Abstract
Fabry disease is a lysosomal disease characterized by globotriaosylceramide (Gb3) accumulation. It may coexist with diabetes mellitus and both cause potentially lethal kidney end-organ damage. However, there is little information on their interaction with kidney disease. We have addressed the interaction between Fabry disease and diabetes in data mining of human kidney transcriptomics databases and in Fabry (Gla-/-) and wild type mice with or without streptozotocin-induced diabetes. Data mining was consistent with differential expression of genes encoding enzymes from the Gb3 metabolic pathway in human diabetic kidney disease, including upregulation of UGCG, the gene encoding the upstream and rate-limiting enzyme glucosyl ceramide synthase. Diabetic Fabry mice displayed the most severe kidney infiltration by F4/80+ macrophages, and a lower kidney expression of kidney protective genes (Pgc1α and Tfeb) than diabetic wild type mice, without a further increase in kidney fibrosis. Moreover, only diabetic Fabry mice developed kidney insufficiency and these mice with kidney insufficiency had a high expression of Ugcg. In conclusion, we found evidence of interaction between diabetes and Fabry disease that may increase the severity of the kidney phenotype through modulation of the Gb3 synthesis pathway and downregulation of kidney protective genes.
1. Introduction
Fabry disease is an X-linked inherited metabolic disorder caused by pathogenic GLA gene variants leading to deficient activity of the lysosomal enzyme alpha-galactosidase A, glycolipid accumulation in multiple cell types and in the circulation, and potentially lethal kidney, heart, or central nervous system end-organ damage [1]. Phenotypic variability in the severity of target organ injury is observed even within families [2,3]. Random X chromosome inactivation results in variable severity in females. However, the phenotypic variability between males having the same GLA variant is poorly understood. Fabry patients are not expected to be protected from the occurrence of common non-transmissible diseases that target the same organs. Diabetes mellitus (DM) is present in up to 13% of Fabry patients, especially among older ones, a prevalence similar to the general population. Older Fabry patients usually have GLA variants associated with later onset disease, such as N215S. A recent report from the UK biobank identified 18 participants with GLA N215S and three had hypertension and DM [4]. In this regard, while severe Fabry nephropathy is uncommon in patients with N215S, the frequency of kidney failure is still around 100-fold higher than in the general population [5,6]. Furthermore, advances in therapy for Fabry disease are expected to increase life expectancy, increasing the risk of age-associated conditions such as DM [7].
Fabry disease and diabetic kidney disease (DKD) can be conceptually defined as proteinuric progressive nephropathies triggered by the accumulation of metabolites: glucose and glycosylated molecules for DM and globotriaosylceramide (Gb3/GL-3) and globotriaosylsphingosine (lyso-Gb3) for Fabry disease [1,8,9,10,11]. We hypothesized that comorbidities such as DM may modulate the phenotypic expression of Fabry disease and vice versa, Fabry disease may modulate the phenotypic expression of diabetic target organ injury. Specifically, the association of DM and Fabry disease may modulate the severity of kidney disease as compared with either disease alone.
We have now studied the impact of DM on the kidney gene expression of enzymes in the Gb3 metabolic pathway in human and murine kidney transcriptomics and the impact of DM on the kidney phenotype of Fabry mice.
2. Results
2.1. Data Mining: Influence of DM on Gb3 Pathway Gene Expression in Human DKD
We explored a potential clinical interaction between DM and Fabry disease through data mining, examining the gene expression of Gb3 metabolic pathway enzymes in human DKD in the Nephroseq database of human kidney transcriptomics [12,13,14,15]. Specifically, we explored the impact of DM on the expression of genes encoding enzymes upstream of Gb3 synthesis, such as glucosylceramide synthase (encoded by UGCG), which is the upstream and rate-limiting enzyme, and Gb3 synthase (encoded by A4GALT) (Figure S1). Additionally, we also explored the impact on acid ceramidase (encoded by ASAH1) that may catalyze the conversion of Gb3 into lyso-Gb3 [16,17,18], and GLA, which encodes alpha-galactosidase A, the enzyme which is defective in Fabry disease and whose lysosomal activity removes the galactose residues that had been added by Gb3 synthase.
The most consistent finding across kidney transcriptomics datasets of human DKD was an increased tubulointerstitial expression of UGCG mRNA encoding the upstream and rate-limiting enzyme glucosylceramide synthase (Table S1a). These changes may be expected to increase Gb3 precursors and in association with increased tubulointerstitial ASAH1, to facilitate the generation of lyso-Gb3, a soluble metabolite that causes kidney cell injury [11,19]. Indeed, glucosylceramide synthase inhibitors decreased Gb3 and lyso-Gb3 in human Fabry disease [20]. The glomerular pattern differed. Glomerular Gb3 synthase gene expression was decreased while glomerular GLA gene expression was increased. Both changes may limit the local generation and accumulation of Gb3 in glomeruli. Additionally, increased UGCG and GLA mRNA were correlated with lower eGFR in human DKD, i.e., the genes appear to be upregulated when kidneys are more severely injured (Table S1b). The expression of ASAH1 decreased as the severity of injury increased. The Kidney Interactive Transcriptomics (KIT) database localized the cells potentially responsible for the changes in gene expression observed in bulk transcriptomics (Table S2). Single-cell transcriptomics data were overall coherent with bulk tissue transcriptomics. They showed a consistent increase in UGCG expression in multiple kidney cell types, ranging from tubular cells to glomerular cells to endothelium and leukocytes. They also identified proximal tubular cells and parietal epithelial cells as sources of increased ASAH1 expression, and decreased expression of A4GALT in parietal epithelial cells and collecting duct intercalated A cells.
Overall, these human DKD data suggest that DKD may modify local Gb3 metabolic pathways in the kidney, and further support a potential interaction between DM and Fabry disease that may modify the kidney phenotype when both conditions coexist. The upregulation of UGCG may increase Gb3 availability, while the coordinated upregulation of GLA may prevent Gb3 accumulation in persons with DKD not having Fabry disease, but in Fabry disease, a lack of GLA activity may result in further Gb3 accumulation in patients with coexistent diabetes and, in the case of missense genetic variants, in the accumulation of abnormal GLA proteins that may cause endoplasmic reticulum stress.
