. is grateful to Camilla degli Scrovegni for continuous support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van der Giezen, M. Mitochondria and the Rise of Eukaryotes. BioScience 2011, 61, 594–601. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Joubert, F.; Puff, N. Mitochondrial Cristae Architecture and Functions: Lessons from Minimal Model Systems. Membranes 2021, 11, 465. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and Structural Differences in Spatially Distinct Subpopulations of Cardiac Mitochondria: Influence of Cardiac Pathologies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef]
- Dorn, G.W.; Vega, R.B.; Kelly, D.P. Mitochondrial Biogenesis and Dynamics in the Developing and Diseased Heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Morciano, G.; Rimessi, A.; Patergnani, S.; Vitto, V.A.M.; Danese, A.; Kahsay, A.; Palumbo, L.; Bonora, M.; Wieckowski, M.R.; Giorgi, C.; et al. Calcium Dysregulation in Heart Diseases: Targeting Calcium Channels to Achieve a Correct Calcium Homeostasis. Pharmacol. Res. 2022, 177, 106119. [Google Scholar] [CrossRef]
- Morciano, G.; Vitto, V.A.M.; Bouhamida, E.; Giorgi, C.; Pinton, P. Mitochondrial Bioenergetics and Dynamism in the Failing Heart. Life 2021, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, H.Z.E.; Zhao, G.; Shah, A.M.; Zhang, M. Role of Oxidative Stress in Calcific Aortic Valve Disease and Its Therapeutic Implications. Cardiovasc. Res. 2022, 118, 1433–1451. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Yamamoto, E.; Tokitsu, T.; Kusaka, H.; Fujisue, K.; Kurokawa, H.; Sugamura, K.; Maeda, H.; Tsujita, K.; Kaikita, K.; et al. Reactive Oxygen Metabolites Are Closely Associated with the Diagnosis and Prognosis of Coronary Artery Disease. J. Am. Heart Assoc. 2015, 4, e001451. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Geck, P.; Werdan, K.; Boekstegers, P. TNF-Alpha and IL-1 Alpha Inhibit Both Pyruvate Dehydrogenase Activity and Mitochondrial Function in Cardiomyocytes: Evidence for Primary Impairment of Mitochondrial Function. Mol. Cell. Biochem. 1997, 177, 61–67. [Google Scholar] [CrossRef]
- Daiber, A. Redox Signaling (Cross-Talk) from and to Mitochondria Involves Mitochondrial Pores and Reactive Oxygen Species. Biochim. Biophys. Acta 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular Identity of the Mitochondrial Permeability Transition Pore and Its Role in Ischemia-Reperfusion Injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Morciano, G.; Naumova, N.; Koprowski, P.; Valente, S.; Sardão, V.A.; Potes, Y.; Rimessi, A.; Wieckowski, M.R.; Oliveira, P.J. The Mitochondrial Permeability Transition Pore: An Evolving Concept Critical for Cell Life and Death. Biol. Rev. 2021, 96, 2489–2521. [Google Scholar] [CrossRef]
- Campo, G.; Morciano, G.; Pavasini, R.; Bonora, M.; Sbano, L.; Biscaglia, S.; Bovolenta, M.; Pinotti, M.; Punzetti, S.; Rizzo, P.; et al. Fo ATP Synthase C Subunit Serum Levels in Patients with ST-Segment Elevation Myocardial Infarction: Preliminary Findings. Int. J. Cardiol. 2016, 221, 993–997. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular Mechanisms and Consequences of Mitochondrial Permeability Transition. Nat. Rev. Mol. Cell Biol. 2021, 4, 266–285. [Google Scholar] [CrossRef]
- Gertz, E.W.; Wisneski, J.A.; Stanley, W.C.; Neese, R.A. Myocardial Substrate Utilization during Exercise in Humans. Dual Carbon-Labeled Carbohydrate Isotope Experiments. J. Clin. Investig. 1988, 82, 2017–2025. [Google Scholar] [CrossRef]
- Wisneski, J.A.; Gertz, E.W.; Neese, R.A.; Gruenke, L.D.; Craig, J.C. Dual Carbon-Labeled Isotope Experiments Using D-[6-14C] Glucose and L-[1,2,3-13C3] Lactate: A New Approach for Investigating Human Myocardial Metabolism during Ischemia. J. Am. Coll. Cardiol. 1985, 5, 1138–1146. [Google Scholar] [CrossRef]
- Ussher, J.R.; Koves, T.R.; Jaswal, J.S.; Zhang, L.; Ilkayeva, O.; Dyck, J.R.B.; Muoio, D.M.; Lopaschuk, G.D. Insulin-Stimulated Cardiac Glucose Oxidation Is Increased in High-Fat Diet-Induced Obese Mice Lacking Malonyl CoA Decarboxylase. Diabetes 2009, 58, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy Metabolic Reprogramming in the Hypertrophied and Early Stage Failing Heart: A Multisystems Approach. Circ. Heart Fail. 2014, 7, 1022–1031. [Google Scholar] [CrossRef]
- Bouhamida, E.; Morciano, G.; Perrone, M.; Kahsay, A.E.; Della Sala, M.; Wieckowski, M.R.; Fiorica, F.; Pinton, P.; Giorgi, C.; Patergnani, S. The Interplay of Hypoxia Signaling on Mitochondrial Dysfunction and Inflammation in Cardiovascular Diseases and Cancer: From Molecular Mechanisms to Therapeutic Approaches. Biology 2022, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Zhabyeyev, P.; Gandhi, M.; Mori, J.; Basu, R.; Kassiri, Z.; Clanachan, A.; Lopaschuk, G.D.; Oudit, G.Y. Pressure-Overload-Induced Heart Failure Induces a Selective Reduction in Glucose Oxidation at Physiological Afterload. Cardiovasc. Res. 2013, 97, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Osorio, J.C.; Stanley, W.C.; Linke, A.; Castellari, M.; Diep, Q.N.; Panchal, A.R.; Hintze, T.H.; Lopaschuk, G.D.; Recchia, F.A. Impaired Myocardial Fatty Acid Oxidation and Reduced Protein Expression of Retinoid X Receptor-Alpha in Pacing-Induced Heart Failure. Circulation 2002, 106, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased Rates of Substrate Oxidation Ex Vivo Predict the Onset of Heart Failure and Contractile Dysfunction in Rats with Pressure Overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef]
- Kowalska, M.; Piekut, T.; Prendecki, M.; Sodel, A.; Kozubski, W.; Dorszewska, J. Mitochondrial and Nuclear DNA Oxidative Damage in Physiological and Pathological Aging. DNA Cell Biol. 2020, 39, 1410–1420. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the Mitochondrial DNA Replication Machinery in Mitochondrial DNA Mutagenesis, Aging and Age-Related Diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef]
- Strauss, K.A.; DuBiner, L.; Simon, M.; Zaragoza, M.; Sengupta, P.P.; Li, P.; Narula, N.; Dreike, S.; Platt, J.; Procaccio, V.; et al. Severity of Cardiomyopathy Associated with Adenine Nucleotide Translocator-1 Deficiency Correlates with MtDNA Haplogroup. Proc. Natl. Acad. Sci. USA 2013, 110, 3453–3458. [Google Scholar] [CrossRef]
- Narula, N.; Zaragoza, M.V.; Sengupta, P.P.; Li, P.; Haider, N.; Verjans, J.; Waymire, K.; Vannan, M.; Wallace, D.C. Adenine Nucleotide Translocase 1 Deficiency Results in Dilated Cardiomyopathy with Defects in Myocardial Mechanics, Histopathological Alterations, and Activation of Apoptosis. JACC Cardiovasc. Imaging 2011, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Santorelli, F.M.; Tanji, K.; Manta, P.; Casali, C.; Krishna, S.; Hays, A.P.; Mancini, D.M.; DiMauro, S.; Hirano, M. Maternally Inherited Cardiomyopathy: An Atypical Presentation of the MtDNA 12S RRNA Gene A1555G Mutation. Am. J. Hum. Genet. 1999, 64, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Pyle, A.; Griffin, H.; Coxhead, J.; Hussain, R.; Braund, P.S.; Li, L.; Burgess, A.; Munroe, P.B.; Little, L.; et al. Heteroplasmic Mitochondrial DNA Variants in Cardiovascular Diseases. PLoS Genet. 2022, 18, e1010068. [Google Scholar] [CrossRef]
- Andreassi, M.G.; Botto, N.; Colombo, M.G.; Biagini, A.; Clerico, A. Genetic Instability and Atherosclerosis: Can Somatic Mutations Account for the Development of Cardiovascular Diseases? Environ. Mol. Mutagen. 2000, 35, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and Dysfunctions of Mitochondrial Dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Knowlton, A.A. Mitochondrial Dynamics in Heart Failure. Congest. Heart Fail. 2011, 17, 257–261. [Google Scholar] [CrossRef]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics in Mammalian Health and Disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W. Mitochondrial Fusion Is Essential for Organelle Function and Cardiac Homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Zhao, F.; Zhang, Z.; Kobayashi, T.; Huang, Y.; Shi, B.; Wu, W.; Liang, Q. Mitochondrial Fission and Mitophagy Coordinately Restrict High Glucose Toxicity in Cardiomyocytes. Front. Physiol. 2020, 11, 604069. [Google Scholar] [CrossRef] [PubMed]
- Horbay, R.; Bilyy, R. Mitochondrial Dynamics during Cell Cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef]
- Silva Ramos, E.; Larsson, N.-G.; Mourier, A. Bioenergetic Roles of Mitochondrial Fusion. Biochim. Biophys. Acta 2016, 1857, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-Mitochondria Contacts Couple MtDNA Synthesis with Mitochondrial Division in Human Cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef]
- Kraus, F.; Ryan, M.T. The Constriction and Scission Machineries Involved in Mitochondrial Fission. J. Cell Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational Changes in Dnm1 Support a Contractile Mechanism for Mitochondrial Fission. Nat. Struct. Mol. Biol. 2011, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff Is an Essential Factor for Mitochondrial Recruitment of Drp1 during Mitochondrial Fission in Mammalian Cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, New Components of the Mitochondrial Fission Machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Ji, W.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin Filaments Target the Oligomeric Maturation of the Dynamin GTPase Drp1 to Mitochondrial Fission Sites. eLife 2015, 4, e11553. [Google Scholar] [CrossRef]
- Zhu, P.-P.; Patterson, A.; Stadler, J.; Seeburg, D.P.; Sheng, M.; Blackstone, C. Intra- and Intermolecular Domain Interactions of the C-Terminal GTPase Effector Domain of the Multimeric Dynamin-like GTPase Drp1. J. Biol. Chem. 2004, 279, 35967–35974. [Google Scholar] [CrossRef]
- Fröhlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural Insights into Oligomerization and Mitochondrial Remodelling of Dynamin 1-like Protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 Recruitment in Mitochondrial Fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.-B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 Regulates Mitochondrial Dynamics through Inhibition of the Fusion Machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Ono, T.; Isobe, K.; Nakada, K.; Hayashi, J.I. Human Cells Are Protected from Mitochondrial Dysfunction by Complementation of DNA Products in Fused Mitochondria. Nat. Genet. 2001, 28, 272–275. [Google Scholar] [CrossRef]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 Is Required for Stress-Induced Mitochondrial Hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of Human Mitofusin 1 Provide Insight into Mitochondrial Tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural Basis for GTP Hydrolysis and Conformational Change of MFN1 in Mediating Membrane Fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A New Mitofusin Topology Places the Redox-Regulated C Terminus in the Mitochondrial Intermembrane Space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar] [CrossRef]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The Human Dynamin-Related Protein OPA1 Is Anchored to the Mitochondrial Inner Membrane Facing the Inter-Membrane Space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic Cleavage of Opa1 Stimulates Mitochondrial Inner Membrane Fusion and Couples Fusion to Oxidative Phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of Mitochondrial Morphology through Proteolytic Cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular Basis of Selective Mitochondrial Fusion by Heterotypic Action between OPA1 and Cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 Links Human Mitochondrial Genome Maintenance to MtDNA Replication and Distribution. Genome Res. 2011, 21, 12–20. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef]
- Morciano, G.; Pedriali, G.; Sbano, L.; Iannitti, T.; Giorgi, C.; Pinton, P. Intersection of Mitochondrial Fission and Fusion Machinery with Apoptotic Pathways: Role of Mcl-1. Biol. Cell 2016, 108, 279–293. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, K.Z.Q.; Chu, C.T. After the Banquet: Mitochondrial Biogenesis, Mitophagy, and Cell Survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial Homeostasis: The Interplay between Mitophagy and Mitochondrial Biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- López-Lluch, G.; Irusta, P.M.; Navas, P.; de Cabo, R. Mitochondrial Biogenesis and Healthy Aging. Exp. Gerontol. 2008, 43, 813–819. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional Integration of Mitochondrial Biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human Mitochondrial Transcription Revisited: Only TFAM and TFB2M Are Required for Transcription of the Mitochondrial Genes In Vitro. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef]
- Wu, Z.; Huang, X.; Feng, Y.; Handschin, C.; Feng, Y.; Gullicksen, P.S.; Bare, O.; Labow, M.; Spiegelman, B.; Stevenson, S.C. Transducer of Regulated CREB-Binding Proteins (TORCs) Induce PGC-1alpha Transcription and Mitochondrial Biogenesis in Muscle Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 14379–14384. [Google Scholar] [CrossRef]
- De Lange, P.; Farina, P.; Moreno, M.; Ragni, M.; Lombardi, A.; Silvestri, E.; Burrone, L.; Lanni, A.; Goglia, F. Sequential Changes in the Signal Transduction Responses of Skeletal Muscle Following Food Deprivation. FASEB J. 2006, 20, 2579–2581. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-Activated Protein Kinase (AMPK) Action in Skeletal Muscle via Direct Phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic Regulation of PGC-1alpha Localization and Turnover Implicates Mitochondrial Adaptation in Calorie Restriction and the Stress Response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef]
- Oka, S.-I.; Sabry, A.D.; Cawley, K.M.; Warren, J.S. Multiple Levels of PGC-1α Dysregulation in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 2. [Google Scholar] [CrossRef]
- Biscaglia, S.; Erriquez, A.; Serenelli, M.; D’Ascenzo, F.; De Ferrari, G.; Ariza Sole, A.; Sanchis, J.; Giannini, F.; Gallo, F.; Scala, A.; et al. Complete versus Culprit-Only Strategy in Older MI Patients with Multivessel Disease. Catheter. Cardiovasc. Interv. 2022, 99, 970–978. [Google Scholar] [CrossRef]
- Wong, N.D. Epidemiological Studies of CHD and the Evolution of Preventive Cardiology. Nat. Rev. Cardiol. 2014, 11, 276–289. [Google Scholar] [CrossRef]
- Campo, G.; Pavasini, R.; Morciano, G.; Lincoff, A.M.; Gibson, C.M.; Kitakaze, M.; Lonborg, J.; Ahluwalia, A.; Ishii, H.; Frenneaux, M.; et al. Clinical Benefit of Drugs Targeting Mitochondrial Function as an Adjunct to Reperfusion in ST-Segment Elevation Myocardial Infarction: A Meta-Analysis of Randomized Clinical Trials. Int. J. Cardiol. 2017, 244, 59–66. [Google Scholar] [CrossRef]
- Campo, G.; Pavasini, R.; Morciano, G.; Lincoff, M.A.; Gibson, M.C.; Kitakaze, M.; Lonborg, J.; Ahluwalia, A.; Ishii, H.; Frenneaux, M.; et al. Data on Administration of Cyclosporine, Nicorandil, Metoprolol on Reperfusion Related Outcomes in ST-Segment Elevation Myocardial Infarction Treated with Percutaneous Coronary Intervention. Data Brief 2017, 14, 197–205. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-Related Protein 1 (Drp1)-Mediated Diastolic Dysfunction in Myocardial Ischemia-Reperfusion Injury: Therapeutic Benefits of Drp1 Inhibition to Reduce Mitochondrial Fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Zhang, P.; Guan, P.; Ye, X.; Lu, Y.; Hang, Y.; Su, Y.; Hu, W. SOCS6 Promotes Mitochondrial Fission and Cardiomyocyte Apoptosis and Is Negatively Regulated by Quaking-Mediated MiR-19b. Oxid. Med. Cell. Longev. 2022, 2022, 1121323. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting Mitochondrial Fission Protects the Heart against Ischemia/Reperfusion Injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Disatnik, M.-H.; Ferreira, J.C.B.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.M.; Qi, X.; Mochly-Rosen, D. Acute Inhibition of Excessive Mitochondrial Fission after Myocardial Infarction Prevents Long-Term Cardiac Dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart against Energy Stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, S.; Jin, Q.; Shi, C.; Zhang, Y.; Zhu, P.; Ma, Q.; Tian, F.; Chen, Y. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of MROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved MPTP Opening. J. Am. Heart Assoc. 2017, 6, e005328. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Balancing Mitochondrial Dynamics via Increasing Mitochondrial Fusion Attenuates Infarct Size and Left Ventricular Dysfunction in Rats with Cardiac Ischemia/Reperfusion Injury. Clin. Sci. 2019, 133, 497–513. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Che, Z.; Meng, X.; Yu, Y.; Li, M.; Yu, Z.; Shi, H.; Yang, D.; Yu, M. MCU Up-Regulation Contributes to Myocardial Ischemia-Reperfusion Injury through Calpain/OPA-1-Mediated Mitochondrial Fusion/Mitophagy Inhibition. J. Cell. Mol. Med. 2019, 23, 7830–7843. [Google Scholar] [CrossRef]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 Processing and Mitochondrial Fragmentation Cause Heart Failure in Mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, Apoptosis, and Heart Failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Javadov, S.; Rajapurohitam, V.; Kilić, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of Mitochondrial Fusion-Fission Proteins during Post-Infarction Remodeling: The Effect of NHE-1 Inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Z.; Qin, X.; Xu, J.; Hou, Z.; Yang, H.; Mao, X.; Xing, W.; Li, X.; Zhang, X.; et al. Enhancing Fatty Acid Utilization Ameliorates Mitochondrial Fragmentation and Cardiac Dysfunction via Rebalancing Optic Atrophy 1 Processing in the Failing Heart. Cardiovasc. Res. 2018, 114, 979–991. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, Z.; Yin, Q.; Fu, C.; Barszczyk, A.; Zhang, X.; Wang, J.; Yang, D. Cardiac-Specific Overexpression of MiR-122 Induces Mitochondria-Dependent Cardiomyocyte Apoptosis and Promotes Heart Failure by Inhibiting Hand2. J. Cell. Mol. Med. 2021, 25, 5326–5334. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a Sodium Glucose Co-Transporter-2 Inhibitor, Alleviates Atrial Remodeling and Improves Mitochondrial Function in High-Fat Diet/Streptozotocin-Induced Diabetic Rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef]
- Montaigne, D.; Marechal, X.; Coisne, A.; Debry, N.; Modine, T.; Fayad, G.; Potelle, C.; El Arid, J.-M.; Mouton, S.; Sebti, Y.; et al. Myocardial Contractile Dysfunction Is Associated with Impaired Mitochondrial Function and Dynamics in Type 2 Diabetic but Not in Obese Patients. Circulation 2014, 130, 554–564. [Google Scholar] [CrossRef]
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-Related Protein 1 Inhibition Attenuates Cardiovascular Calcification in the Presence of Oxidative Stress. Circ. Res. 2017, 121, 220–233. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Pedriali, G.; Cimaglia, P.; Mikus, E.; Calvi, S.; Albertini, A.; Giorgi, C.; Campo, G.; Ferrari, R.; et al. Impairment of Mitophagy and Autophagy Accompanies Calcific Aortic Valve Stenosis Favoring Cell Death and the Severity of Disease. Cardiovasc. Res. 2021, 118, 2548–2559. [Google Scholar] [CrossRef]
- Abudupataer, M.; Zhu, S.; Yan, S.; Xu, K.; Zhang, J.; Luo, S.; Ma, W.; Alam, M.F.; Tang, Y.; Huang, H.; et al. Aorta Smooth Muscle-on-a-Chip Reveals Impaired Mitochondrial Dynamics as a Therapeutic Target for Aortic Aneurysm in Bicuspid Aortic Valve Disease. eLife 2021, 10, e69310. [Google Scholar] [CrossRef]
- Cahill, T.J.; Leo, V.; Kelly, M.; Stockenhuber, A.; Kennedy, N.W.; Bao, L.; Cereghetti, G.M.; Harper, A.R.; Czibik, G.; Liao, C.; et al. Resistance of Dynamin-Related Protein 1 Oligomers to Disassembly Impairs Mitophagy, Resulting in Myocardial Inflammation and Heart Failure. J. Biol. Chem. 2015, 290, 25907–25919. [Google Scholar] [CrossRef]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of Mitochondrial DNA Nucleoids Regulated by Mitochondrial Fission Is Essential for Maintenance of Homogeneously Active Mitochondria during Neonatal Heart Development. Mol. Cell. Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 Are Essential for Postnatal Metabolic Remodeling in Heart. Circ. Res. 2012, 111, 1012–1026. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.-H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef]
- Chehaitly, A.; Guihot, A.-L.; Proux, C.; Grimaud, L.; Aurrière, J.; Legouriellec, B.; Rivron, J.; Vessieres, E.; Tétaud, C.; Zorzano, A.; et al. Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis. Antioxidants 2022, 11, 1078. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.-H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1-Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Salabei, J.K.; Hill, B.G. Mitochondrial Fission Induced by Platelet-Derived Growth Factor Regulates Vascular Smooth Muscle Cell Bioenergetics and Cell Proliferation. Redox Biol. 2013, 1, 542–551. [Google Scholar] [CrossRef]
- Guo, X.; Chen, K.-H.; Guo, Y.; Liao, H.; Tang, J.; Xiao, R.-P. Mitofusin 2 Triggers Vascular Smooth Muscle Cell Apoptosis via Mitochondrial Death Pathway. Circ. Res. 2007, 101, 1113–1122. [Google Scholar] [CrossRef]
- Guo, Y.-H.; Chen, K.; Gao, W.; Li, Q.; Chen, L.; Wang, G.-S.; Tang, J. Overexpression of Mitofusin 2 Inhibited Oxidized Low-Density Lipoprotein Induced Vascular Smooth Muscle Cell Proliferation and Reduced Atherosclerotic Lesion Formation in Rabbit. Biochem. Biophys. Res. Commun. 2007, 363, 411–417. [Google Scholar] [CrossRef]
- He, L.; Zhou, Q.; Huang, Z.; Xu, J.; Zhou, H.; Lv, D.; Lu, L.; Huang, S.; Tang, M.; Zhong, J.; et al. PINK1/Parkin-Mediated Mitophagy Promotes Apelin-13-Induced Vascular Smooth Muscle Cell Proliferation by AMPKα and Exacerbates Atherosclerotic Lesions. J. Cell. Physiol. 2019, 234, 8668–8682. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, J.; Liu, X.; Zhang, Q. MicroRNA-138 Attenuates Myocardial Ischemia Reperfusion Injury through Inhibiting Mitochondria-Mediated Apoptosis by Targeting HIF1-α. Exp. Ther. Med. 2019, 18, 3325–3332. [Google Scholar] [CrossRef]
- Aishwarya, R.; Alam, S.; Abdullah, C.S.; Morshed, M.; Nitu, S.S.; Panchatcharam, M.; Miriyala, S.; Kevil, C.G.; Bhuiyan, M.S. Pleiotropic Effects of Mdivi-1 in Altering Mitochondrial Dynamics, Respiration, and Autophagy in Cardiomyocytes. Redox Biol. 2020, 36, 101660. [Google Scholar] [CrossRef]
- Luo, J.-Y.; Liu, F.; Fang, B.-B.; Tian, T.; Li, Y.-H.; Zhang, T.; Li, X.-M.; Yang, Y.-N. NFKB1 Gene Mutant Was Associated with Prognosis of Coronary Artery Disease and Exacerbated Endothelial Mitochondrial Fission and Dysfunction. Oxid. Med. Cell. Longev. 2022, 2022, 9494926. [Google Scholar] [CrossRef]
- Jin, Q.; Li, R.; Hu, N.; Xin, T.; Zhu, P.; Hu, S.; Ma, S.; Zhu, H.; Ren, J.; Zhou, H. DUSP1 Alleviates Cardiac Ischemia/Reperfusion Injury by Suppressing the Mff-Required Mitochondrial Fission and Bnip3-Related Mitophagy via the JNK Pathways. Redox Biol. 2018, 14, 576–587. [Google Scholar] [CrossRef]
- Rodríguez-Graciani, K.M.; Chapa-Dubocq, X.R.; MacMillan-Crow, L.A.; Javadov, S. Association Between L-OPA1 Cleavage and Cardiac Dysfunction During Ischemia-Reperfusion Injury in Rats. Cell. Physiol. Biochem. 2020, 54, 1101–1114. [Google Scholar] [CrossRef]
- Kulek, A.R.; Undyala, V.V.R.; Anzell, A.R.; Raghunayakula, S.; MacMillan-Crow, L.A.; Sanderson, T.H.; Przyklenk, K. Does Disruption of Optic Atrophy-1 (OPA1) Contribute to Cell Death in HL-1 Cardiomyocytes Subjected to Lethal Ischemia-Reperfusion Injury? Cells 2022, 11, 3083. [Google Scholar] [CrossRef]
- Pisano, A.; Cerbelli, B.; Perli, E.; Pelullo, M.; Bargelli, V.; Preziuso, C.; Mancini, M.; He, L.; Bates, M.G.D.; Lucena, J.R.; et al. Impaired Mitochondrial Biogenesis Is a Common Feature to Myocardial Hypertrophy and End-Stage Ischemic Heart Failure. Cardiovasc. Pathol. 2016, 25, 103–112. [Google Scholar] [CrossRef]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1alpha and ERRalpha Target Gene Downregulation Is a Signature of the Failing Human Heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA Replication Impairs Mitochondrial Biogenesis in Human Failing Hearts. Circ. Res. 2010, 106, 1541–1548. [Google Scholar] [CrossRef]
- Garnier, A.; Fortin, D.; Deloménie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed Mitochondrial Transcription Factors and Oxidative Capacity in Rat Failing Cardiac and Skeletal Muscles. J. Physiol. 2003, 551, 491–501. [Google Scholar] [CrossRef]
- Fabregat-Andrés, Ó.; Ridocci-Soriano, F.; Estornell-Erill, J.; Corbí-Pascual, M.; Valle-Muñoz, A.; Berenguer-Jofresa, A.; Barrabés, J.A.; Mata, M.; Monsalve, M. Blood PGC-1α Concentration Predicts Myocardial Salvage and Ventricular Remodeling After ST-Segment Elevation Acute Myocardial Infarction. Rev. Esp. Cardiol. 2015, 68, 408–416. [Google Scholar] [CrossRef]
- Fabregat-Andrés, Ó.; Tierrez, A.; Mata, M.; Estornell-Erill, J.; Ridocci-Soriano, F.; Monsalve, M. Induction of PGC-1α Expression Can Be Detected in Blood Samples of Patients with ST-Segment Elevation Acute Myocardial Infarction. PLoS ONE 2011, 6, e26913. [Google Scholar] [CrossRef]
- Fabregat-Andres, O.; Paredes, F.; Monsalve, M.; Milara, J.; Ridocci-Soriano, F.; Gonzalez-Hervas, S.; Mena, A.; Facila, L.; Hornero, F.; Morell, S.; et al. MRNA PGC-1α Levels in Blood Samples Reliably Correlates with Its Myocardial Expression: Study in Patients Undergoing Cardiac Surgery. Anatol. J. Cardiol. 2016, 16, 622–629. [Google Scholar] [CrossRef]
- Ramaccini, D.; Pedriali, G.; Perrone, M.; Bouhamida, E.; Modesti, L.; Wieckowski, M.R.; Giorgi, C.; Pinton, P.; Morciano, G. Some Insights into the Regulation of Cardiac Physiology and Pathology by the Hippo Pathway. Biomedicines 2022, 10, 726. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.; Li, J.; Zhou, P.; Zhao, X.; Chen, R.; Song, L.; Zhao, H.; Yan, H. LATS2 Deletion Attenuates Myocardial Ischemia-Reperfusion Injury by Promoting Mitochondrial Biogenesis. Oxid. Med. Cell. Longev. 2021, 2021, 1058872. [Google Scholar] [CrossRef]
- Guariento, A.; Blitzer, D.; Doulamis, I.; Shin, B.; Moskowitzova, K.; Orfany, A.; Ramirez-Barbieri, G.; Staffa, S.J.; Zurakowski, D.; Del Nido, P.J.; et al. Preischemic Autologous Mitochondrial Transplantation by Intracoronary Injection for Myocardial Protection. J. Thorac. Cardiovasc. Surg. 2020, 160, e15–e29. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The Emerging Role of Nrf2 in Mitochondrial Function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef]
- Jia, L.; Yang, L.; Tian, Y.; Yang, L.; Wu, D.; Zhang, H.; Li, M.; Wu, N. Nrf2 Participates in the Protective Effect of Exogenous Mitochondria against Mitochondrial Dysfunction in Myocardial Ischaemic and Hypoxic Injury. Cell. Signal. 2022, 92, 110266. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Costantino, S.; Paneni, F. MicroRNA-122 in Heart Failure with Reduced Ejection Fraction: Epiphenomenon or Causal? Int. J. Cardiol. 2020, 303, 66–67. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of Mitochondrial Dynamics in the Failing Heart: Normalization Following Long-Term Therapy with Elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328. [Google Scholar] [CrossRef]
- Chen, H.; Ren, S.; Clish, C.; Jain, M.; Mootha, V.; McCaffery, J.M.; Chan, D.C. Titration of Mitochondrial Fusion Rescues Mff-Deficient Cardiomyopathy. J. Cell Biol. 2015, 211, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse Aortic Constriction Leads to Accelerated Heart Failure in Mice Lacking PPAR-Gamma Coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Wende, A.R.; Zaha, V.G.; Pires, K.M.; Wayment, B.; Olsen, C.; Bugger, H.; Buchanan, J.; Wang, X.; Moreira, A.B.; et al. PGC-1β Deficiency Accelerates the Transition to Heart Failure in Pressure Overload Hypertrophy. Circ. Res. 2011, 109, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional Role of Mitochondria in Arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191–202. [Google Scholar] [CrossRef]
- Yang, K.-C.; Bonini, M.G.; Dudley, S.C. Mitochondria and Arrhythmias. Free Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef]
- Westerman, S.; Wenger, N. Gender Differences in Atrial Fibrillation: A Review of Epidemiology, Management, and Outcomes. Curr. Cardiol. Rev. 2019, 15, 136–144. [Google Scholar] [CrossRef]
- Thiedemann, K.U.; Ferrans, V.J. Left Atrial Ultrastructure in Mitral Valvular Disease. Am. J. Pathol. 1977, 89, 575–604. [Google Scholar]
- Wiersma, M.; van Marion, D.M.S.; Wüst, R.C.I.; Houtkooper, R.H.; Zhang, D.; de Groot, N.M.S.; Henning, R.H.; Brundel, B.J.J.M. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef]
- Dong, J.; Zhao, J.; Zhang, M.; Liu, G.; Wang, X.; Liu, Y.; Yang, N.; Liu, Y.; Zhao, G.; Sun, J.; et al. Β3-Adrenoceptor Impairs Mitochondrial Biogenesis and Energy Metabolism During Rapid Atrial Pacing-Induced Atrial Fibrillation. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 114–126. [Google Scholar] [CrossRef]
- Bell, D.S.H.; Goncalves, E. Atrial Fibrillation and Type 2 Diabetes: Prevalence, Etiology, Pathophysiology and Effect of Anti-Diabetic Therapies. Diabetes Obes. Metab. 2019, 21, 210–217. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Meng, L.; Gong, M.; Li, J.; Shi, W.; Qiu, J.; Yang, Y.; Zhao, J.; Suo, Y.; et al. Pioglitazone Inhibits Diabetes-Induced Atrial Mitochondrial Oxidative Stress and Improves Mitochondrial Biogenesis, Dynamics, and Function Through the PPAR-γ/PGC-1α Signaling Pathway. Front. Pharmacol. 2021, 12, 658362. [Google Scholar] [CrossRef]
- Jeganathan, J.; Saraf, R.; Mahmood, F.; Pal, A.; Bhasin, M.K.; Huang, T.; Mittel, A.; Knio, Z.; Simons, R.; Khabbaz, K.; et al. Mitochondrial Dysfunction in Atrial Tissue of Patients Developing Postoperative Atrial Fibrillation. Ann. Thorac. Surg. 2017, 104, 1547–1555. [Google Scholar] [CrossRef]
- Kumar, A.; Avishay, D.M.; Jones, C.R.; Shaikh, J.D.; Kaur, R.; Aljadah, M.; Kichloo, A.; Shiwalkar, N.; Keshavamurthy, S. Sudden Cardiac Death: Epidemiology, Pathogenesis and Management. Rev. Cardiovasc. Med. 2021, 22, 147–158. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms Underlying Acute Protection from Cardiac Ischemia-Reperfusion Injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Devalla, H.D.; Gélinas, R.; Aburawi, E.H.; Beqqali, A.; Goyette, P.; Freund, C.; Chaix, M.-A.; Tadros, R.; Jiang, H.; Le Béchec, A.; et al. TECRL, a New Life-Threatening Inherited Arrhythmia Gene Associated with Overlapping Clinical Features of Both LQTS and CPVT. EMBO Mol. Med. 2016, 8, 1390–1408. [Google Scholar] [CrossRef]
- Hou, C.; Jiang, X.; Zhang, H.; Zheng, J.; Qiu, Q.; Zhang, Y.; Sun, X.; Xu, M.; Chang, A.C.Y.; Xie, L.; et al. TECRL Deficiency Results in Aberrant Mitochondrial Function in Cardiomyocytes. Commun. Biol. 2022, 5, 470. [Google Scholar] [CrossRef]
- Valli, H.; Ahmad, S.; Chadda, K.R.; Al-Hadithi, A.B.A.K.; Grace, A.A.; Jeevaratnam, K.; Huang, C.L.-H. Age-Dependent Atrial Arrhythmic Phenotype Secondary to Mitochondrial Dysfunction in Pgc-1β Deficient Murine Hearts. Mech. Ageing Dev. 2017, 167, 30–45. [Google Scholar] [CrossRef]
- Valli, H.; Ahmad, S.; Fraser, J.A.; Jeevaratnam, K.; Huang, C.L.-H. Pro-Arrhythmic Atrial Phenotypes in Incrementally Paced Murine Pgc1β-/- Hearts: Effects of Age. Exp. Physiol. 2017, 102, 1619–1634. [Google Scholar] [CrossRef]
- Ahmad, S.; Valli, H.; Chadda, K.R.; Cranley, J.; Jeevaratnam, K.; Huang, C.L.-H. Ventricular Pro-Arrhythmic Phenotype, Arrhythmic Substrate, Ageing and Mitochondrial Dysfunction in Peroxisome Proliferator Activated Receptor-γ Coactivator-1β Deficient (Pgc-1β-/-) Murine Hearts. Mech. Ageing Dev. 2018, 173, 92–103. [Google Scholar] [CrossRef]
- Iung, B.; Vahanian, A. Epidemiology of Acquired Valvular Heart Disease. Can. J. Cardiol. 2014, 30, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, M.; Lu, L.; Zheng, Y.; Zhang, P. Congenital Heart Disease: Causes, Diagnosis, Symptoms, and Treatments. Cell Biochem. Biophys. 2015, 72, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Iung, B.; Vahanian, A. Epidemiology of Valvular Heart Disease in the Adult. Nat. Rev. Cardiol. 2011, 8, 162–172. [Google Scholar] [CrossRef]
- Lincoln, J.; Garg, V. Etiology of Valvular Heart Disease-Genetic and Developmental Origins. Circ. J. 2014, 78, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An Overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules 2019, 9, 878. [Google Scholar] [CrossRef]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving Concepts in Dilated Cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 228–239. [Google Scholar] [CrossRef]
- Pedriali, G.; Morciano, G.; Patergnani, S.; Cimaglia, P.; Morelli, C.; Mikus, E.; Ferrari, R.; Gasbarro, V.; Giorgi, C.; Wieckowski, M.R.; et al. Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives. Int. J. Mol. Sci. 2020, 21, 4899. [Google Scholar] [CrossRef]
- Liu, F.; Chen, J.; Hu, W.; Gao, C.; Zeng, Z.; Cheng, S.; Yu, K.; Qian, Y.; Xu, D.; Zhu, G.; et al. PTP1B Inhibition Improves Mitochondrial Dynamics to Alleviate Calcific Aortic Valve Disease via Regulating OPA1 Homeostasis. JACC Basic Transl. Sci. 2022, 7, 697–712. [Google Scholar] [CrossRef]
- Wang, L.; Ming Wang, L.; Chen, W.; Chen, X. Bicuspid Aortic Valve: A Review of Its Genetics and Clinical Significance. J. Heart Valve Dis. 2016, 25, 568–573. [Google Scholar]
- Ashrafian, H.; Docherty, L.; Leo, V.; Towlson, C.; Neilan, M.; Steeples, V.; Lygate, C.A.; Hough, T.; Townsend, S.; Williams, D.; et al. A Mutation in the Mitochondrial Fission Gene Dnm1l Leads to Cardiomyopathy. PLoS Genet. 2010, 6, e1001000. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 Mutation and Late-Onset Cardiomyopathy: Mitochondrial Dysfunction and MtDNA Instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef]
- Hsiao, Y.T.; Shimizu, I.; Wakasugi, T.; Yoshida, Y.; Ikegami, R.; Hayashi, Y.; Suda, M.; Katsuumi, G.; Nakao, M.; Ozawa, T.; et al. Cardiac Mitofusin-1 Is Reduced in Non-Responding Patients with Idiopathic Dilated Cardiomyopathy. Sci. Rep. 2021, 11, 6722. [Google Scholar] [CrossRef]
- Ahuja, P.; Wanagat, J.; Wang, Z.; Wang, Y.; Liem, D.A.; Ping, P.; Antoshechkin, I.A.; Margulies, K.B.; Maclellan, W.R. Divergent Mitochondrial Biogenesis Responses in Human Cardiomyopathy. Circulation 2013, 127, 1957–1967. [Google Scholar] [CrossRef]
- Sebastiani, M.; Giordano, C.; Nediani, C.; Travaglini, C.; Borchi, E.; Zani, M.; Feccia, M.; Mancini, M.; Petrozza, V.; Cossarizza, A.; et al. Induction of Mitochondrial Biogenesis Is a Maladaptive Mechanism in Mitochondrial Cardiomyopathies. J. Am. Coll. Cardiol. 2007, 50, 1362–1369. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Liu, Y.; Zhou, W.; Sun, R.; Xia, M. Retinol Binding Protein 4 Induces Mitochondrial Dysfunction and Vascular Oxidative Damage. Atherosclerosis 2015, 240, 335–344. [Google Scholar] [CrossRef]
- Wei, Z.; Chong, H.; Jiang, Q.; Tang, Y.; Xu, J.; Wang, H.; Shi, Y.; Cui, L.; Li, J.; Zhang, Y.; et al. Smooth Muscle Overexpression of PGC1α Attenuates Atherosclerosis in Rabbits. Circ. Res. 2021, 129, e72–e86. [Google Scholar] [CrossRef]
- Stein, S.; Lohmann, C.; Handschin, C.; Stenfeldt, E.; Borén, J.; Lüscher, T.F.; Matter, C.M. ApoE-/- PGC-1α-/- Mice Display Reduced IL-18 Levels and Do Not Develop Enhanced Atherosclerosis. PLoS ONE 2010, 5, e13539. [Google Scholar] [CrossRef]
- Vandenbeek, R.; Khan, N.P.; Estall, J.L. Linking Metabolic Disease With the PGC-1α Gly482Ser Polymorphism. Endocrinology 2018, 159, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, W.; Li, X.; Tang, Y.; Xie, P.; Ji, Y.; Fan, L.; Chen, Q. Association between PPARGC1A Gene Polymorphisms and Coronary Artery Disease in a Chinese Population. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Guo, D.; Li, Y.; Liang, S.; Wu, Y. The Impact of Severity of Hypertension on Association of PGC-1alpha Gene with Blood Pressure and Risk of Hypertension. BMC Cardiovasc. Disord. 2007, 7, 33. [Google Scholar] [CrossRef]
- Fitchett, D.; Inzucchi, S.E.; Cannon, C.P.; McGuire, D.K.; Scirica, B.M.; Johansen, O.E.; Sambevski, S.; Kaspers, S.; Pfarr, E.; George, J.T.; et al. Empagliflozin Reduced Mortality and Hospitalization for Heart Failure Across the Spectrum of Cardiovascular Risk in the EMPA-REG OUTCOME Trial. Circulation 2019, 139, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Cardioprotective Effects of Sirtuin-1 and Its Downstream Effectors: Potential Role in Mediating the Heart Failure Benefits of SGLT2 (Sodium-Glucose Cotransporter 2) Inhibitors. Circ. Heart Fail. 2020, 13, e007197. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Zhuravlev, A.D.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. Mitochondria-Mediated Cardiovascular Benefits of Sodium-Glucose Co-Transporter 2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 5371. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.R.; Zhou, Y.J.; Sha, Y.; Wu, X.P.; Yang, J.Q.; Liu, F. Melatonin Attenuates Vascular Calcification by Inhibiting Mitochondria Fission via an AMPK/Drp1 Signalling Pathway. J. Cell. Mol. Med. 2020, 24, 6043–6054. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.R.; Zhou, Y.J.; Yang, J.Q.; Liu, F.; Wu, X.P.; Sha, Y. Melatonin Attenuates Calcium Deposition from Vascular Smooth Muscle Cells by Activating Mitochondrial Fusion and Mitophagy via an AMPK/OPA1 Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 5298483. [Google Scholar] [CrossRef]
- Domínguez-Rodríguez, A.; Abreu-González, P.; Báez-Ferrer, N.; Reiter, R.J.; Avanzas, P.; Hernández-Vaquero, D. Melatonin and Cardioprotection in Humans: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Cardiovasc. Med. 2021, 8, 635083. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef]
- Vögtle, F.-N. Open Questions on the Mitochondrial Unfolded Protein Response. FEBS J. 2021, 288, 2856–2869. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Ono, K.; Nagao, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; et al. Autophagy Limits Acute Myocardial Infarction Induced by Permanent Coronary Artery Occlusion. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2261–H2271. [Google Scholar] [CrossRef]
- Zhang, W.; Siraj, S.; Zhang, R.; Chen, Q. Mitophagy Receptor FUNDC1 Regulates Mitochondrial Homeostasis and Protects the Heart from I/R Injury. Autophagy 2017, 13, 1080–1081. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-Inducible Kinase 1 (PINK1)/Park6 Is Indispensable for Normal Heart Function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.-H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Liu, L.; McKeehan, W.L.; Wang, F. The Fibroblast Growth Factor Signaling Axis Controls Cardiac Stem Cell Differentiation through Regulating Autophagy. Autophagy 2012, 8, 690–691. [Google Scholar] [CrossRef]
- Cadete, V.J.J.; Vasam, G.; Menzies, K.J.; Burelle, Y. Mitochondrial Quality Control in the Cardiac System: An Integrative View. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 782–796. [Google Scholar] [CrossRef]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; González, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef]
- Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Reche-López, D.; Álvarez-Córdoba, M.; Munuera-Cabeza, M.; Talaverón-Rey, M.; Povea-Cabello, S.; Suárez-Carrillo, A.; Romero-González, A.; et al. MtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).