
Understanding the Molecular Progression of Chronic Traumatic Encephalopathy in Traumatic Brain Injury, Aging and Neurodegenerative Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
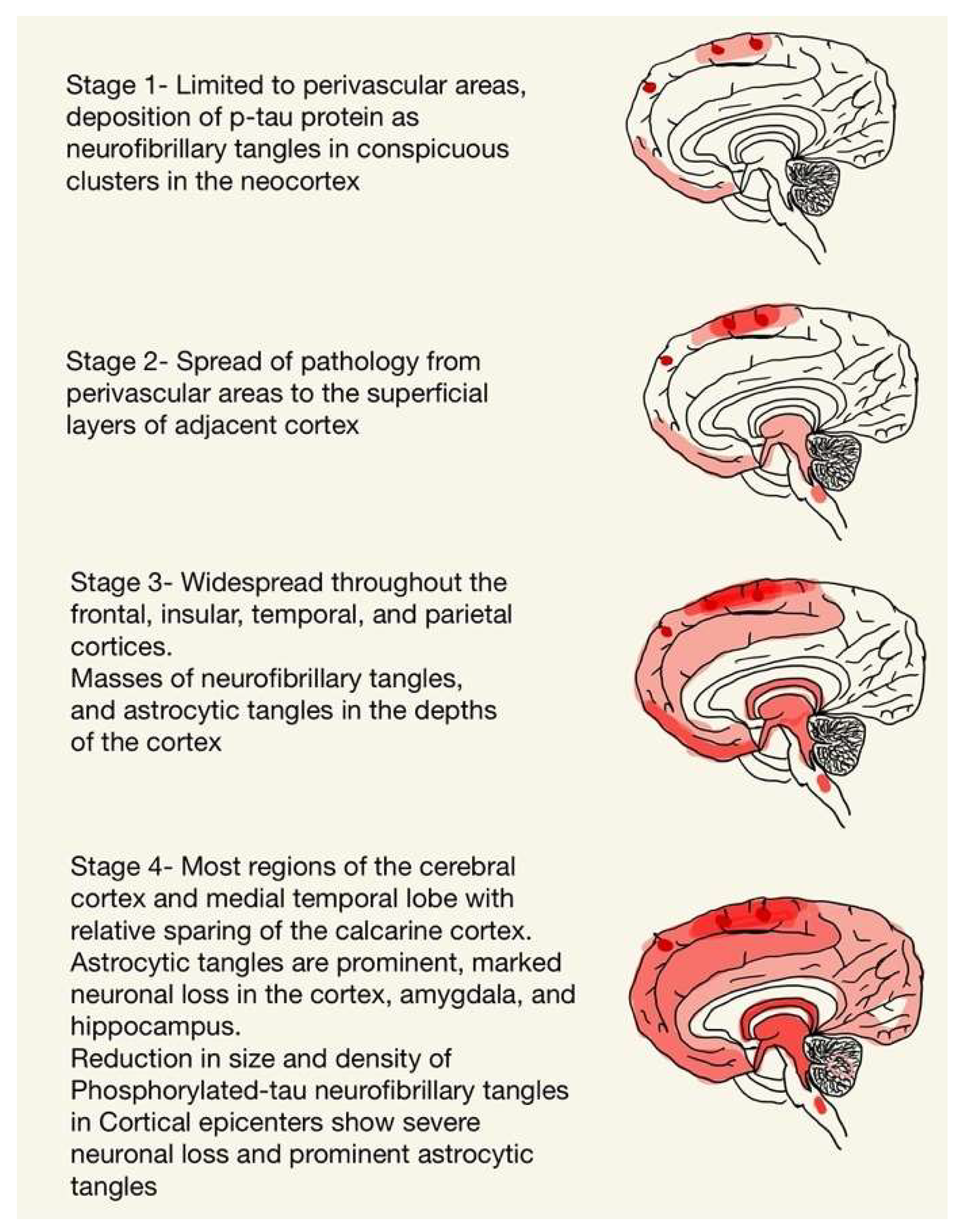
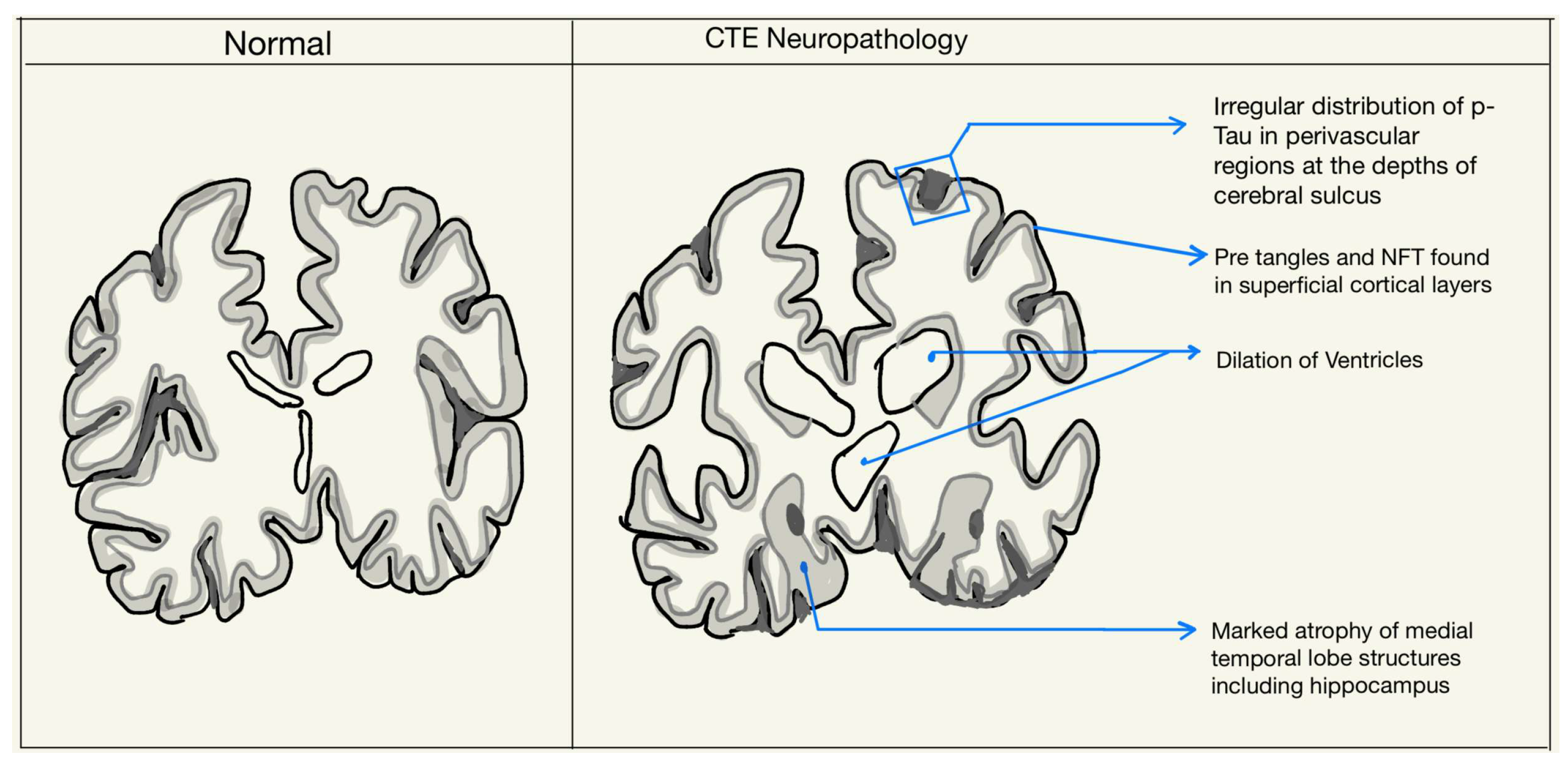
2. Neuropathology of CTE
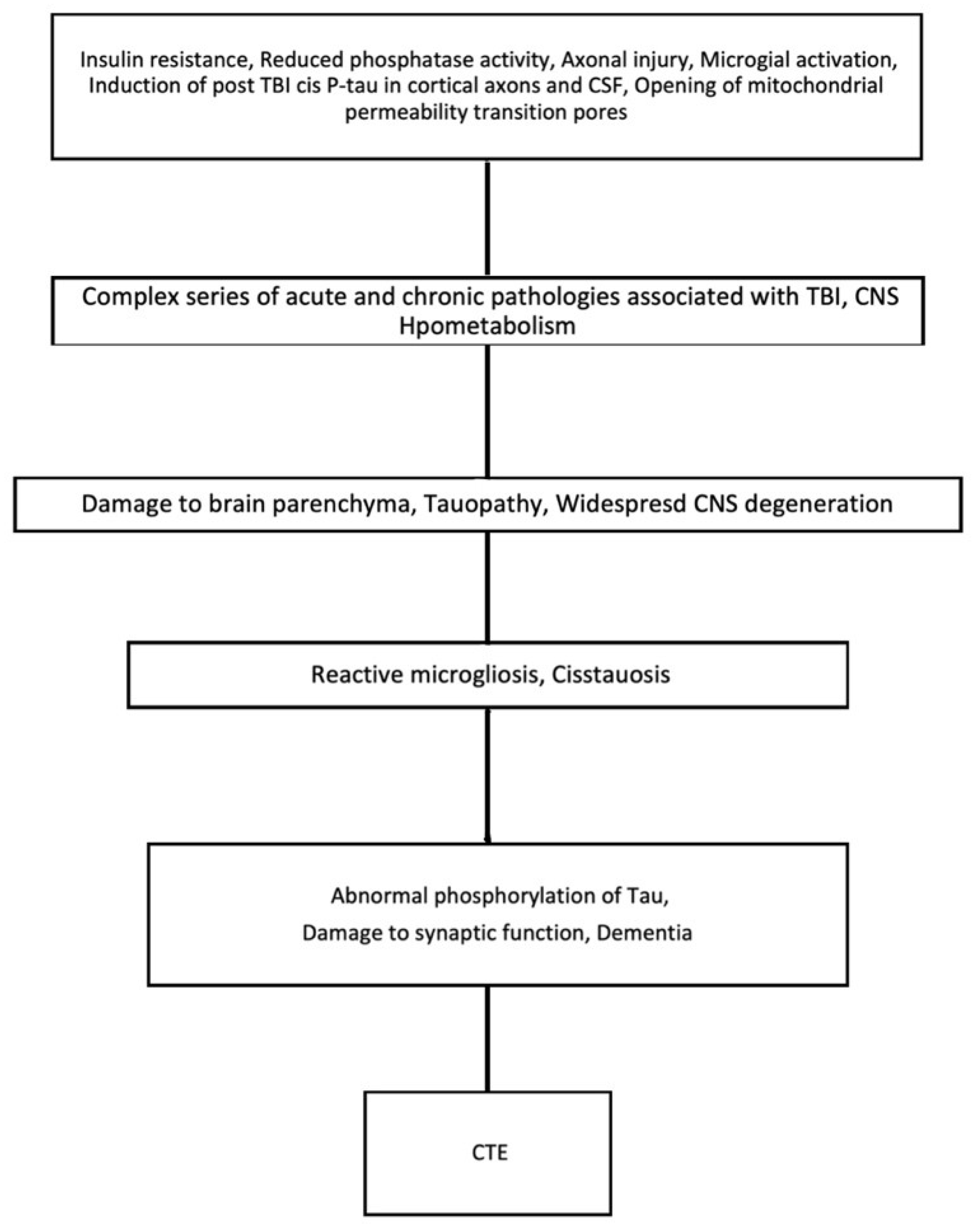
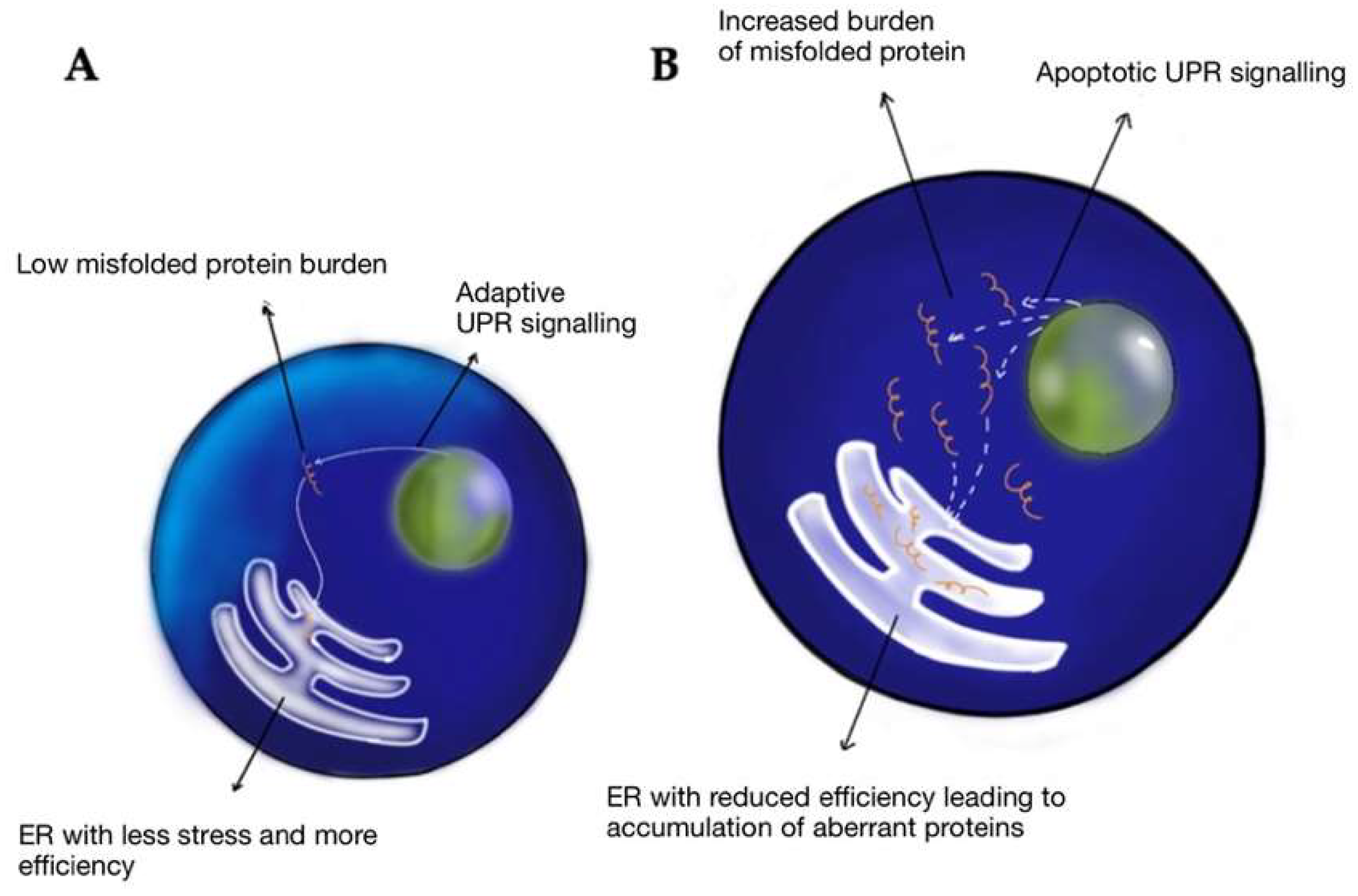
3. TBI and CTE
4. Aging and CTE
5. Neurodegenerative Diseases and CTE
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McKee, A.C. The Neuropathology of Chronic Traumatic Encephalopathy: The Status of the Literature. Semin. Neurol. 2020, 40, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Mckee, A.C.; Abdolmohammadi, B.; Stein, T.D. The neuropathology of chronic traumatic encephalopathy. Handb. Clin. Neurol. 2018, 158, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.; Gardner, A.J.; Schneider, K.J.; Guskiewicz, K.M.; Bailes, J.; Cantu, R.C.; Castellani, R.J.; Turner, M.; Jordan, B.D.; Randolph, C.; et al. A systematic review of potential long-term effects of sport-related concussion. Br. J. Sport. Med. 2017, 51, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Baugh, C.M.; Stamm, J.M.; Riley, D.O.; Gavett, B.E.; Shenton, M.E.; Lin, A.; Nowinski, C.J.; Cantu, R.C.; McKee, A.C.; Stern, R.A. Chronic traumatic encephalopathy: Neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav. 2012, 6, 244–254. [Google Scholar] [CrossRef]
- Smith, D.H.; Johnson, V.E.; Trojanowski, J.Q.; Stewart, W. Chronic traumatic encephalopathy—Confusion and controversies. Nat. Rev. Neurol. 2019, 15, 179–183. [Google Scholar] [CrossRef]
- Shively, S.B.; Priemer, D.S.; Stein, M.B.; Perl, D.P. Pathophysiology of Traumatic Brain Injury, Chronic Traumatic Encephalopathy, and Neuropsychiatric Clinical Expression. Psychiatr. Clin. N. Am. 2021, 44, 443–458. [Google Scholar] [CrossRef]