2.2. Interaction of DM and Fabry Disease in Mice
Overall, human kidney transcriptomics data supported the hypothesis that coexistent DM and Fabry disease may interact to modify the kidney phenotype. Next, we evaluated this hypothesis in vivo by inducing DM by administering streptozotocin (STZ) to Fabry and wild type (WT) mice and comparing them to controls administered vehicle.
Following induction of DM, no differences were apparent between diabetic WT and diabetic Fabry mice in mean glycemia values over follow-up (WT-DM 434 ± 36 vs. Fabry-DM 417 ± 36 mg/dL, p-value = 0.75), insulin requirements (1.76 ± 0.33 vs. 1.60 ± 0.27 IU/day, p-value = 0.70) or mortality. All WT mice surviving longer than 7 days required insulin and this was the case for 12/15 (80%) diabetic Fabry mice. There was one early (before day 7 after the last streptozotocin injection) death in each group and one late (beyond day 24 after the last streptozotocin injection) death in the diabetic WT group. Two (12.5%) diabetic Fabry were sacrificed because they looked sick and were found to have kidney insufficiency for a total of 2/12 (17%) and 3/16 (19%) dead or sacrificed mice in the diabetic WT and diabetic Fabry groups, respectively.
2.3. Impact of DM on the Kidney Expression of Genes in the Gb3 Metabolic Pathway in Mice
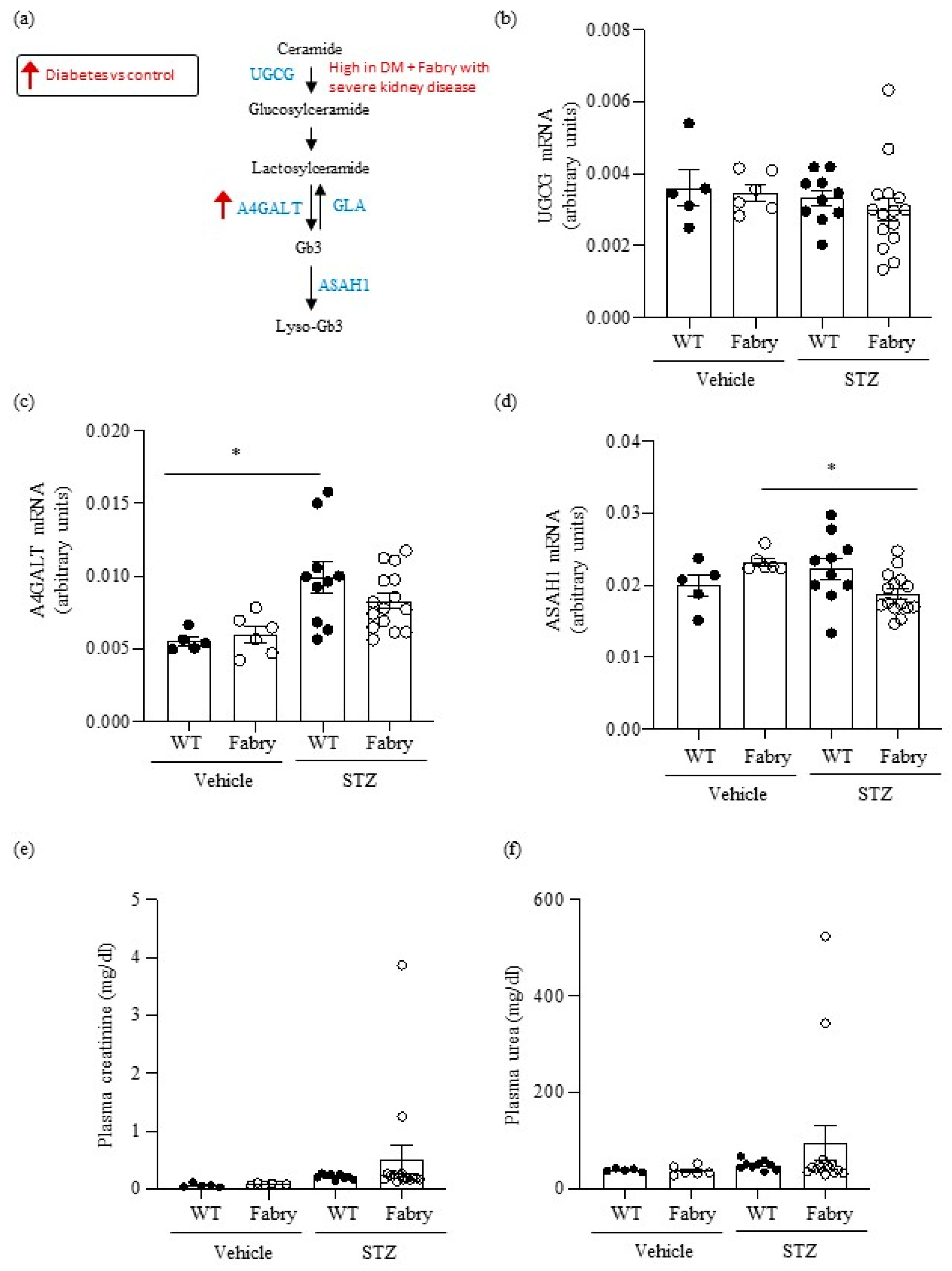
First, we explored the regulation of metabolic pathways involved in the generation of metabolites that accumulate in Fabry disease (Figure 1a). Although short-term DM in mice did not influence overall kidney Ugcg gene expression, very high levels were observed in two Fabry mice with DM that had developed severe kidney disease (Figure 1b), as detailed below (Section 2.7). DM also increased A4galt gene expression (Figure 1c), potentially further contributing to increased Gb3 availability. Diabetic Fabry mice had lower kidney expression of Asah1 than non-diabetic Fabry mice (Figure 1d).
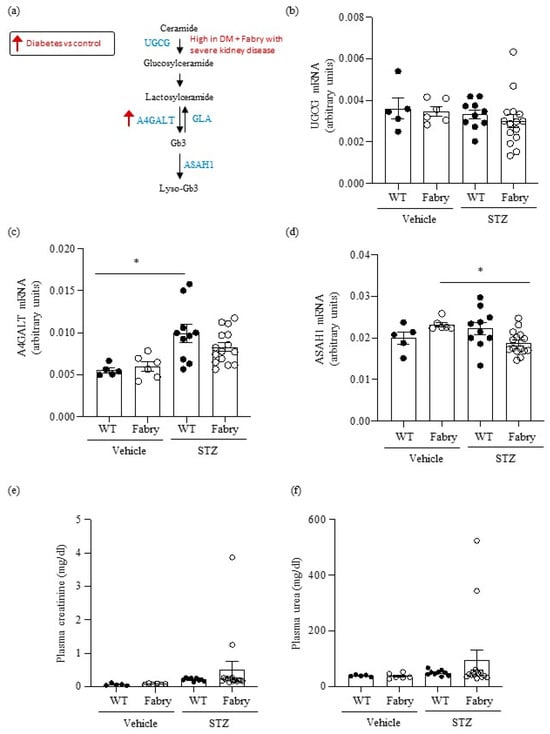
Figure 1.
Impact of diabetes on genes encoding enzymes in the Gb3 pathway and on kidney function in wild type and Fabry mice. (a) Scheme of Gb3 synthesis and metabolism: glucosylceramide synthase encoded by Ugcg, Gb3 synthase encoded by A4galt, acid ceramidase encoded by Asah1, and alpha-galactosidase A encoded by Gla. Arrows present a summary of data shown in panels (b–d). (b–d) Kidney mRNA was measured by real time RT-PCR in diabetic wild-type or Fabry mice or vehicle controls. (b) Ugcg, (c) A4galt, (d) Asah1. (e) Plasma creatinine and (f) plasma urea. Data expressed as mean ± SEM of 5–15 animals per group. * p < 0.05 vs. respective vehicle group. Significance (p < 0.05) was assessed by Student’s t-test for two groups of data and ANOVA for three or more groups with Bonferroni post-hoc correction.
2.4. Interaction of DM and Fabry Disease on Kidney Function in Mice
Next, we addressed the potential impact of coexistent Fabry disease and DM on kidney injury. For that, we analyzed kidney function, changes in kidney gene expression and histology in diabetic WT and diabetic Fabry mice.
No differences in mean values of plasma urea and creatinine were observed between the groups (Figure 1f,e). However, 2/15 (13%) surviving diabetic Fabry mice developed kidney insufficiency characterized by high serum urea and creatinine levels, while no diabetic WT mice developed kidney insufficiency. There were no differences in median (IQR) proteinuria values at the last follow-up: 100 (30–200) mg/dL and 100 (30–200) mg/dL for diabetic WT and diabetic Fabry mice, respectively.
Consistent with prior reports [21], non-diabetic Fabry and WT mice did not show significant differences in kidney function, histology, or gene expression, with the exception of a mild inflammation evidenced by F4/80 immunohistochemistry (Figure 2, Figure 3 and Figure 4). By contrast, DM promoted gene expression and/or histological evidence of kidney inflammation and fibrosis, which were observed in both WT and Fabry mice, as discussed below.
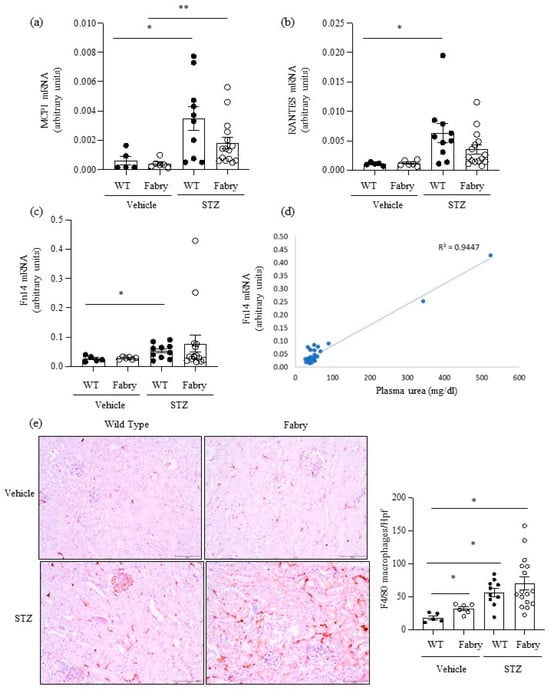
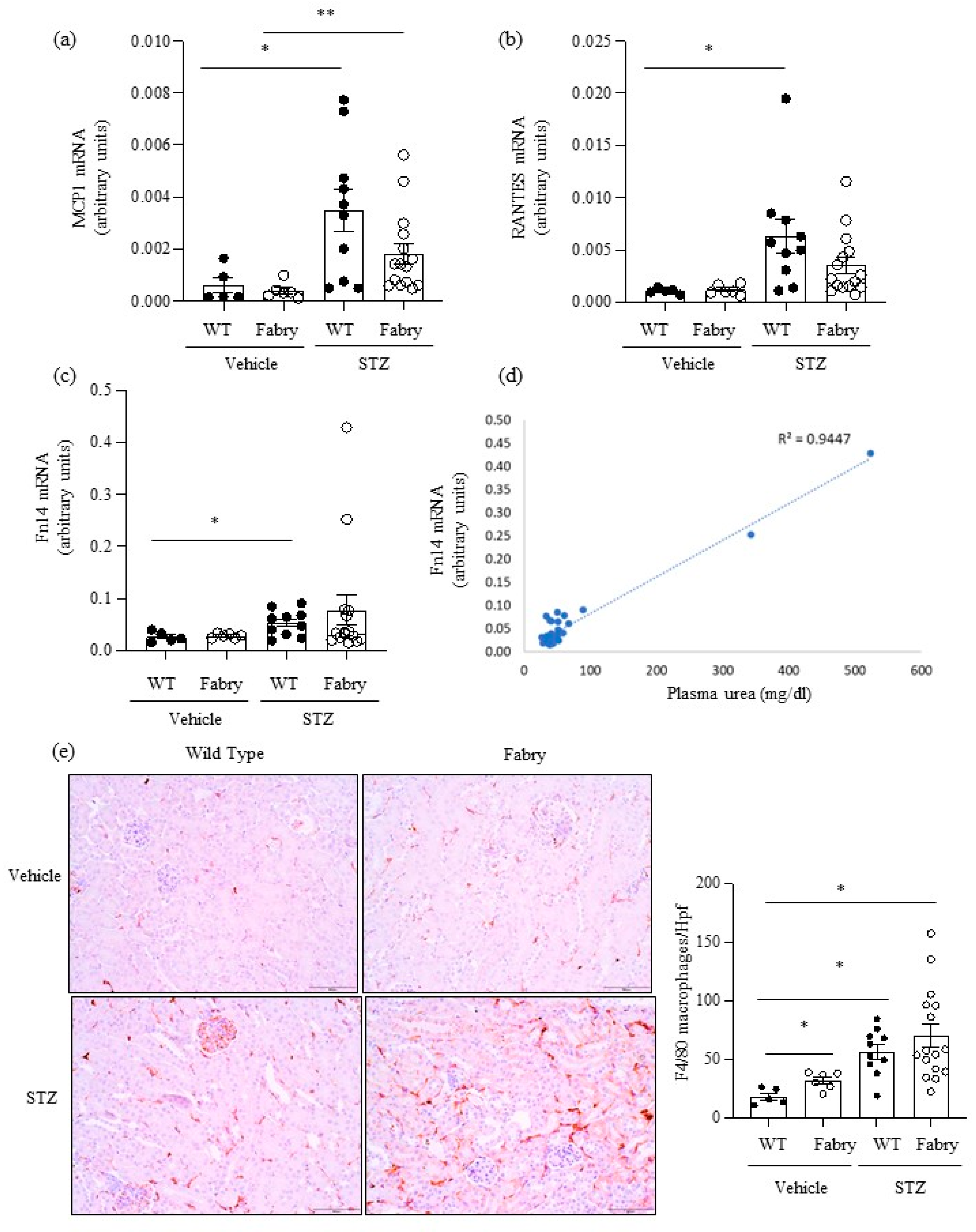
Figure 2.
Impact of diabetes on kidney inflammation in Fabry mice. (a) Mcp1, (b) Rantes and (c) Fn14 mRNA expression in kidneys was measured by real time RT-PCR. (d) Correlation between serum urea and kidney Fn14 mRNA expression as assessed by linear regression. (e) Immunohistochemistry assessment of F4/80+ macrophages: representative images and quantitation. * p < 0.05 vs. vehicle WT. ** p < 0.05 vs. vehicle Fabry.Original magnification ×20. Data expressed as mean ± SEM of 5–15 animals per group. Significance (p < 0.05) was assessed by Student’s t-test for two groups of data and ANOVA for three or more groups with Bonferroni post-hoc correction.
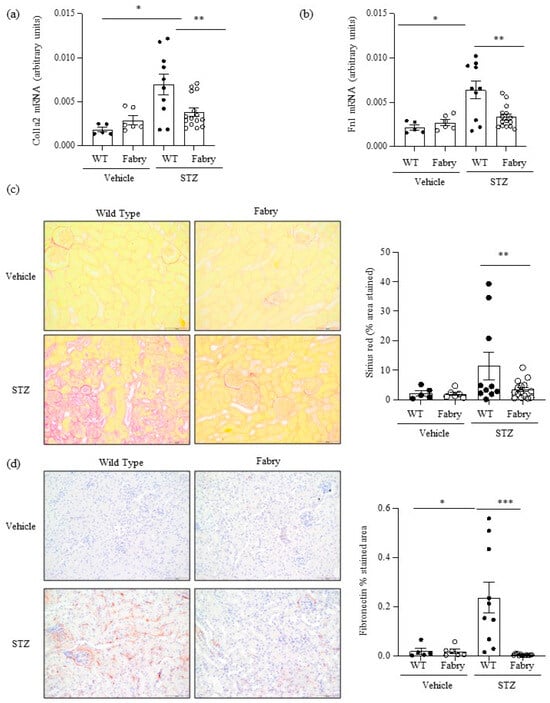
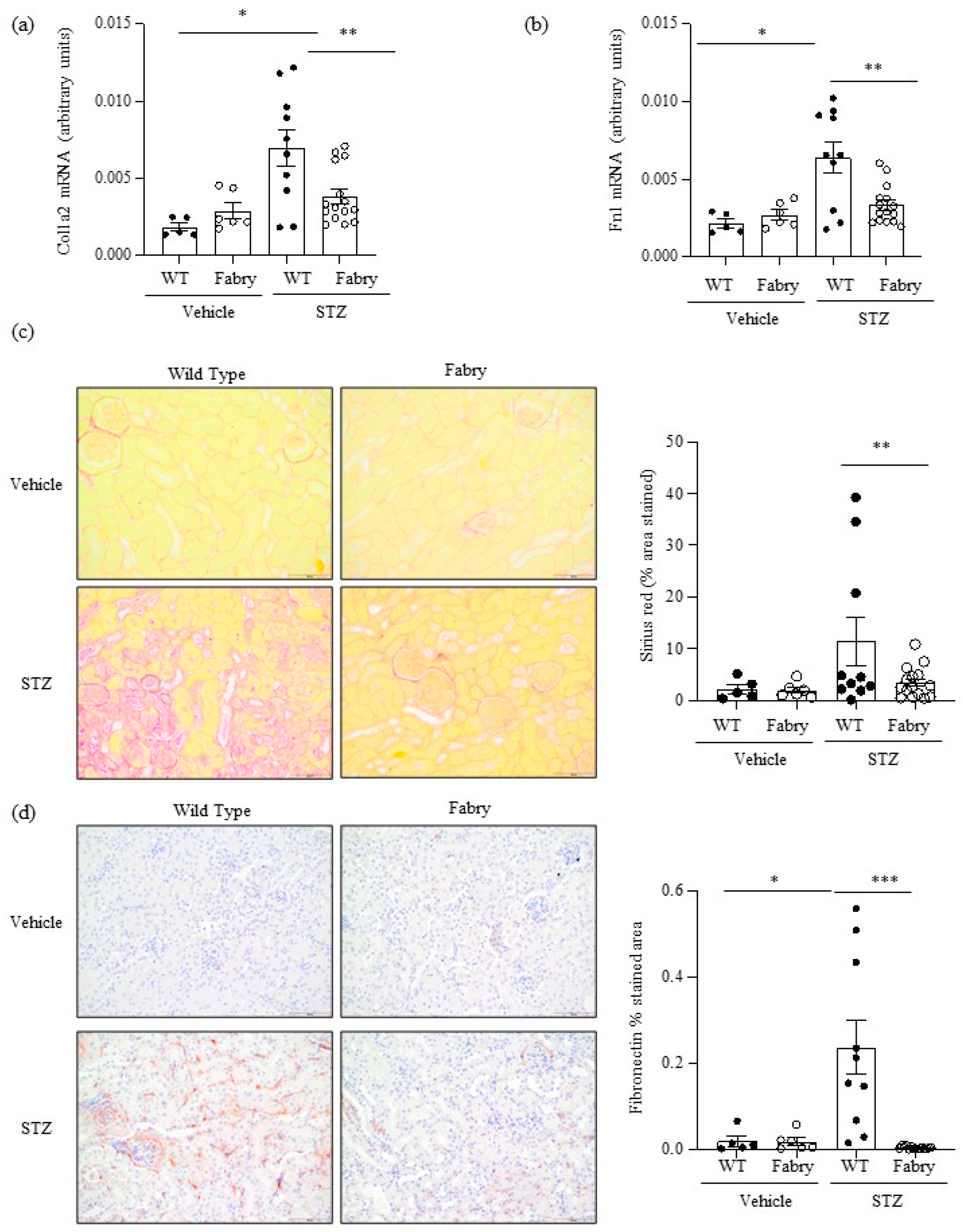
Figure 3.
Impact of diabetes on kidney fibrosis in Fabry mice. (a) Kidney Col1a2 and (b) Fn1 mRNA expression was measured by real time RT-PCR. (c) Sirius red staining for collagen deposition: representative images and quantitation. (d) Immunohistochemistry disclosed milder fibronectin deposition in diabetic Fabry mice than in diabetic WT mice. Original magnification ×20. * p < 0.05 vs. vehicle WT. ** p < 0.01 vs. WT diabetic, *** p < 0.0005 vs. WT diabetic. Data expressed as mean ± SEM of 5–15 animals per group. Significance (p < 0.05) was assessed by Student’s t-test for two groups of data and ANOVA for three or more groups with Bonferroni post-hoc correction.
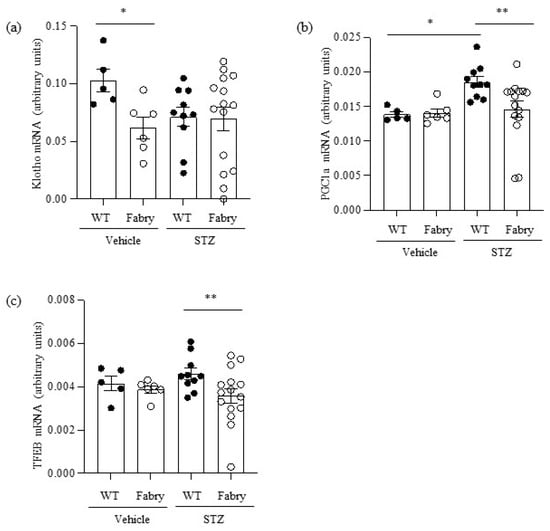
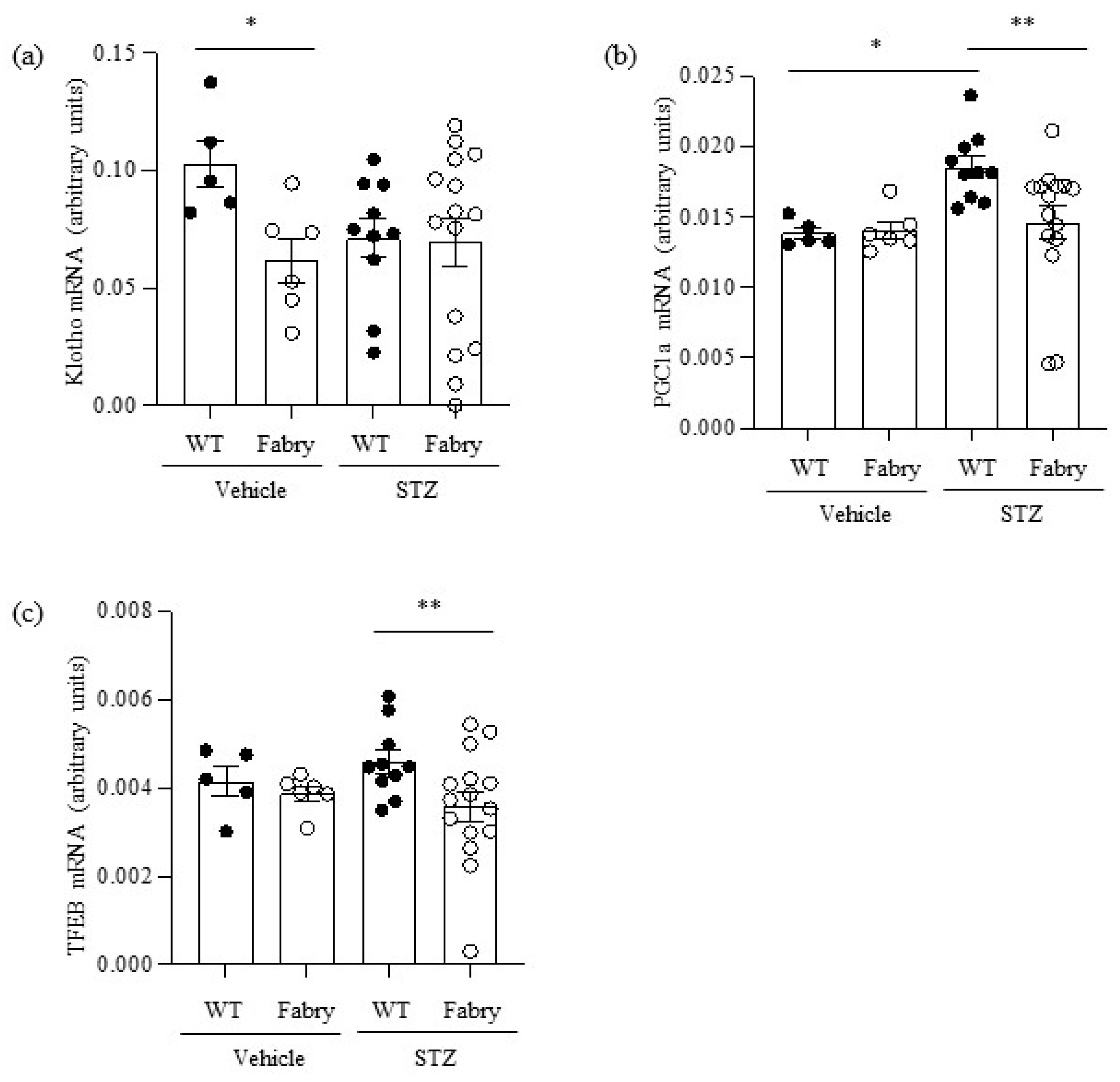
Figure 4.
Impact of diabetes on the expression of kidney nephroprotective genes in Fabry mice. (a) Kidney Klotho mRNA, (b) Pgc1α mRNA, (c) Tfeb mRNA. * p < 0.05 vs. WT vehicle, ** p < 0.05 vs. WT diabetic. Data expressed as mean ± SEM of 5–15 animals per group. Significance (p < 0.05) was assessed by Student’s t-test for two groups of data and ANOVA for three or more groups with Bonferroni post-hoc correction.
2.5. Interaction of DM and Fabry Disease on Kidney Inflammation and Fibrosis in Mice
Next, we explored the impact of DM on kidney inflammation and fibrosis in Fabry mice. MCP1 and RANTES are key chemokines upregulated during kidney injury. Kidney Mcp1 and Rantes mRNA were significantly upregulated in diabetic WT mice compared to WT mice (Figure 2a,b), while values in diabetic Fabry mice did not significantly differ from those in diabetic WT mice (Figure 2a,b). The gene expression of the proinflammatory cytokine receptor Fn14 was also significantly increased in diabetic WT mice compared to non-diabetic WT mice (Figure 2c) while diabetic Fabry mice did not differ from either non-diabetic Fabry mice or diabetic WT mice (Figure 2c). Of interest, kidney Fn14 mRNA expression was very high in the two diabetic Fabry mice that developed kidney insufficiency and correlated with kidney function (Figure 2c,d). Kidney inflammation was assessed as infiltration by F4/80+ macrophages (Figure 2e). Both DM and Fabry disease promoted kidney infiltration by macrophages and the combination of DM and Fabry disease resulted in the highest inflammatory infiltrates: over 100 F4/80+ cells per hpf were only observed in mice with both conditions. Both kidney Mcp1 and Rantes gene expression correlated with inflammatory infiltrates and between themselves (Figure S2).
Kidney fibrosis is a characteristic feature of both DKD and Fabry disease [22]. At the mRNA level, there was evidence of activation of fibrosis in diabetic WT mice and this was significantly milder in diabetic Fabry mice, as assessed by the kidney expression of the Col1a2 and Fn1 genes encoding interstitial fibrosis collagen and fibronectin, respectively (Figure 3a,b). Within the time frame studied, kidney fibrosis assessed by Sirius red staining, denoting collagen deposits, and interstitial fibronectin, representing extracellular matrix deposition, was only observed in some diabetic WT mice (Figure 3c,d). Similar to fibrosis gene expression changes, fibrosis was significantly milder in diabetic Fabry mice than in diabetic WT mice (Figure 3c,d). This contrasts with the level/expression of inflammatory markers (Fn14 and F4/80 macrophage infiltration) which were more severe in the diabetic Fabry mice. As was the case for chemokine expression, there was a good correlation between the kidney expression of Col1a2 and Fn1 and between these two genes and Sirius red quantification of fibrosis (Figure S3).
2.6. Interaction of DM and Fabry Disease on the Expression of Kidney Protective Genes in Mice
So far, a lack of association has been observed between the severity of kidney inflammation and the severity of kidney fibrosis in diabetic Fabry and diabetic WT mice. The more severe kidney infiltration by leukocytes in diabetic Fabry mice than in diabetic WT mice could not be explained by differences in chemokine gene expression. Recently, attention has been focused on the role of the downregulation of kidney protective genes in the pathogenesis of kidney inflammation and disease [22,23]. Stress-inducing mediators such as high glucose levels or inflammatory cytokines may amplify kidney injury through further recruitment of inflammatory cells and downregulation of kidney protective factors such as Klotho or PGC1α [23,24,25]. Klotho has anti-inflammatory antifibrotic and antiaging properties and Klotho administration was protective in several experimental nephropathies [26,27,28,29,30,31,32,33,34]. PGC1α is a transcription factor master regulator of mitochondrial biogenesis that is downregulated during acute kidney injury [35,36,37,38,39,40]. PGC1α downregulation promotes kidney inflammation and sensitizes to kidney injury [41]. Finally, transcription factor EB (TFEB) is a master regulator of lysosomal biogenesis, autophagy, lysosomal exocytosis, lipid catabolism, energy metabolism and immune response [39,42,43,44,45].
Kidney Klotho mRNA was decreased in non-diabetic Fabry mice (Figure 4a). However, there was heterogeneity regarding kidney Klotho expression in diabetic Fabry mice, which included the two mice with strikingly low kidney Klotho expression. Western blot confirmed mRNA findings (Figure S4).
PGC1α mRNA was upregulated in diabetic WT mice compared to controls in what may be interpreted as an adaptive response to the additional energy stress imposed on proximal tubular cells to reabsorb large amounts of filtered glucose (Figure 4b). Interestingly, diabetic Fabry mice failed to upregulate PGC1α: kidney PGC1α mRNA was lower in diabetic Fabry mice than in diabetic WT mice and, additionally, it was strikingly decreased in two diabetic Fabry mice (Figure 4b). TFEB mRNA followed a similar pattern to PGC1α: a trend towards higher expression in diabetic WT mice and significant downregulation in diabetic Fabry mice compared to diabetic WT mice (Figure 4c).
Overall, kidneys from diabetic Fabry mice appear defective in the compensatory upregulation of kidney protective genes such as PGC1α and TFEB that may be upregulated in diabetic WT kidneys.
2.7. Diabetic Fabry Mice with Kidney Insufficiency
Two (12.5%) diabetic Fabry mice developed kidney insufficiency. Unlike other mice that died suddenly during follow-up, likely as a result of severe serum glucose excursions, these two mice looked sick and were sacrificed prematurely as per animal ethics committee guidelines and found to have developed kidney insufficiency. Interestingly, these two mice were outliers for certain features in the combination of both diabetic groups. This may provide insight into pathogenic mechanisms (Figure S5). Specifically, diabetic Fabry mice that developed kidney insufficiency had very high kidney gene expression for the upstream Gb3 pathway enzyme Ugcg and for the TWEAK receptor Fn14 (Figure S5a,b), and very suppressed expression of the kidney protective factor PGC1α (Figure S5c).
3. Discussion
The main findings are that DM and Fabry disease may interact resulting in the modulation of kidney Gb3 synthesis pathways that may increase Gb3 availability through increased expression of UGCG, the gene encoding the upstream and rate-limiting enzyme glucosylceramide synthase, and limit the recruitment of compensatory kidney protective genes, potentially causing more severe inflammation and accelerated kidney failure.
In humans, diabetes is associated with changes in the kidney expression of genes encoding enzymes in the Gb3 pathway, including upregulation of UGCG, a therapeutic target in Fabry disease. Thus, glucosyl ceramide synthase inhibitors dramatically decrease Gb3 and lyso-Gb3 in Fabry patients [20]. Indeed, the increasing kidney UGCG expression in diabetic persons with more severe kidney disease was apparently “compensated” by increasing GLA expression as kidney disease got more severe, a “compensation” that would be absent in Fabry patients with defective GLA genes in which enzymatic activity would be absent or very reduced. Indeed, induction of DM in Fabry mice also resulted in increased kidney Ugcg gene expression in those mice that developed kidney insufficiency.
Insufficient activation of tissue protective genes such as Klotho, PGC1α and TFEB may favor end-organ damage [46,47]. Induction of DM in Fabry mice reproduced the kidney proinflammatory responses observed in diabetic WT mice. Additionally, Fabry mice failed to upregulate the nephroprotective genes PGC1α and TFEB in response to DM, potentially rendering the kidneys more sensitive to Fabry or DM-induced injury. While increased kidney inflammatory cell infiltration was observed in diabetic Fabry mice, this did not appear to be the result of increased kidney gene expression of the chemokines that were studied. However, PGC1α has anti-inflammatory actions in the kidney and was more severely downregulated when DM and Fabry disease coexisted, potentially accounting for the increased inflammation [41]. In this regard, the adverse impact of combining Fabry disease and DM appeared to be larger on inflammation than on fibrosis, potentially identifying a pathogenic molecular pathway to be explored in more detail: inflammation driven by loss of kidney protective factors.
While there were no significant differences in serum urea, creatinine, or proteinuria between diabetic Fabry and diabetic WT mice, 12% of diabetic Fabry mice developed kidney insufficiency which, in addition to increased plasma urea and creatinine, was characterized by a shared gene expression pattern of increased Ugcg and Fn14 and suboptimal recruitment of the kidney protective factor Pgc1α. Given the design of the study, it remains unclear whether this implies that the combined gene expression pattern drives accelerated kidney disease in a subset of mice and what the trigger might be. Several potential links between these genes and tissue injury may be hypothesized (Figure S6) [23,41]. Interestingly, Fn14 and PGC1α have been previously linked in the pathogenesis of kidney injury, as TWEAK activation of Fn14 downregulates PGC1α leading to spontaneous inflammation and an increased severity of kidney injury [23,41]. Although there is less information on the interaction between UGCG and Fn14, both have been linked in biomarker panels in diverse clinical conditions [48,49]. Further studies are required to characterize how common is this phenotype in a longer follow-up or when older mice are studied. Indeed, older mice are prone to severe kidney injury, and this has been related to decreased expression of kidney protective factors, such as PGC1α and to exacerbated proinflammatory responses [50], i.e., the pattern observed in diabetic Fabry mice. Only a minority of male Fabry individuals develop severe kidney disease with late-onset genetic variants, such as N215S, in whom kidney failure is 100-fold more common than in the general population but only occurs in 3% of men with this variant [5]. Thus, the interaction with DM observed in mice with Fabry disease, which usually do not develop spontaneous kidney disease but may develop kidney insufficiency when DM is present, may be relevant to understanding the natural history of kidney disease in late-onset Fabry disease.
The kidney phenotype of Gla deficient mice is histologically normal up to 48 weeks, despite lysosomal glycolipid deposits [21,50,51]. This is consistent with the long natural history of Fabry nephropathy in humans, of around 40 years from birth to kidney failure [52]. Although many biological processes are accelerated in short-lived animals, this is not the case for Fabry disease. However, mice are sensitive to Fabry nephropathy, as demonstrated when they overexpress Gb3 synthase on top of Gla deficiency, i.e., when glycolipid accumulation is larger [53].
Several limitations should be acknowledged. The short follow-up may not have fully grasped the full impact of the combination of Fabry disease and DM on the development of kidney insufficiency. However, ethics considerations precluded a longer experiment, given the mortality from kidney insufficiency of diabetic Fabry mice. Moreover, type 1 DM in young mice may not fully reflect type 2 DM in older individuals, i.e., the most common clinical scenario, which may result in a more severe phenotype. Backcrossing Fabry mice with db/db mice or other type 2 DM models and a long (>48 weeks) follow-up is beyond the scope of the current project but may provide further information in the future. Finally, gene expression may differ from protein expression. Despite the limitations, our work offers valuable multilevel (human systems biology, in vivo mouse studies) information on the interaction between DM and Fabry disease, uncovering evidence supporting that the combination may increase kidney disease severity in certain scenarios and having identified molecules that merit specific studies.
In conclusion, the combined presence of DM and Fabry disease may increase the severity of kidney disease through upregulation of the gene encoding the rate-limiting enzyme in Gb3 synthesis (UGCG encoding glucosyl ceramide synthase), downregulation of kidney protective factors and more severe inflammation. The emerging field of boosting kidney protection may be applicable to Fabry nephropathy developing in the presence of additional metabolic kidney stressors such as DM. In this regard, SGLT2 inhibitors decrease the urinary excretion of inflammatory cytokines and increase kidney protective molecules such as Klotho in patients with DM [25,54].
4. Materials and Methods
4.1. Animal Model
Procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the animal ethics committee of IIS-FJD (PROEX 036/16). We studied whether DM modifies the kidney phenotype of Gla knockout mice (Fabry mice, Jackson Lab, Bar Harbor, ME, USA) that contain a neo cassette replacing exon 3 and intron 3 of the Gla gene, abolishing gene expression. Insulin-deficient diabetes was induced in male Fabry or WT mice by administering intraperitoneally 125 mg/kg per day streptozotocin (STZ, Merck, Darmstadt, Alemania) for two consecutive days or vehicle [55]. Male mice were studied because Fabry is an X-linked disease. This means that because of random X chromosome inactivation, females are a mosaic of Fabry and healthy cells and the Fabry phenotype is not reproducible. Streptozotocin was dissolved in citrate buffer at pH 4.5. All streptozotocin-injected mice developed glycemia above 200 mg/dL. Severely hyperglycemic (blood glucose > 520 mg/dL) mice received NPH insulin (1.0–1.5 IU, Lilly USA, Indianapolis, IN, USA) daily to prevent weight loss and death [55]. After 30 days of DM, 16 h–fasted mice were anesthetized (100 mg/kg ketamine, (Ketalar, Pfizer, New York, NY, USA) and 15 mg/kg xylazine, (Xilagesic, Laboratorios Calier, Barcelona, Spain)). The intracardiac puncture was performed to collect blood in heparinized tubes which were subsequently centrifuged at 2500 rpm for 10 min to obtain plasma for biochemistry (urea, creatinine). Kidneys were perfused in situ with cold saline before removal. One kidney was snap-frozen in liquid nitrogen for RNA and protein studies and the other was fixed and paraffin-embedded for histological studies. Proteinuria was assessed by dipstick (Albustix, Siemens Healthcare, Erlangen, Germany). Two diabetic Fabry mice were sacrificed 17 and 27 days after administering the last streptozotocin dose because they looked sick and were found to have kidney insufficiency as defined by very high plasma creatinine and urea levels.
4.2. RT-qPCR
One microgram of RNA was isolated using Trizol (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed with High Capacity cDNA Archive Kit.( Applied Biosystems, Foster City, CA, USA) Real-time quantitative PCR (RT-qPCR) was performed on an ABI Prism 7500 PCR system with pre-developed primer and probe assays (Applied Biosystems, Foster City, CA, USA) using the DeltaDelta Ct method, as previously described [56]. Expression levels are given as ratios to GAPDH expression.
4.3. Immunohistochemistry
Paraffin-embedded 3 µm thick tissue sections were processed for conventional histology or immunohistochemistry using the Envision detection kit (Dako, Glostrup, Denmark) according to the manufacturer’s instructions. For immunohistochemistry, sections were counterstained with Carazzi’s hematoxylin (PanReacAppliChem GmbH, Darmstadt, Germany). Primary antibodies were rat polyclonal anti-F4/80 antigen (1:50; Serotec, Oxford, UK) and mouse anti-fibronectin monoclonal (1:200, Chemicon, Temecula, CA, USA) antibodies. Negative controls included incubation with a non-specific immunoglobulin of the same isotype as the primary antibody. The total number of F4/80-positive macrophages was quantitated in 15 randomly chosen fields (200×) per kidney using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA) and reported as number of positive cells/hpf. Staining was quantified in cortical tissue, as described [56].
For Sirius red staining, tissue sections were deparaffinized with xylene and graded concentrations of ethanol up to 70%, where slides stayed for 5 days at 4 °C. Direct Red 80 (Sigma-Aldrich, Merck, Darmstadt, Alemania, 365548) was dissolved in picrosirius acid (Sigma-Aldrich, Merck, Darmstadt, Germany, P6744) and incubated with tissue sections for 30 min at room temperature. Samples were dehydrated with a 100% ethanol wash and xylene. Slides were mounted in DPX medium (Merck, Darmstadt, Germany, 100579). The image was quantified with ImageProPlus software (Media Cybernetics, