

Enhanced NSAIDs Solubility in Drug–Drug Formulations with Ciprofloxacin †

 ,
,  ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
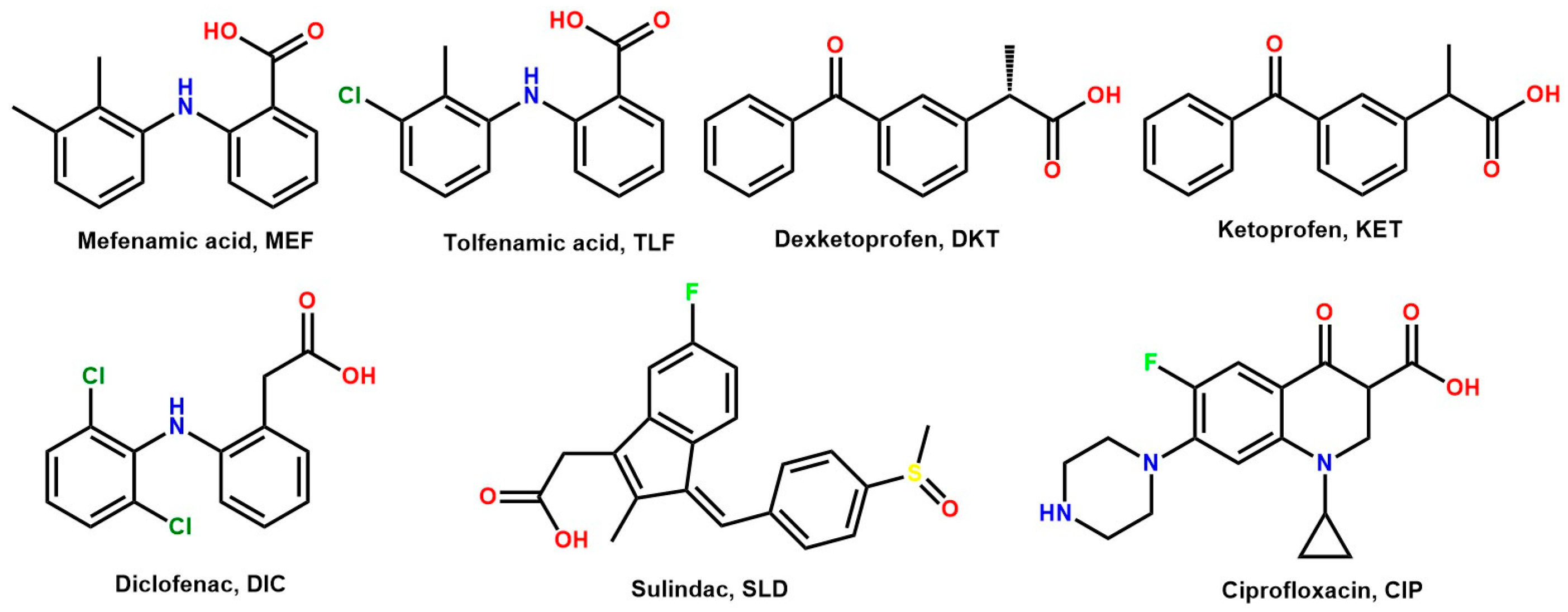
2.1. Design of Molecular Salts
2.2. Salt Synthesis
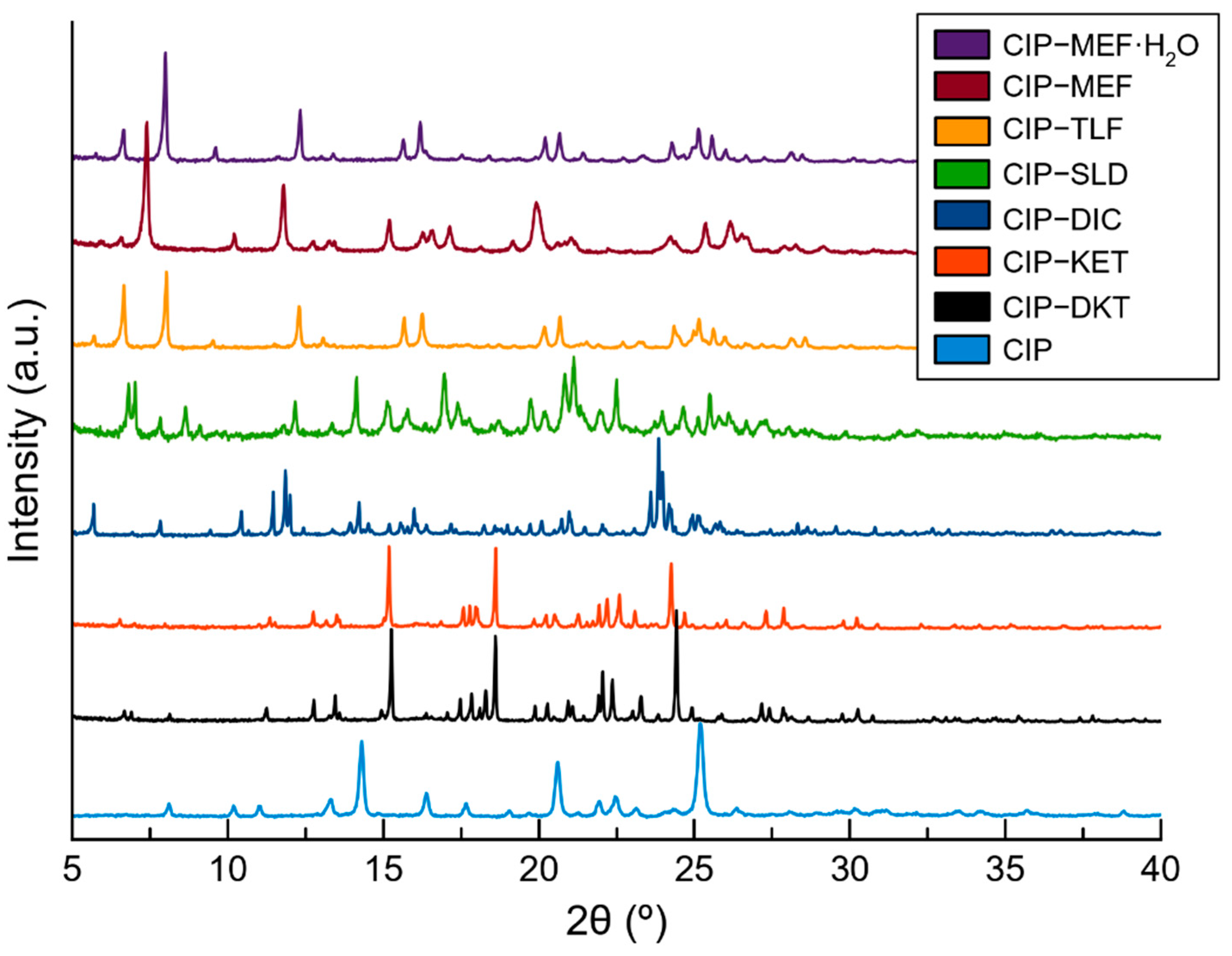
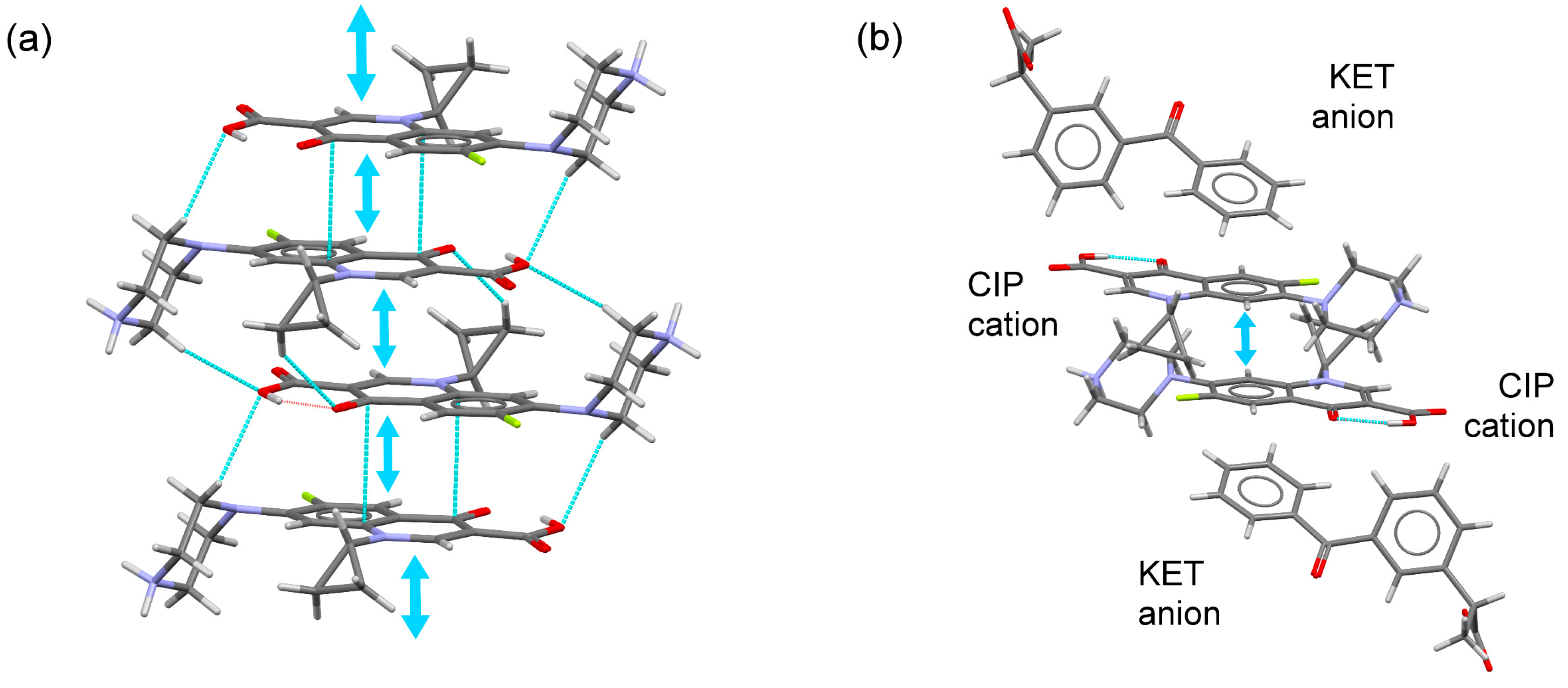
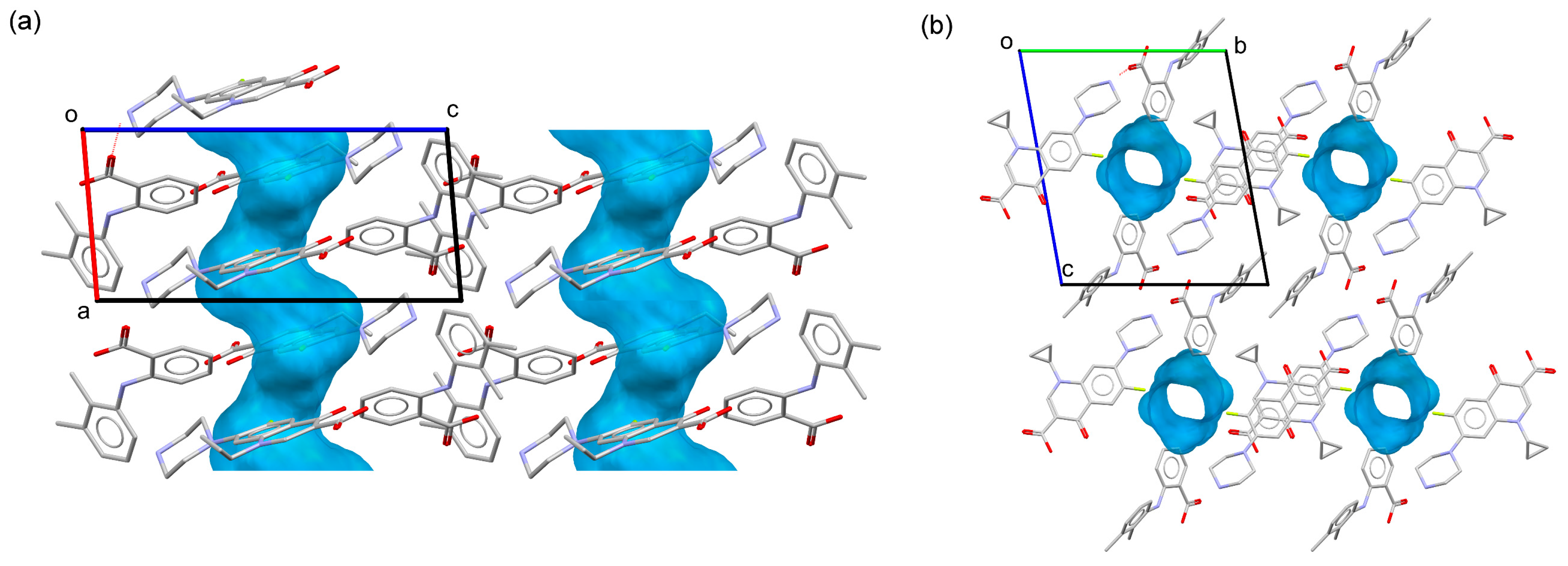
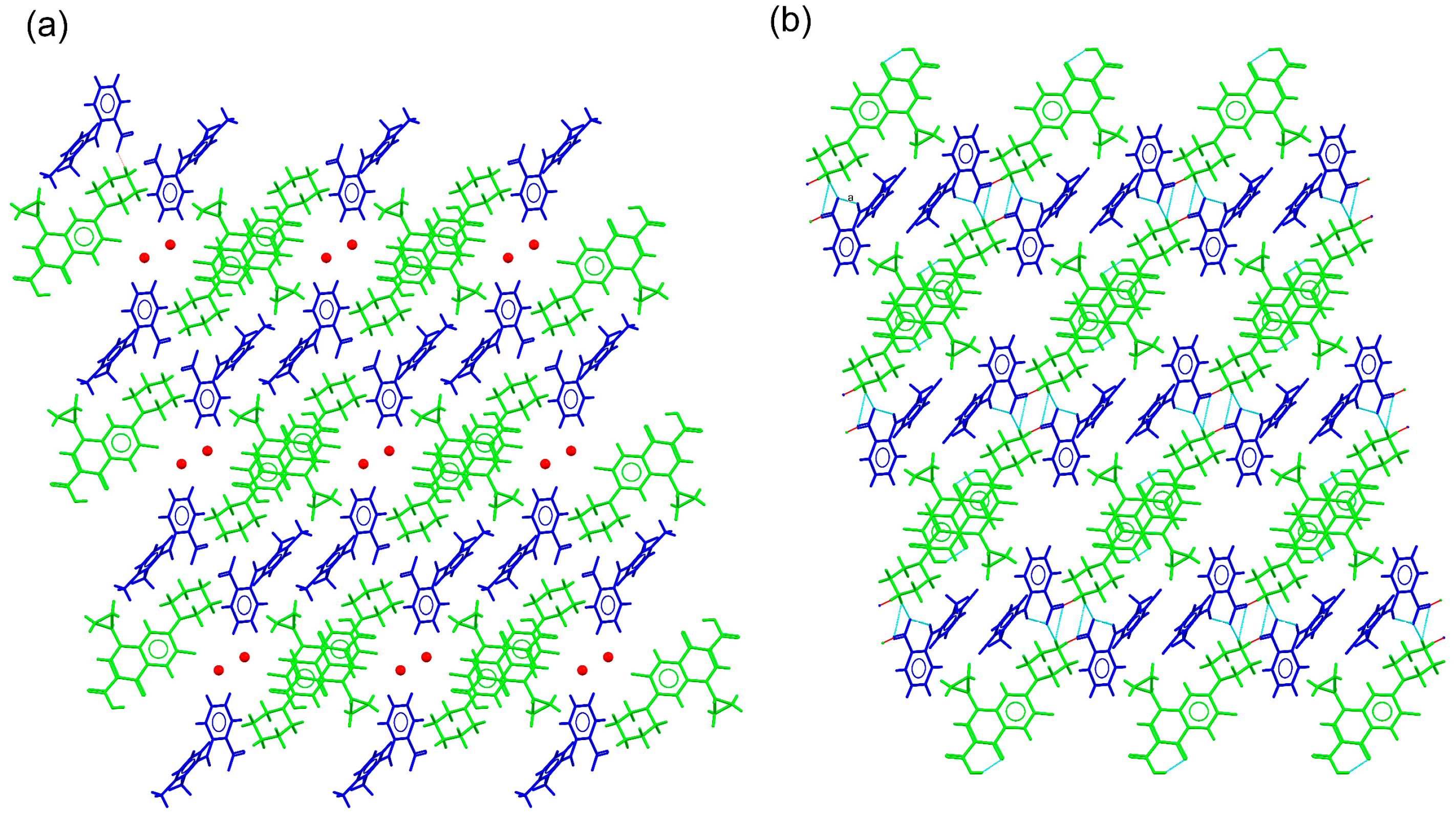