- VanItallie, T.B. Traumatic brain injury (TBI) in collision sports: Possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism 2019, 100, 153943. [Google Scholar] [CrossRef]
- Blumbergs, P.C.; Scott, G.; Manavis, J.; Wainwright, H.; Simpson, D.A.; McLean, A.J. Stalning af amyloid percursor protein to study axonal damage in mild head Injury. Lancet 1994, 344, 1055–1056. [Google Scholar] [CrossRef]
- McKee, A.C.; Daneshvar, D.H.; Alvarez, V.E.; Stein, T.D. The neuropathology of sport. Acta Neuropathol. 2014, 127, 29–51. [Google Scholar] [CrossRef]
- Oppenheimer, D.R. Microscopic lesions in the brain following head injury. J. Neurol. Neurosurg. Psychiatry 1968, 31, 299–306. [Google Scholar] [CrossRef]
- Mckee, A.C.; Stein, T.D.; Nowinski, C.J.; Stern, R.A.; Daneshvar, D.H.; Alvarez, V.E.; Lee, H.-S.; Hall, G.; Wojtowicz, S.M.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef]
- Genis, L.; Chen, Y.; Shohami, E.; Michaelson, D.M. Tau hyperphosphorylation in apolipoprotein E-deficient and control mice after closed head injury. J. Neurosci. Res. 2000, 60, 559–564. [Google Scholar] [CrossRef]
- Amadoro, G.; Ciotti, M.T.; Costanzi, M.; Cestari, V.; Calissano, P.; Canu, N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2892–2897. [Google Scholar] [CrossRef] [PubMed]
- Daneshvar, D.H.; Goldstein, L.E.; Kiernan, P.T.; Stein, T.D.; McKee, A.C. Post-traumatic neurodegeneration and chronic traumatic encephalopathy. Mol. Cell. Neurosci. 2015, 66, 81–90. [Google Scholar] [CrossRef]
- Schmidt, M.; Zhukareva, V.; Newell, K.; Lee, V.; Trojanowski, J. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001, 101, 518–524. [Google Scholar] [CrossRef]
- Stein, T.D.; Alvarez, V.E.; McKee, A.C. Chronic traumatic encephalopathy: A spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimer’s Res. Ther. 2014, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kwong, L.K.; Sampathu, D.M.; Trojanowski, J.Q.; Lee, V.M.-Y. TDP-43 Proteinopathy in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Arch. Neurol. 2007, 64, 1388. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Trojanowski, J.Q.; Smith, D.H. Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol. 2011, 122, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.W.; Allsop, D.; Bruton, C. The occult aftermath of boxing. J. Neurol. Neurosurg. Psychiatry 1990, 53, 373–378. [Google Scholar] [CrossRef]
- Mez, J.; Daneshvar, D.H.; Kiernan, P.T.; Abdolmohammadi, B.; Alvarez, V.E.; Huber, B.R.; Alosco, M.L.; Solomon, T.M.; Nowinski, C.J.; McHale, L.; et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 2017, 318, 360. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Asken, B.M. Injury cascades in TBI-related neurodegeneration. Brain Inj. 2017, 31, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, S.M.; Leclercq, P.D.; Moyes, L.; Graham, D.I.; Smith, C.; Griffin, W.S.T.; Nicoll, J.A.R. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci. Int. 2004, 146, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gentleman, S.M.; Leclercq, P.D.; Murray, L.S.; Griffin, W.S.T.; Graham, D.I.; Nicoll, J.A.R. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol. Appl. Neurobiol. 2013, 39, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo-Pereira, M.E.; Rockwell, P.; Schmidt-Glenewinkel, T.; Serrano, P. Neuroinflammation and J2 prostaglandins: Linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 2015, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Giza, C.C.; Hovda, D.A. The Neurometabolic Cascade of Concussion. J. Athl. Train. 2001, 36, 228–235. [Google Scholar]
- Saulle, M.; Greenwald, B.D. Chronic Traumatic Encephalopathy: A Review. Rehabil. Res. Pract. 2012, 2012, 816069. [Google Scholar] [CrossRef]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R.A. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy After Repetitive Head Injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- Ren, Z.; Iliff, J.J.; Yang, L.; Yang, J.; Chen, X.; Chen, M.J.; Giese, R.N.; Wang, B.; Shi, X.; Nedergaard, M. ‘Hit & Run’ Model of Closed-Skull Traumatic Brain Injury (TBI) Reveals Complex Patterns of Post-Traumatic AQP4 Dysregulation. J. Cereb. Blood Flow Metab. 2013, 33, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of Glymphatic Pathway Function Promotes Tau Pathology after Traumatic Brain Injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Graham, D.I. βA4 amyloid protein deposition in brain after head trauma. Lancet 1991, 338, 1422–1423. [Google Scholar] [CrossRef]
- Smith, D.H.; Johnson, V.E.; Stewart, W. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat. Rev. Neurol. 2013, 9, 211–221. [Google Scholar] [CrossRef]
- Olney, J.W.; Sharpe, L.G. Brain Lesions in an Infant Rhesus Monkey Treated with Monosodium Glutamate. Science 1969, 166, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Blaylock, R.; Maroon, J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis. Surg. Neurol. Int. 2011, 2, 107. [Google Scholar] [CrossRef] [PubMed]
- Mckee, A.C.; Cairns, N.J.; Dickson, D.; Folkerth, R.D.; Dirk, K.C.; Litvan, I.; Perl, D.; Stein, T.D.; Vonsattel, J.-P.; Stewart, W.; et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016, 131, 75–86. [Google Scholar] [CrossRef]
- Ling, H.; Holton, J.L.; Shaw, K.; Davey, K.; Lashley, T.; Revesz, T. Histological evidence of chronic traumatic encephalopathy in a large series of neurodegenerative diseases. Acta Neuropathol. 2015, 3, 891–893. [Google Scholar] [CrossRef]
- Hay, J.; Johnson, V.E.; Young, A.M.H.; Smith, D.H.; Stewart, W. Blood brain barrier disruption is an early event that may persist for many years following traumatic brain injury in humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar] [CrossRef]
- Hay, J.; Johnson, V.E.; Smith, D.H.; Stewart, W. Chronic Traumatic Encephalopathy: The Neuropathological Legacy of Traumatic Brain Injury. Annu. Rev. Pathol. 2016, 11, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.P.; Wallace, E.; BCh, M.; Loftus, T.; Keaney, J.; Kealy, J.; Humphries, M.M.; Molloy, M.G.; Meaney, J.F.; Farrell, M.; et al. Blood-Brain Barrier Dysfunction as a Hallmark Pathology in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2016, 75, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Merlo Pich, M.; Genova, M.; Ventura, B.; Bovina, C.; Formiggini, G.; Parenti Castelli, G. Mitochondrial bioenergetics in aging. Mitochondrial Bioenerg. Aging Biochim. Biophys. Acta 2000, 1459, 397–404. Available online: www.elsevier.com/locate/bba (accessed on 10 October 2022). [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Cell Physiol. 2007, 292, 670–686. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in Inflammatory and Degenerative Brain Diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. Available online: https://academic.oup.com/jnen/article/63/9/901/2916516 (accessed on 10 October 2022). [CrossRef]
- Barrientos, A.; Casademont, J.; Cardellach, F.; Estivill, X.; Urbano-Marquez, A.; Nunes, V. Reduced steady-state levels of mitochondrial RNA and increased mitochondrial DNA amount in human brain with aging. Mol. Brain Res. 1997, 52, 284–289. [Google Scholar] [CrossRef]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2013, 14, 434–456. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Scheuner, D.; Schröder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef]
- Verkhratsky, A. Physiology and Pathophysiology of the Calcium Store in the Endoplasmic Reticulum of Neurons. Physiol. Rev. 2005, 85, 201–279. [Google Scholar] [CrossRef]
- Mattson, M.P. apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic Microglia in the Aging Human Brain. GLIA 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Huang, Y.; Wynne, A.M.; Godbout, J.P. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 2009, 23, 309–317. [Google Scholar] [CrossRef]
- Palmer, A.L.; Ousman, S.S. Astrocytes and Aging. Front. Aging Neurosci. 2018, 10, 337. [Google Scholar] [CrossRef]
- Koellhoffer, E.C.; Mccullough, L.D.; Ritzel, R.M. Molecular Sciences Old Maids: Aging and Its Impact on Microglia Function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef]
- Aoyama, K.; Sang, W.S.; Hamby, A.M.; Liu, J.; Wai, Y.C.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Hazrati, L.-N.; Tartaglia, M.C.; Diamandis, P.; Davis, K.D.; Green, R.E.; Wennberg, R.; Wong, J.C.; Ezerins, L.; Tator, C.H.; Pascual-Leone, A.; et al. Absence of chronic traumatic encephalopathy in retired football players with multiple concussions and neurological symptomatology. Front. Hum. Neurosci. 2013, 7, 222. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Gavett, B.E.; Stern, R.A.; Nowinski, C.J.; Cantu, R.C.; Kowall, N.W.; Perl, D.P.; Tessa Hedley-Whyte, E.; Price, B.; Sullivan, C.; et al. TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 918–929. Available online: www.jneuropath.com (accessed on 7 October 2022). [CrossRef] [PubMed]
- Gu, D.; Ou, S.; Liu, G. Traumatic Brain Injury and Risk of Dementia and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Syst. Rev. Neuroepidemiol. 2021, 56, 4–16. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Zou, J.; Caoid, F. A meta-analysis of cohort studies: Traumatic brain injury and risk of Alzheimer’s Disease. PLoS ONE 2021, 16, e0253206. [Google Scholar] [CrossRef]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 28, 8. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Yaffe, K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell. Neurosci. 2015, 66, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Rabaneda-Lombarte, N.; Manuel Vidal-Taboada, J.; Valente, T.; Ezquerra, M.; Fernández-Santiago, R.; Martí, M.J.; Compta, Y.; Saura, J.; Solà, C. ARTICLE Altered expression of the immunoregulatory ligand-receptor pair CD200-CD200R1 in the brain of Parkinson’s disease patients. Npj Park. Dis. 2022, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial Activation and Chronic Neurodegeneration. Neurother. J. Am. Soc. Exp. Neurother. 2010, 7, 345–365. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Murasugi, T.; Oda, T. Acute neuroinflammation exacerbates excitotoxicity in rat hippocampus in vivo. Exp. Neurol. 2002, 177, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.P.K.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E.H. Parkin ubiquitinates tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruchika, F.; Shah, S.; Neupane, D.; Vijay, R.; Mehkri, Y.; Lucke-Wold, B. Understanding the Molecular Progression of Chronic Traumatic Encephalopathy in Traumatic Brain Injury, Aging and Neurodegenerative Disease. Int. J. Mol. Sci. 2023, 24, 1847. https://doi.org/10.3390/ijms24031847
Ruchika F, Shah S, Neupane D, Vijay R, Mehkri Y, Lucke-Wold B. Understanding the Molecular Progression of Chronic Traumatic Encephalopathy in Traumatic Brain Injury, Aging and Neurodegenerative Disease. International Journal of Molecular Sciences. 2023; 24(3):1847. https://doi.org/10.3390/ijms24031847
Chicago/Turabian StyleRuchika, FNU, Siddharth Shah, Durga Neupane, Ruddra Vijay, Yusuf Mehkri, and Brandon Lucke-Wold. 2023. "Understanding the Molecular Progression of Chronic Traumatic Encephalopathy in Traumatic Brain Injury, Aging and Neurodegenerative Disease" International Journal of Molecular Sciences 24, no. 3: 1847. https://doi.org/10.3390/ijms24031847
APA StyleRuchika, F., Shah, S., Neupane, D., Vijay, R., Mehkri, Y., & Lucke-Wold, B. (2023). Understanding the Molecular Progression of Chronic Traumatic Encephalopathy in Traumatic Brain Injury, Aging and Neurodegenerative Disease. International Journal of Molecular Sciences, 24(3), 1847. https://doi.org/10.3390/ijms24031847