
Adenosinergic System and BDNF Signaling Changes as a Cross-Sectional Feature of RTT: Characterization of Mecp2 Heterozygous Mouse Females
 ,
,  , ,
, , 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
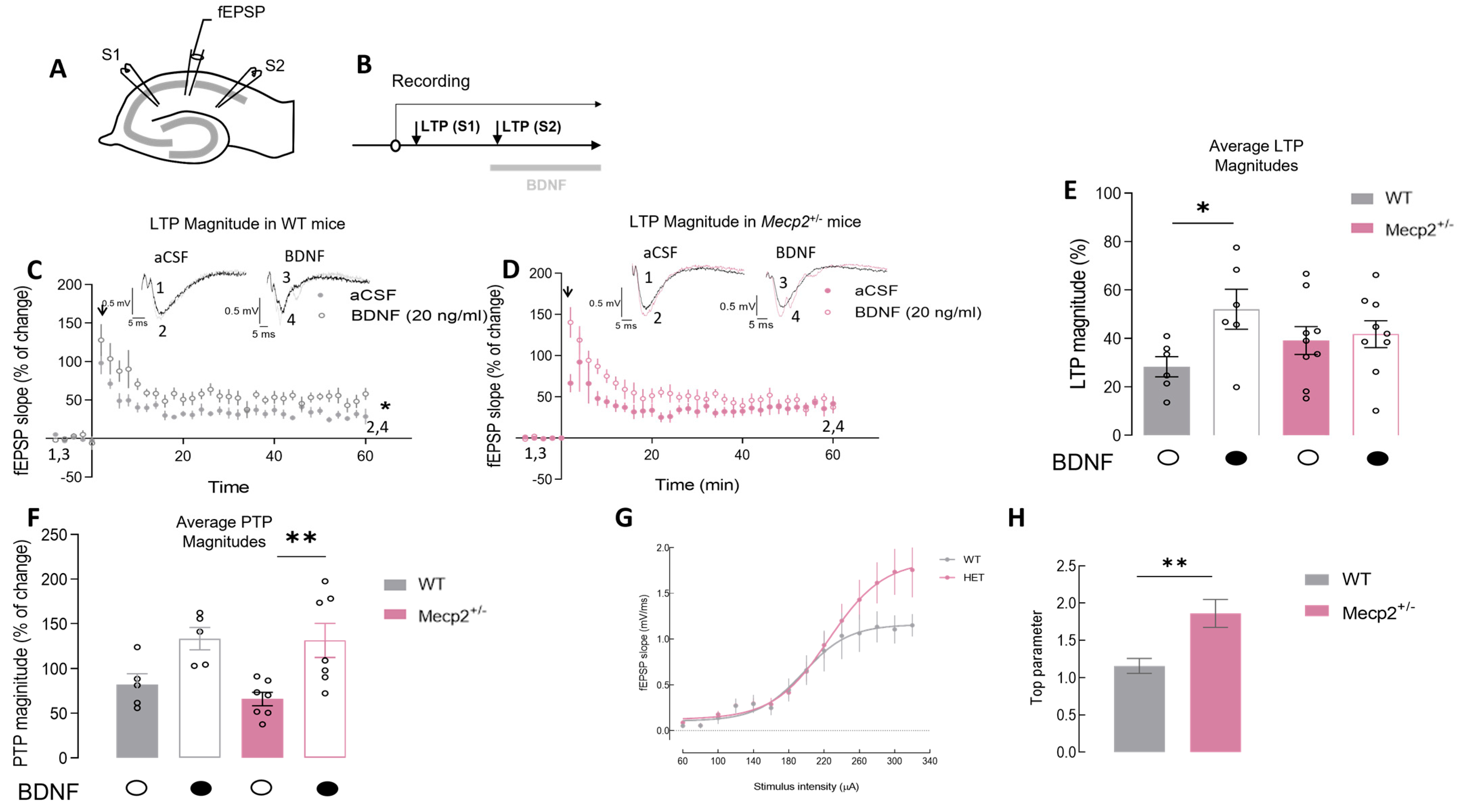
2.1. The Magnitude of Hippocampal LTP Is Maintained in Mecp2+/− Female Mice but BDNF Loses Its Effect on LTP Magnitude
2.2. BDNF Protein Levels Are Decreased in the Hippocampus and Cortex of Symtomatic Heterozygous Mecp2+/− Female Mice
2.3. TrkB-FL Protein Levels Are Not Changed in the Hippocampus of Mecp2+/− Mice
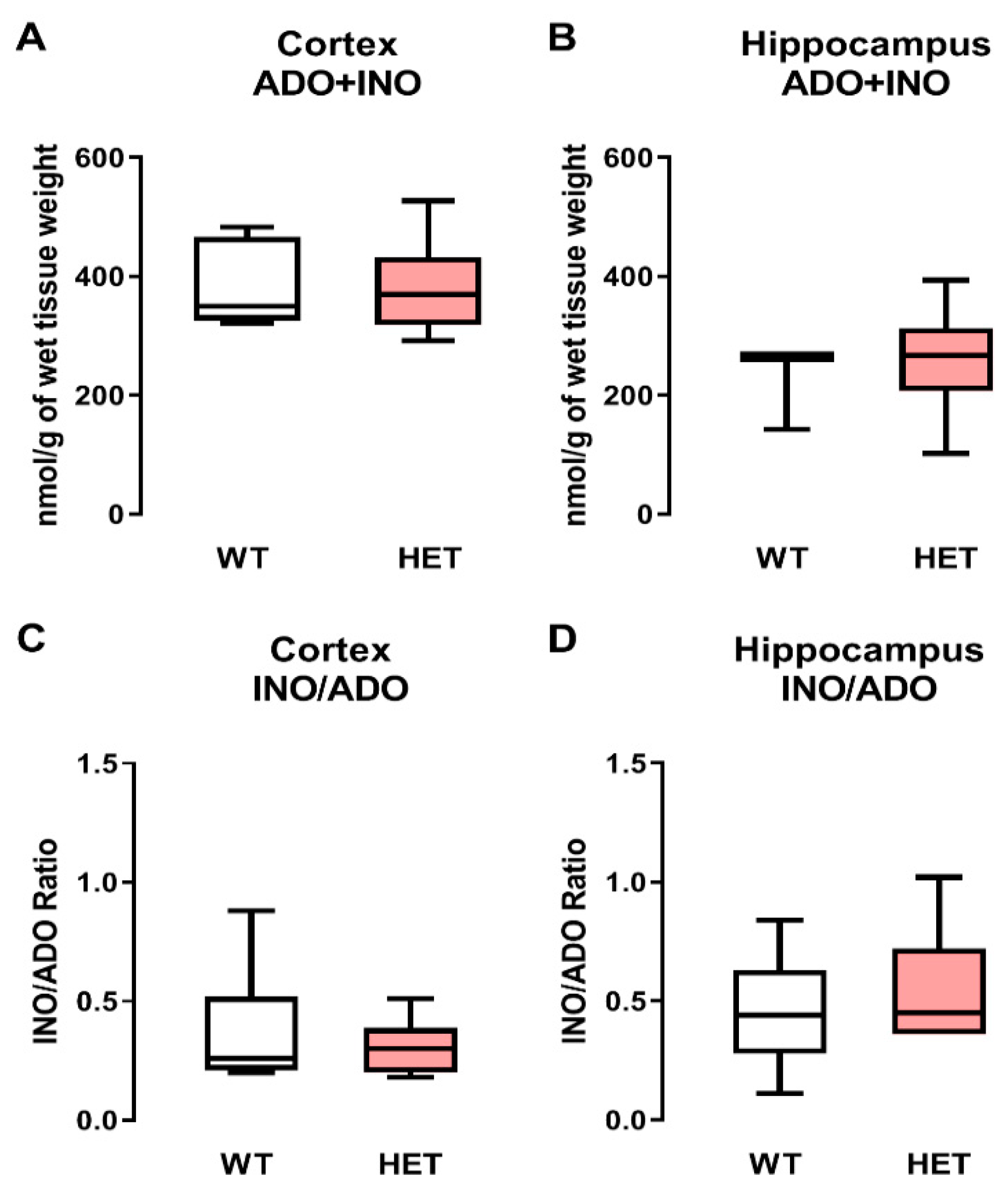
2.4. Adenosine (Plus Inosine) Levels Remain Unaltered in the Cortex and Hippocampus of Heterozygous Mecp2+/− Female Mice
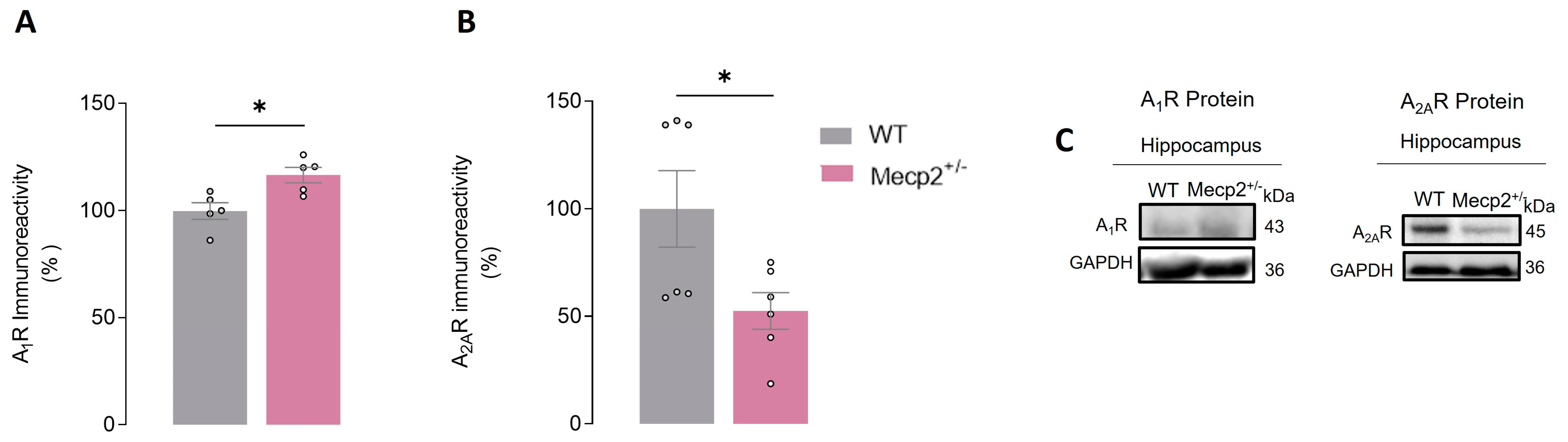
2.5. Mecp2+/− Females Display Alterations in Hippocampal A2AR and A1R Adenosine Receptor Levels
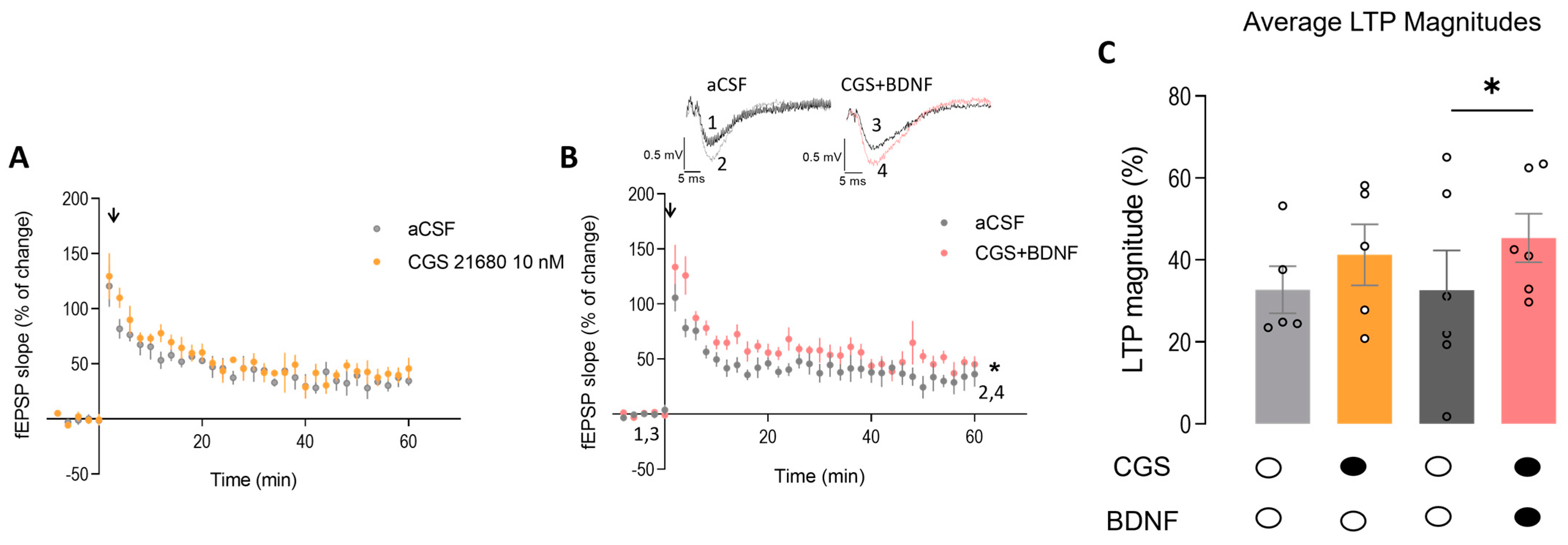
2.6. Exogenous Activation of A2AR Restores BDNF-Induced LTP Facilitation in the Hippocampus of Heterozygous Mecp2+/− Female Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Ex Vivo Electrophysiological Recordings
4.3. Extraction and Analysis of Adenosine and Inosine by Liquid Chromatography with Diode Array Detection (HPLC/DAD)
4.4. Western Blot
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl- CpG-Binding Protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Petriti, U.; Dudman, D.C.; Scosyrev, E.; Lopez-Leon, S. Global Prevalence of Rett Syndrome: Systematic Review and Meta-Analysis. Syst. Rev. 2023, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Clarke, A.J.; Leonard, H.; Bailey, M.E.S.; Carolyn, N.; Zappella, M.; Renieri, A. Rett Syndrome: Revised Diagnostic Criteria and Nomenclature. Ann. Neurol. 2011, 68, 944–950. [Google Scholar] [CrossRef]
- Banerjee, A.; Miller, M.T.; Li, K.; Sur, M.; Kaufmann, W.E. Towards a Better Diagnosis and Treatment of Rett Syndrome: A Model Synaptic Disorder. Brain 2019, 142, 239–248. [Google Scholar] [CrossRef]
- Gold, W.A.; Krishnarajy, R.; Ellaway, C.; Christodoulou, J. Rett Syndrome: A Genetic Update and Clinical Review Focusing on Comorbidities. ACS Chem. Neurosci. 2018, 9, 167–176. [Google Scholar] [CrossRef]
- Bedogni, F.; Rossi, R.L.; Galli, F.; Cobolli Gigli, C.; Gandaglia, A.; Kilstrup-Nielsen, C.; Landsberger, N. Rett Syndrome and the Urge of Novel Approaches to Study MeCP2 Functions and Mechanisms of Action. Neurosci. Biobehav. Rev. 2014, 46, 187–201. [Google Scholar] [CrossRef]
- Sharifi, O.; Yasui, D.H. The Molecular Functions of MeCP2 in Rett Syndrome Pathology. Front. Genet. 2021, 12, 624290. [Google Scholar] [CrossRef]
- Li, W.; Pozzo-Miller, L. BDNF Deregulation in Rett Syndrome. Neuropharmacology 2014, 76, 737–746. [Google Scholar] [CrossRef]
- Binder, D.K.; Scharfman, H.E. Brain-Derived Neurotrophic Factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Eide, F.F. Naturally Occuring Truncated Trkb Receptors Have Dominant Inhibitory Effects on Brain-Derived Neurotrophic Factor Signaling. J. Neurosci. 1996, 16, 3123–3129. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Blum, R.; Pichler, B.; Lepier, A.; Kafitz, K.W.; Konnerth, A. Truncated TrkB-T1 Mediates Neurotrophin-Evoked Calcium Signalling in Glia Cells. Nature 2003, 426, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Lourenço, C.; Duarte, S.T.; Palminha, C.; Gaspar, C.; Rodrigues, T.M.; Magalhães-Cardosof, T.; Rei, N.; Colino-Oliveira, M.; Gomes, R.; Ferreira, S.; et al. Impairment of Adenosinergic System in Rett Syndrome: Novel Therapeutic Target to Boost BDNF Signalling. Neurobiol. Dis. 2020, 145, 105043. [Google Scholar] [CrossRef]
- Chang, Q.; Khare, G.; Dani, V.; Nelson, S.; Jaenisch, R. The Disease Progression of Mecp2 Mutant Mice Is Affected by the Level of BDNF Expression. Neuron 2006, 49, 341–348. [Google Scholar] [CrossRef]
- Wang, H.; Chan, S.; Ogier, M.; Hellard, D.; Wang, Q.; Smith, C.; Katz, D.M. Dysregulation of Brain-Derived Neurotrophic Factor Expression and Neurosecretory Function in Mecp2 Null Mice. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 10911–10915. [Google Scholar] [CrossRef]
- Ogier, M.; Wang, H.; Hong, E.; Wang, Q.; Greenberg, M.E.; Katz, D.M. Brain-Derived Neurotrophic Factor Expression and Respiratory Function Improve after Ampakine Treatment in a Mouse Model of Rett Syndrome. J. Neurosci. 2007, 27, 10912–10917. [Google Scholar] [CrossRef]
- Diógenes, M.J.; Costenla, A.R.; Lopes, L.V.; Jerónimo-Santos, A.; Sousa, V.C.; Fontinha, B.M.; Ribeiro, J.A.; Sebastião, A.M. Enhancement of LTP in Aged Rats Is Dependent on Endogenous BDNF. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2011, 36, 1823–1836. [Google Scholar] [CrossRef]
- Rajagopal, R.; Chen, Z.-Y.; Lee, F.S.; Chao, M.V. Transactivation of Trk Neurotrophin Receptors by G-Protein-Coupled Receptor Ligands Occurs on Intracellular Membranes. J. Neurosci. 2004, 24, 6650. [Google Scholar] [CrossRef]
- Jerónimo-Santos, A.; Fonseca-Gomes, J.; Guimarães, D.A.; Tanqueiro, S.R.; Ramalho, R.M.; Ribeiro, J.A.; Sebastiaõ, A.M.; Diógenes, M.J. Brain-Derived Neurotrophic Factor Mediates Neuroprotection against A β-Induced Toxicity through a Mechanism Independent on Adenosine 2A Receptor Activation. Growth Factors 2015, 33, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Vaz, S.H.; Lérias, S.R.; Parreira, S.; Diógenes, M.J.; Sebastião, A.M. Adenosine A2A Receptor Activation Is Determinant for BDNF Actions upon GABA and Glutamate Release from Rat Hippocampal Synaptosomes. Purinergic Signal. 2015, 11, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.F.; Ferreira, F.; Rodrigues, R.S.; Soares, R.; Pedro, D.M.; Duarte-Samartinho, M.; Aroeira, R.I.; Ferreiro, E.; Valero, J.; Solá, S.; et al. Regulation of Hippocampal Postnatal and Adult Neurogenesis by Adenosine A2A Receptor: Interaction with Brain-Derived Neurotrophic Factor. Stem Cells 2021, 39, 1362–1381. [Google Scholar] [CrossRef] [PubMed]
- Tebano, M.T.; Martire, A.; Potenza, R.L.; Grò, C.; Pepponi, P.; Armida, M.; Domenici, M.R.; Schwarzschild, M.A.; Chen, J.F.; Popoli, P. Adenosine A(2A) Receptors Are Required for Normal BDNF Levels and BDNF-Induced Potentiation of Synaptic Transmission in the Mouse Hippocampus. J. Neurochem. 2008, 104, 279–286. [Google Scholar] [CrossRef]
- Jeon, S.J.; Rhee, S.Y.; Ryu, J.H.; Cheong, J.H.; Kwon, K.; Yang, S.-I.; Park, S.H.; Lee, J.; Kim, H.Y.; Han, S.-H.; et al. Activation of Adenosine A2A Receptor Up-Regulates BDNF Expression in Rat Primary Cortical Neurons. Neurochem. Res. 2011, 36, 2259. [Google Scholar] [CrossRef]
- Bliss, T.V.; Collingridge, G.L. A Synaptic Model of Memory: Long-Term Potentiation in the Hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G. Synaptic Plasticity and Memory: An Evaluation of the Hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef]
- Calfa, G.; Percy, A.; Pozzo-Miller, L. On Experimental Models of Rett Syndrome Based on Mecp2 Dysfunction. Exp. Biol. Med. 2011, 236, 3–19. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; MacDonald, J.L. Sex Differences in Mecp2-Mutant Rett Syndrome Model Mice and the Impact of Cellular Mosaicism on Phenotype Development. Brain Res. 2020, 1729, 146644. [Google Scholar] [CrossRef]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett Syndrome: A Neurological Disorder with Metabolic Components. Open Biol. 2018, 8, 170216. [Google Scholar] [CrossRef]
- Vidal, S.; Xiol, C.; Pascual-Alonso, A.; O’Callaghan, M.; Pineda, M.; Armstrong, J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int. J. Mol. Sci. 2019, 20, 3925. [Google Scholar] [CrossRef] [PubMed]
- Good, K.V.; Vincent, J.B.; Ausió, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.; Cobb, S.; Downs, J. Clinical and Biological Progress over 50 Years in Rett Syndrome. Nat. Rev. Neurol. 2016, 13, 37–51. [Google Scholar] [CrossRef]
- Li, W.; Bellot-Saez, A.; Phillips, M.L.; Yang, T.; Longo, F.M.; Pozzo-Miller, L. A Small-Molecule TrkB Ligand Restores Hippocampal Synaptic Plasticity and Object Location Memory in Rett Syndrome Mice. Dis. Models Mech. 2017, 10, 837–845. [Google Scholar] [CrossRef]
- Schmid, D.A.; Yang, T.; Ogier, M.; Adams, I.; Mirakhur, Y.; Wang, Q.; Massa, S.M.; Longo, F.M.; Katz, D.M. A TrkB Small Molecule Partial Agonist Rescues TrkB Phosphorylation Deficits and Improves Respiratory Function in a Mouse Model of Rett Syndrome. J. Neurosci. 2012, 32, 1803–1810. [Google Scholar] [CrossRef]
- Sebastião, A.M.; Assaife-Lopes, N.; Diógenes, M.J.; Vaz, S.H.; Ribeiro, J.A. Modulation of Brain-Derived Neurotrophic Factor (BDNF) Actions in the Nervous System by Adenosine A2A Receptors and the Role of Lipid Rafts. Biochim. Biophys. Acta (BBA)-Biomembr. 2011, 1808, 1340–1349. [Google Scholar] [CrossRef]
- Cristalli, G.; Costanzi, S.; Lambertucci, C.; Lupidi, G.; Vittori, S.; Volpini, R.; Camaioni, E. Adenosine Deaminase: Functional Implications and Different Classes of Inhibitors. Med. Res. Rev. 2001, 21, 105–128. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Schulz, R.; Hutchison, A.J.; Do, U.H.; Sills, M.A.; Williams, M. [3H]CGS 21680, a Selective A2 Adenosine Receptor Agonist Directly Labels A2 Receptors in Rat Brain. J. Pharmacol. Exp. Ther. 1989, 251, 888–893. [Google Scholar]
- Hoffbuhr, K.C.; Moses, L.M.; Jerdonek, M.A.; Naidu, S.; Hoffman, E.P. Associations between MeCP2 Mutations, X-Chromosome Inactivation, and Phenotype. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 99–105. [Google Scholar] [CrossRef]
- Ishii, T.; Makita, Y.; Ogawa, A.; Amamiya, S.; Yamamoto, M.; Miyamoto, A.; Oki, J. The Role of Different X-Inactivation Pattern on the Variable Clinical Phenotype with Rett Syndrome. Brain Dev. 2001, 23, S161–S164. [Google Scholar] [CrossRef]
- Xiol, C.; Vidal, S.; Pascual-Alonso, A.; Blasco, L.; Brandi, N.; Pacheco, P.; Gerotina, E.; O’Callaghan, M.; Pineda, M.; Armstrong, J.; et al. X Chromosome Inactivation Does Not Necessarily Determine the Severity of the Phenotype in Rett Syndrome Patients. Sci. Rep. 2019, 9, 11983. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.M.; Baker, S.A.; Zoghbi, H.Y. MECP2 Disorders: From the Clinic to Mice and Back. J. Clin. Investig. 2015, 125, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, G.; Angelucci, A.; Morando, L.; Boggio, E.M.; Giustetto, M.; Pizzorusso, T. Early Environmental Enrichment Moderates the Behavioral and Synaptic Phenotype of MeCP2 Null Mice. Biol. Psychiatry 2010, 67, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Boggio, E.M.; Lonetti, G.; Pizzorusso, T.; Giustetto, M. Boggio Synaptic Determinants of Rett Syndrome. Front. Synaptic Neurosci. 2010, 2. [Google Scholar] [CrossRef]
- Weng, S.-M.; McLeod, F.; Bailey, M.E.S.; Cobb, S.R. Synaptic Plasticity Deficits in an Experimental Model of Rett Syndrome: Long-Term Potentiation Saturation and Its Pharmacological Reversal. Neuroscience 2011, 180, 314–321. [Google Scholar] [CrossRef]
- Asaka, Y.; Jugloff, D.G.M.; Zhang, L.; Eubanks, J.H.; Fitzsimonds, R.M. Hippocampal Synaptic Plasticity Is Impaired in the Mecp2-Null Mouse Model of Rett Syndrome. Neurobiol. Dis. 2006, 21, 217–227. [Google Scholar] [CrossRef]
- Chao, H.-T.; Zoghbi, H.Y.; Rosenmund, C. MeCP2 Controls Excitatory Synaptic Strength by Regulating Glutamatergic Synapse Number. Neuron 2007, 56, 58–65. [Google Scholar] [CrossRef]
- Assaife-Lopes, N.; Sousa, V.C.; Pereira, D.B.; Ribeiro, J.A.; Sebastião, A.M. Regulation of TrkB Receptor Translocation to Lipid Rafts by Adenosine A(2A) Receptors and Its Functional Implications for BDNF-Induced Regulation of Synaptic Plasticity. Purinergic Signal 2014, 10, 251–267. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A Mouse Mecp2-Null Mutation Causes Neurological Symptoms That Mimic Rett Syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.W.; Collingridge, G.L. The LTP Program: A Data Acquisition Program for on-Line Analysis of Long-Term Potentiation and Other Synaptic Events. J. Neurosci. Methods 2001, 108, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Diogenes, M.J.; Dias, R.B.; Rombo, D.M.; Vicente Miranda, H.; Maiolino, F.; Guerreiro, P.; Nasstrom, T.; Franquelim, H.G.; Oliveira, L.M.A.; Castanho, M.A.R.B.; et al. Extracellular Alpha-Synuclein Oligomers Modulate Synaptic Transmission and Impair LTP Via NMDA-Receptor Activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Costa, A.F.; Moreira, S.; Ferreirinha, F.; Magalhães-Cardoso, M.T.; Calejo, I.; Silva-Ramos, M.; Correia-de-Sá, P. Inhibition of Cholinergic Neurotransmission by Β3-Adrenoceptors Depends on Adenosine Release and A1-Receptor Activation in Human and Rat Urinary Bladders. Am. J. Physiol.-Ren. Physiol. 2017, 313, F388–F403. [Google Scholar] [CrossRef]
- Vieira, C.; Ferreirinha, F.; Magalhães-Cardoso, M.T.; Silva, I.; Marques, P.; Correia-de-Sá, P. Post-Inflammatory Ileitis Induces Non-Neuronal Purinergic Signaling Adjustments of Cholinergic Neurotransmission in the Myenteric Plexus. Front. Pharmacol. 2017, 8, 811. [Google Scholar] [CrossRef]
- Silva, I.; Magalhães-Cardoso, M.T.; Ferreirinha, F.; Moreira, S.; Costa, A.F.; Silva, D.; Vieira, C.; Silva-Ramos, M.; Correia-de-Sá, P. Β3 Adrenoceptor-Induced Cholinergic Inhibition in Human and Rat Urinary Bladders Involves the Exchange Protein Directly Activated by Cyclic AMP 1 Favoring Adenosine Release. Br. J. Pharmacol. 2020, 177, 1589–1608. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda-Lourenço, C.; Rosa, J.; Rei, N.; Belo, R.F.; Lopes, A.L.; Silva, D.; Vieira, C.; Magalhães-Cardoso, T.; Viais, R.; Correia-de-Sá, P.; et al. Adenosinergic System and BDNF Signaling Changes as a Cross-Sectional Feature of RTT: Characterization of Mecp2 Heterozygous Mouse Females. Int. J. Mol. Sci. 2023, 24, 16249. https://doi.org/10.3390/ijms242216249
Miranda-Lourenço C, Rosa J, Rei N, Belo RF, Lopes AL, Silva D, Vieira C, Magalhães-Cardoso T, Viais R, Correia-de-Sá P, et al. Adenosinergic System and BDNF Signaling Changes as a Cross-Sectional Feature of RTT: Characterization of Mecp2 Heterozygous Mouse Females. International Journal of Molecular Sciences. 2023; 24(22):16249. https://doi.org/10.3390/ijms242216249
Chicago/Turabian StyleMiranda-Lourenço, Catarina, Jéssica Rosa, Nádia Rei, Rita F. Belo, Ana Luísa Lopes, Diogo Silva, Cátia Vieira, Teresa Magalhães-Cardoso, Ricardo Viais, Paulo Correia-de-Sá, and et al. 2023. "Adenosinergic System and BDNF Signaling Changes as a Cross-Sectional Feature of RTT: Characterization of Mecp2 Heterozygous Mouse Females" International Journal of Molecular Sciences 24, no. 22: 16249. https://doi.org/10.3390/ijms242216249
APA StyleMiranda-Lourenço, C., Rosa, J., Rei, N., Belo, R. F., Lopes, A. L., Silva, D., Vieira, C., Magalhães-Cardoso, T., Viais, R., Correia-de-Sá, P., Sebastião, A. M., & Diógenes, M. J. (2023). Adenosinergic System and BDNF Signaling Changes as a Cross-Sectional Feature of RTT: Characterization of Mecp2 Heterozygous Mouse Females. International Journal of Molecular Sciences, 24(22), 16249. https://doi.org/10.3390/ijms242216249