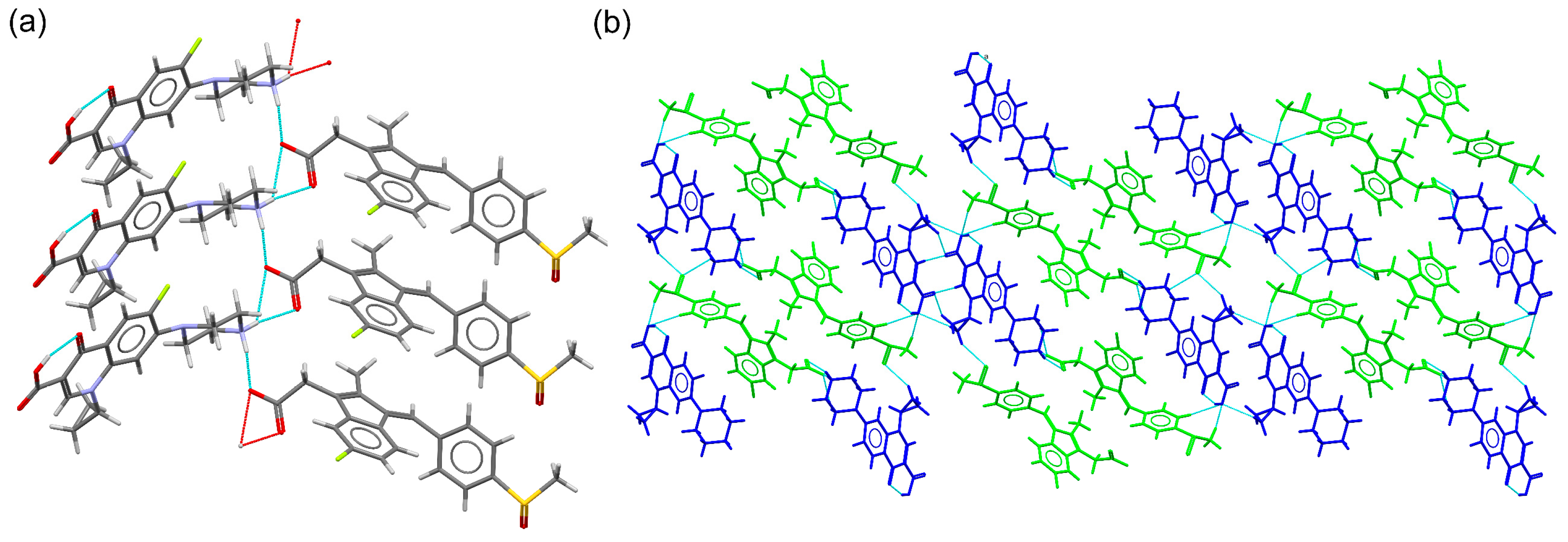
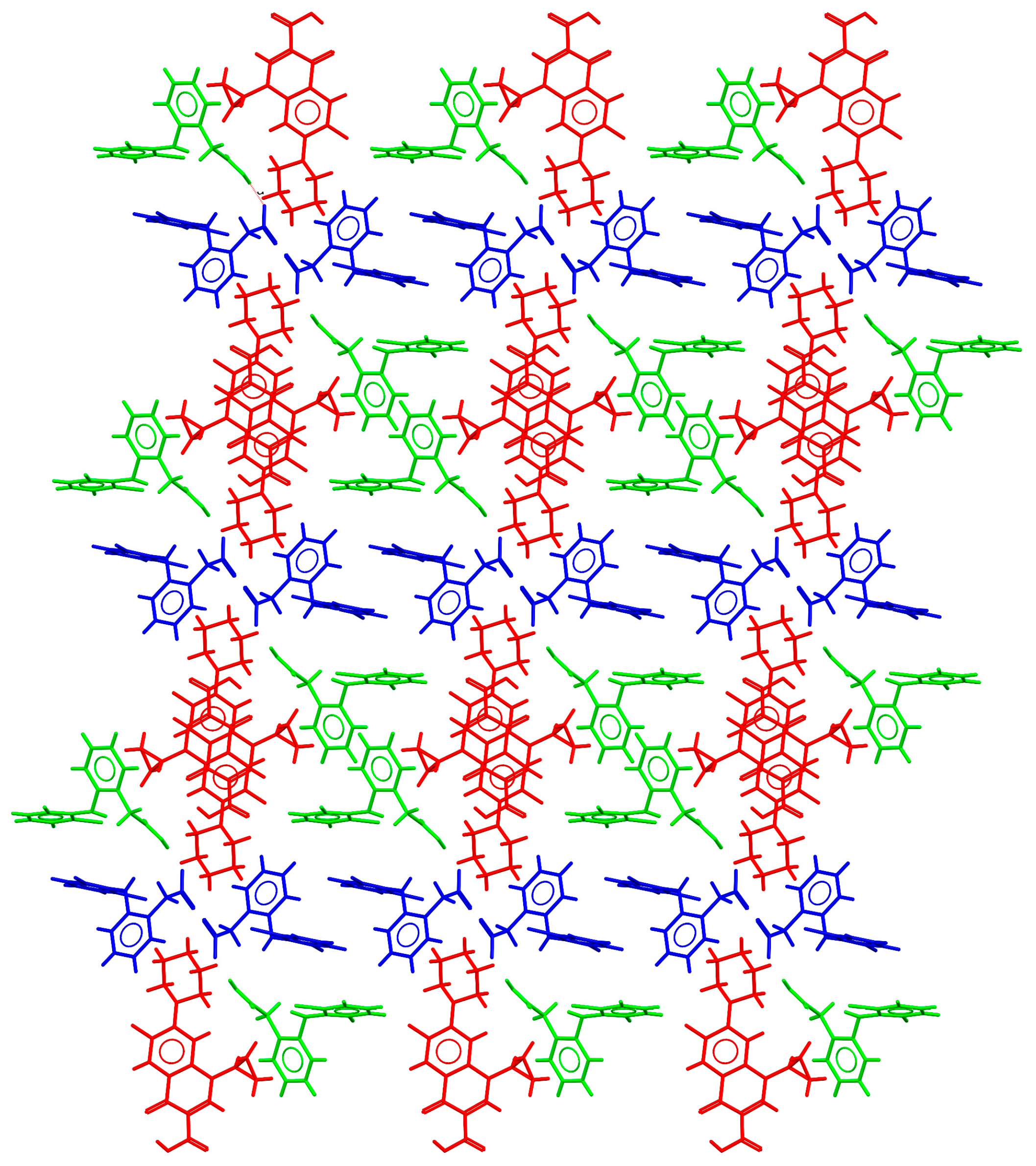
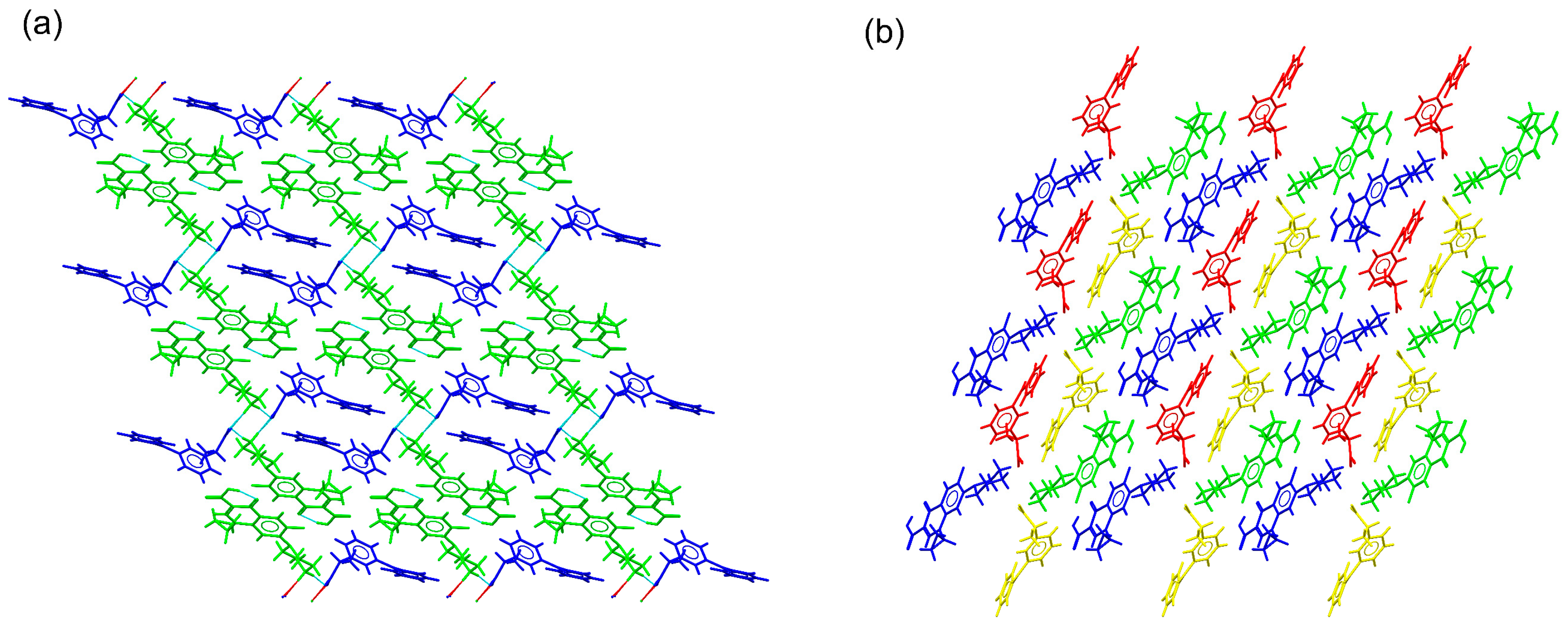
2.3. Structural Studies of Molecular Salts
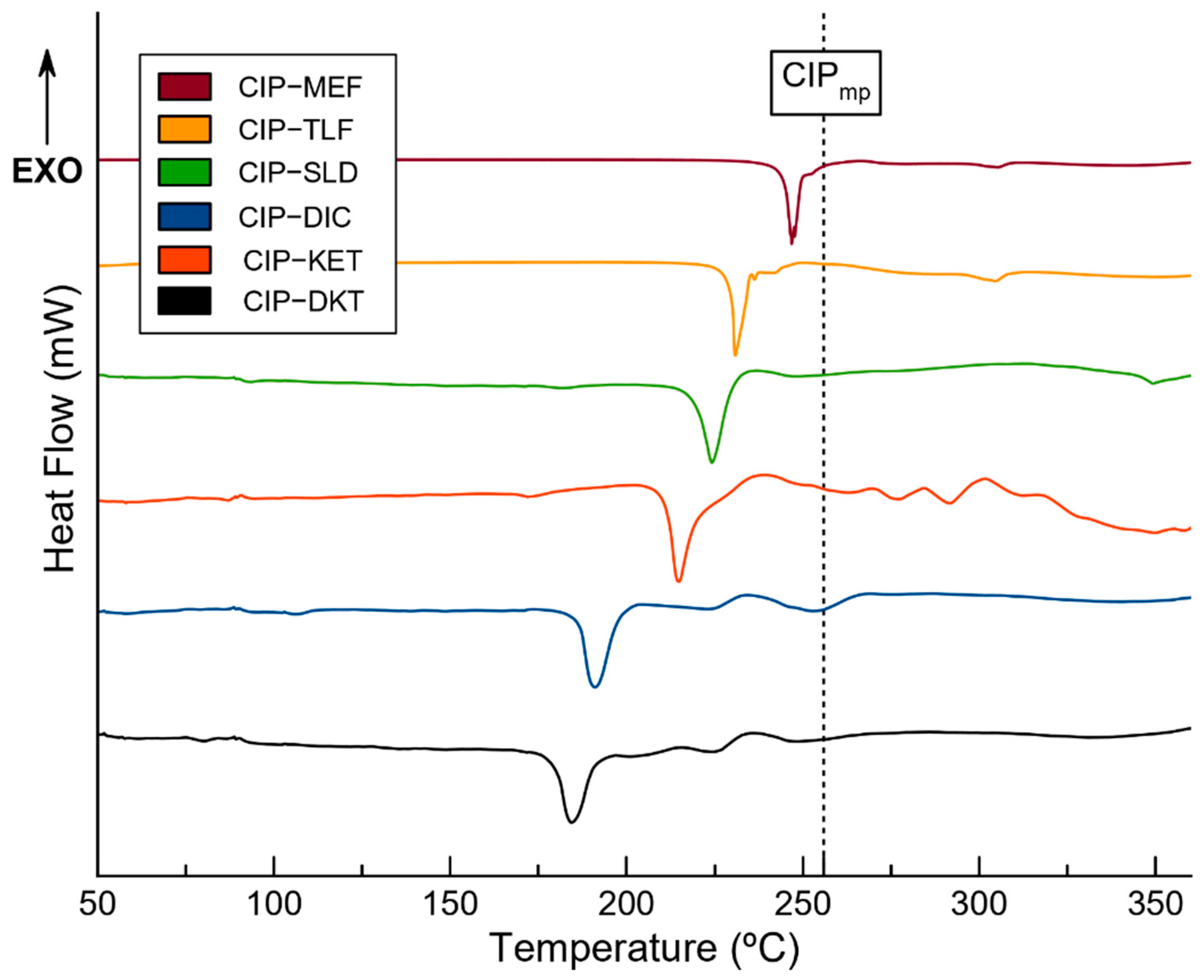
2.4. Thermal Stability
2.5. Thermodynamic Stability
2.6. Solubility Studies
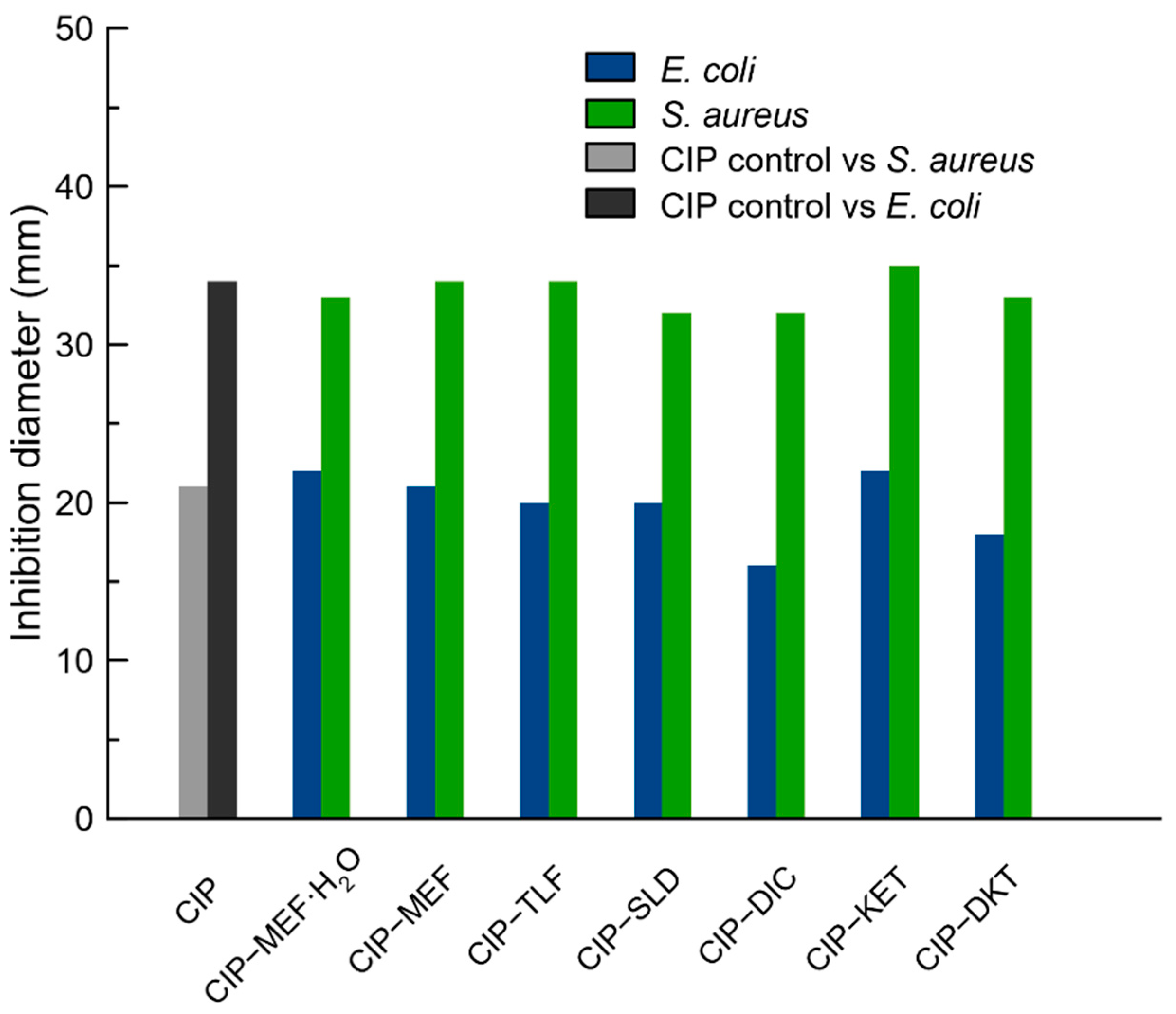
2.7. Antimicrobial Activity
3. Materials and Methods
3.1. Materials
3.2. Mechanochemical Syntheses
3.3. Preparation of Single Crystals
3.4. Powder X-ray Diffraction (PXRD)
3.5. Single-Crystal X-ray Diffraction (SCXRD)
3.6. Differential Scanning Calorimetry and Thermogravimetric Analysis
3.7. Slurry and Ageing Condition Experiments
3.8. Solubility Studies
3.9. Evaluation of Antimicrobial Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in vitro Drug Product Dissolution and in vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.J.; Steed, J.W. Pharmaceutical Cocrystals, Salts and Multicomponent Systems; Intermolecular Interactions and Property Based Design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Bighley, L.D.; Berge, S.M.; Monkhouse, D.C. Salt Forms of Drugs and Absorption. In Encyclopaedia of Pharmaceutical Technology; Swarbrick, J., Boylan, J., Eds.; Dekker: New York, NY, USA, 1996; Volume 13, pp. 453–499. [Google Scholar]
- Serajuddin, A.T.M. Salt Formation to Improve Drug Solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J. Acid-Base Crystalline Complexes and the PKa Rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- NHS Non-Steroidal Anti-Inflammmatory Drugs. Available online: https://www.nhs.uk/conditions/nsaids/ (accessed on 22 December 2022).
- U.S. Food and Drug Administration PONSTEL® (Mefenamic Acid Capsules, USP). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/015034s040lbl.pdf (accessed on 22 December 2022).
- Pentikäinen, P.J.; Neuvonen, P.J.; Backman, C. Human Pharmacokinetics of Tolfenamic Acid, a New Anti-Inflammatory Agent. Eur. J. Clin. Pharmacol. 1981, 19, 359–365. [Google Scholar] [CrossRef]
- Kuczyńska, J.; Pawlak, A.; Nieradko-Iwanicka, B. The Comparison of Dexketoprofen and Other Painkilling Medications (Review from 2018 to 2021). Biomed. Pharmacother. 2022, 149, 112819. [Google Scholar] [CrossRef]
- Jamali, F.; Brocks, D.R. Clinical Pharmacokinetics of Ketoprofen and Its Enantiomers. Clin. Pharmacokinet. 1990, 19, 197–217. [Google Scholar] [CrossRef]
- Todd, P.A.; Sorkin, E.M. Diclofenac Sodium. Drugs 1988, 35, 244–285. [Google Scholar] [CrossRef]
- Altman, R.; Bosch, B.; Brune, K.; Patrignani, P.; Young, C. Advances in NSAID Development: Evolution of Diclofenac Products Using Pharmaceutical Technology. Drugs 2015, 75, 859–877. [Google Scholar] [CrossRef]
- Davies, N.M.; Watson, M.S. Clinical Pharmacokinetics of Sulindac. Clin. Pharmacokinet. 1997, 32, 437–459. [Google Scholar] [CrossRef]
- Campoli-Richards, D.M.; Monk, J.P.; Price, A.; Benfield, P.; Todd, P.A.; Ward, A. Ciprofloxacin. Drugs 1988, 35, 373–447. [Google Scholar] [CrossRef] [PubMed]
- FDA Highlights of Prescribing Information of Ciprofloxacin. Available online: www.fda.gov/medwatch (accessed on 28 October 2022).
- Tehler, U.; Fagerberg, J.H.; Svensson, R.; Larhed, M.; Artursson, P.; Bergström, C.A.S. Optimizing Solubility and Permeability of a Biopharmaceutics Classification System (BCS) Class 4 Antibiotic Drug Using Lipophilic Fragments Disturbing the Crystal Lattice. J. Med. Chem. 2013, 56, 2690–2694. [Google Scholar] [CrossRef] [PubMed]
- Mesallati, H.; Mugheirbi, N.A.; Tajber, L. Two Faces of Ciprofloxacin: Investigation of Proton Transfer in Solid State Transformations. Cryst. Growth Des. 2016, 16, 6574–6585. [Google Scholar] [CrossRef]
- Bag, P.P.; Ghosh, S.; Khan, H.; Devarapalli, R.; Reddy, C.M. Drug-Drug Salt Forms of Ciprofloxacin with Diflunisal and Indoprofen. CrystEngComm 2014, 16, 7393–7396. [Google Scholar] [CrossRef]
- Surov, A.O.; Manin, A.N.; Voronin, A.P.; Drozd, K.V.; Simagina, A.A.; Churakov, A.V.; Perlovich, G.L. Pharmaceutical Salts of Ciprofloxacin with Dicarboxylic Acids. Eur. J. Pharm. Sci. 2015, 77, 112–121. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, L.; Yang, D.; Zhang, N.; He, L.; Du, G.; Lu, Y. Salt Screening and Characterization of Ciprofloxacin. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 20–28. [Google Scholar] [CrossRef]
- Rollof, J.; Vinge, E. Neurologic Adverse Effects during Concomitant Treatment with Ciprofloxacin, NSAIDS, and Chloroquine: Possible Drug Interaction. Ann. Pharmacother. 1993, 27, 1058–1059. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The Salt-Cocrystal Continuum: The Influence of Crystal Structure on Ionization State. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef]
- Thomas, L.H.; Klapwijk, A.R.; Wales, C.; Wilson, C.C. Intermolecular Hydrogen Transfer and Solubility Tuning in Multi-Component Molecular Crystals of the API Piroxicam. CrystEngComm 2014, 16, 5924–5932. [Google Scholar] [CrossRef]
- Gopi, S.P.; Ganguly, S.; Desiraju, G.R. A Drug-Drug Salt Hydrate of Norfloxacin and Sulfathiazole: Enhancement of in vitro Biological Properties via Improved Physicochemical Properties. Mol. Pharm. 2016, 13, 3590–3594. [Google Scholar] [CrossRef]
- Verdugo-Escamilla, C.; Alarcón-Payer, C.; Acebedo-Martínez, F.J.; Domínguez-Martín, A.; Choquesillo-Lazarte, D. New Metformin-Citric Acid Pharmaceutical Molecular Salt: Improving Metformin Physicochemical Properties. Crystals 2022, 12, 1748. [Google Scholar] [CrossRef]
- Surov, A.O.; Vasilev, N.A.; Voronin, A.P.; Churakov, A.V.; Emmerling, F.; Perlovich, G.L. Ciprofloxacin Salts with Benzoic Acid Derivatives: Structural Aspects, Solid-State Properties and Solubility Performance. CrystEngComm 2020, 22, 4238–4249. [Google Scholar] [CrossRef]
- Settimo, L.; Bellman, K.; Knegtel, R.M.A. Comparison of the Accuracy of Experimental and Predicted PKa Values of Basic and Acidic Compounds. Pharm. Res. 2014, 31, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Yuvali, D.; Yilmaz, E.; Narin, İ. A New Liquid Phase Microextraction Method-Based Reverse Micelle for Analysis of Dexketoprofen in Human Plasma by HPLC-DAD. J. Anal. Sci. Technol. 2020, 11, 53. [Google Scholar] [CrossRef]
- Pérez-Merino, L.; Casajuana, M.C.; Bernal, G.; Faba, J.; Astilleros, A.E.; González, R.; Giralt, M.; Romeu, M.; Nogués, M.R. Evaluation of the Effectiveness of Three Physiotherapeutic Treatments for Subacromial Impingement Syndrome: A Randomised Clinical Trial. Physiotherapy 2016, 102, 7–63. [Google Scholar] [CrossRef]
- Avdeef, A. Solubility of Sparingly-Soluble Ionizable Drugs. Adv. Drug Deliv. Rev. 2007, 59, 568–590. [Google Scholar] [CrossRef]
- Pentikäinen, P.J.; Tokola, O.; Alhava, E.; Penttilä, A. Pharmacokinetics of Tolfenamic Acid: Disposition in Bile, Blood and Urine after Intravenous Administration to Man. Eur. J. Clin. Pharmacol. 1984, 27, 349–354. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Tsume, Y.; Langguth, P.; Garcia-Arieta, A.; Amidon, G.L. In silico Prediction of Drug Dissolution and Absorption with Variation in Intestinal PH for BCS Class II Weak Acid Drugs: Ibuprofen and Ketoprofen. Biopharm. Drug Dispos. 2012, 33, 366–377. [Google Scholar] [CrossRef]
- Fillet, M.; Bechet, I.; Piette, V.; Crommen, J. Separation of Nonsteroidal Anti-Inflammatory Drugs by Capillary Electrophoresis Using Nonaqueous Electrolytes. Electrophor. Int. J. 1999, 20, 1907–1915. [Google Scholar] [CrossRef]
- Jones, O.A.H.; Voulvoulis, N.; Lester, J.N. Aquatic Environmental Assessment of the Top 25 English Prescription Pharmaceuticals. Water Res. 2002, 36, 5013–5022. [Google Scholar] [CrossRef]
- Lemmens, G.; Brouwers, J.; Snoeys, J.; Augustijns, P.; Vanuytsel, T. Insight into the Colonic Disposition of Sulindac in Humans. J. Pharm. Sci. 2021, 110, 259–267. [Google Scholar] [CrossRef] [PubMed]
- ChemAxon MarvinSketch 2020. Available online: https://chemaxon.com/marvin (accessed on 22 December 2022).
- Loschen, C.; Klamt, A. Solubility Prediction, Solvate and Cocrystal Screening as Tools for Rational Crystal Engineering. J. Pharm. Pharmacol. 2015, 67, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.N.; Choquesillo-Lazarte, D.; Cuffini, S.L.; Pidcock, E.; Infantes, L. Optimization and Comparison of Statistical Tools for the Prediction of Multicomponent Forms of a Molecule: The Antiretroviral Nevirapine as a Case Study. CrystEngComm 2020, 22, 7460–7474. [Google Scholar] [CrossRef]
- Acebedo-Martínez, F.J.; Alarcón-Payer, C.; Rodríguez-Domingo, L.; Domínguez-Martín, A.; Gómez-Morales, J.; Choquesillo-Lazarte, D. Furosemide/Non-Steroidal Anti-Inflammatory Drug-Drug Pharmaceutical Solids: Novel Opportunities in Drug Formulation. Crystals 2021, 11, 1339. [Google Scholar] [CrossRef]
- Baptista, J.A.; Rosado, M.T.S.; Castro, R.A.E.; Évora, A.O.L.; Maria, T.M.R.; Silva, M.R.; Canotilho, J.; Eusébio, M.E.S. Dihydrofolate Reductase Inhibitors: The Pharmacophore as a Guide for Co-Crystal Screening. Molecules 2021, 26, 6721. [Google Scholar] [CrossRef]
- Mohammady, M.; Hadidi, M.; Ghetmiri, S.I.; Yousefi, G. Design of Ultra-Fine Carvedilol Nanococrystals: Development of a Safe and Stable Injectable Formulation. Eur. J. Pharm. Biopharm. 2021, 168, 139–151. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Grepioni, F. Mechanochemical Preparation of Co-Crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. [Google Scholar] [CrossRef]
- Eshtiagh-Hosseini, H.; Aghabozorg, H.; Mirzaei, M.; Beyramabadi, S.A.; Eshghi, H.; Morsali, A.; Shokrollahi, A.; Aghaei, R. Hydrothermal Synthesis, Experimental and Theoretical Characterization of a Novel Cocrystal Compound in the 2:1 Stoichiometric Ratio Containing 6-Methyluracil and Dipicolinic Acid. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 78, 1392–1396. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-Set Analysis of Hydrogen-Bond Patterns in Organic Crystals. Acta Crystallogr. Sect. B Struct. Sci. 1990, 46, 256–262. [Google Scholar] [CrossRef] [PubMed]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Shunnar, A.F.; Dhokale, B.; Karothu, D.P.; Bowskill, D.H.; Sugden, I.J.; Hernandez, H.H.; Naumov, P.; Mohamed, S. Efficient Screening for Ternary Molecular Ionic Cocrystals Using a Complementary Mechanosynthesis and Computational Structure Prediction Approach. Chem. Eur. J. 2020, 26, 4752–4765. [Google Scholar] [CrossRef] [PubMed]
- Perlovich, G. Melting Points of One- and Two-Component Molecular Crystals as Effective Characteristics for Rational Design of Pharmaceutical Systems. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2020, 76, 696–706. [Google Scholar] [CrossRef]
- O’Neil, M.J. (Ed.) The Merck Index—An Encyclopedia of Chemicals, Drugs, and Biologicals; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Sathisaran, I.; Dalvi, S.V. Engineering Cocrystals of Poorlywater-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef]
- Vasoya, J.M.; Shah, A.V.; Serajuddin, A.T.M. Investigation of Possible Solubility and Dissolution Advantages of Cocrystals, I: Aqueous Solubility and Dissolution Rates of Ketoconazole and Its Cocrystals as Functions of PH. ADMET DMPK 2019, 7, 106–130. [Google Scholar] [CrossRef]
- Sareen, S.; Joseph, L.; Mathew, G. Improvement in Solubility of Poor Water-Soluble Drugs by Solid Dispersion. Int. J. Pharm. Investig. 2012, 2, 12. [Google Scholar] [CrossRef]
- Varshosaz, J.; Ghassami, E.; Ahmadipour, S. Crystal Engineering for Enhanced Solubility and Bioavailability of Poorly Soluble Drugs. Curr. Pharm. Des. 2018, 24, 2473–2496. [Google Scholar] [CrossRef]
- Gould, P.L. Salt Selection for Basic Drugs. Int. J. Pharm. 1986, 33, 201–217. [Google Scholar] [CrossRef]
- Devi, S.; Kumar, A.; Kapoor, A.; Verma, V.; Yadav, S.; Bhatia, M. Ketoprofen–FA Co-Crystal: In vitro and in vivo Investigation for the Solubility Enhancement of Drug by Design of Expert. AAPS PharmSciTech 2022, 23, 101. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A.; Berger, C.M.; Brownell, C. PH-Metric Solubility. 2: Correlation between the Acid-Base Titration and the Saturation Shake-Flask Solubility-PH Methods. Pharm. Res. 2000, 17, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Fini, A.; Fazio, G.; Feroci, G. Solubility and Solubilization Properties of Non-Steroidal Anti-Inflammatory Drugs. Int. J. Pharm. 1995, 126, 95–102. [Google Scholar] [CrossRef]
- Herzfeldt, C.D.; Kümmel, R. Dissociation Constants, Solubilities and Dissolution Rates of Some Selected Nonsteroidal Antiinflammatories. Drug Dev. Ind. Pharm. 1983, 9, 767–793. [Google Scholar] [CrossRef]
- Fitriani, L.; Firdaus, W.A.; Sidadang, W.; Rosaini, H.; Putra, O.D.; Oyama, H.; Uekusa, H.; Zaini, E. Improved Solubility and Dissolution Rate of Ketoprofen by the Formation of Multicomponent Crystals with Tromethamine. Crystals 2022, 12, 275. [Google Scholar] [CrossRef]
- Avdeef, A.; Bendels, S.; Tsinman, O.; Tsinman, K.; Kansy, M. Solubility-Excipient Classification Gradient Maps. Pharm. Res. 2007, 24, 530–545. [Google Scholar] [CrossRef]
- Gao, L.; Zheng, W.Y.; Yang, W.L.; Zhang, X.R. Drug-Drug Salt Forms of Vortioxetine with Mefenamic Acid and Tolfenamic Acid. J. Mol. Struct. 2022, 1268, 133725. [Google Scholar] [CrossRef]
- Chiarini, A.; Tartarini, A.; Fini, A. PH-Solubility Relationship and Partition Coefficients for Some Anti-Inflammatory Arylaliphatic Acids. Arch. Pharm. 1984, 317, 268–273. [Google Scholar] [CrossRef]
- Ledwidge, M.T.; Corrigan, O.I. Effects of Surface Active Characteristics and Solid State Forms on the PH Solubility Profiles of Drug-Salt Systems. Int. J. Pharm. 1998, 174, 187–200. [Google Scholar] [CrossRef]
- Nugrahani, I.; Tjengal, B.; Gusdinar, T.; Horikawa, A.; Uekusa, H. A Comprehensive Study of a New 1.75 Hydrate of Ciprofloxacin Salicylate: SCXRD Structure Determination, Solid Characterization, Water Stability, Solubility, and Dissolution Study. Crystals 2020, 10, 349. [Google Scholar] [CrossRef]
- Ràfols, C.; Fael, H.; Fuguet, E.; Outhwaite, B.; Lee, S.; Ruiz, R. Dissolution Rates of Ciprofloxacin and Its Cocrystal with Resorcinol. ADMET DMPK 2018, 6, 61–70. [Google Scholar] [CrossRef]
- Anderson, B.D.; Conradi, R.A. Predictive Relationships in the Water Solubility of Salts of a Nonsteroidal Anti-Inflammatory Drug. J. Pharm. Sci. 1985, 74, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Bruker-AXS. Bruker APEX4. In APEX4 V2022.1; Bruker-AXS: Madison, WI, USA, 2022. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| API | Reported pKa Values | Ref. | Calculated pKa Values * | ΔpKa |
|---|---|---|---|---|
| Ciprofloxacin | 8.74 (−NH) | [27] | 8.77 | -- |
| Dexketoprofen | 3.88; 4.55; 5.02 | [28,29,30] | 3.88 | 4.86 |
| Mefenamic acid | 3.93; 4.64 | [31] | 3.89 | 4.81 |
| Tolfenamic acid | 4.3; 5.11 | [32,33] | 3.88 | 4.44 |
| Ketoprofen | 3.8; 3.7; 4; 4.30 | [31,33,34,35] | 3.88 | 4.94 |
| Diclofenac | 4.15; 4.2 | [33,36] | 4.00 | 4.59 |
| Sulindac | 4.7 | [30,37] | 4.09 | 4.04 |
| Coformer | H_ex (kcal/mol) | Reference |
|---|---|---|
| Diflunisal | −3.80753 | [17] |
| Tolfenamic acid | −2.718395 | This work |
| Diclofenac | −3.069495 | This work |
| Mefenamic acid | −2.540215 | This work |
| Indoprofen | −1.520345 | [17] |
| Ketoprofen | −1.530325 | This work |
| Dexketoprofen | −1.530325 | This work |
| Sulindac | −0.260615 | This work |
| Compound | Experimental (HPLC) CIP Solubility pH 6.8 (mg/mL) | Experimental (HPLC) CIP Solubility pH 1.2 (mg/mL) | Solubility Enhancement Compared to CIP/NSAIDs (pH 6.8) | Reported NSAID Solubility (mg/mL) | Ref. |
|---|---|---|---|---|---|
| CIP–DKT | 0.465 | 9.980 | 5.5x/- | Not reported | |
| CIP–KET | 0.207 | 9.533 | 2.4x/0.8x | <0.2 | [58,59,60,61,62] |
| CIP–MEF | 0.140 | 1.182 | 1.7x/2x | <0.05 | [63,64] |
| CIP–TLF | 0.047 | 6.567 | 0.6x/0.74x | <0.05 | [63] |
| CIP–DIC | 0.035 | 5.946 | 0.4x/20.8x | <0.003 | [60,61,65,66] |
| CIP | 0.084 | 14.671 | 0.08 | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acebedo-Martínez, F.J.; Domínguez-Martín, A.; Alarcón-Payer, C.; Sevillano-Páez, A.; Verdugo-Escamilla, C.; González-Pérez, J.M.; Martínez-Checa, F.; Choquesillo-Lazarte, D. Enhanced NSAIDs Solubility in Drug–Drug Formulations with Ciprofloxacin. Int. J. Mol. Sci. 2023, 24, 3305. https://doi.org/10.3390/ijms24043305
Acebedo-Martínez FJ, Domínguez-Martín A, Alarcón-Payer C, Sevillano-Páez A, Verdugo-Escamilla C, González-Pérez JM, Martínez-Checa F, Choquesillo-Lazarte D. Enhanced NSAIDs Solubility in Drug–Drug Formulations with Ciprofloxacin. International Journal of Molecular Sciences. 2023; 24(4):3305. https://doi.org/10.3390/ijms24043305
Chicago/Turabian StyleAcebedo-Martínez, Francisco Javier, Alicia Domínguez-Martín, Carolina Alarcón-Payer, Alejandro Sevillano-Páez, Cristóbal Verdugo-Escamilla, Josefa María González-Pérez, Fernando Martínez-Checa, and Duane Choquesillo-Lazarte. 2023. "Enhanced NSAIDs Solubility in Drug–Drug Formulations with Ciprofloxacin" International Journal of Molecular Sciences 24, no. 4: 3305. https://doi.org/10.3390/ijms24043305
APA StyleAcebedo-Martínez, F. J., Domínguez-Martín, A., Alarcón-Payer, C., Sevillano-Páez, A., Verdugo-Escamilla, C., González-Pérez, J. M., Martínez-Checa, F., & Choquesillo-Lazarte, D. (2023). Enhanced NSAIDs Solubility in Drug–Drug Formulations with Ciprofloxacin. International Journal of Molecular Sciences, 24(4), 3305. https://doi.org/10.3390/ijms24043305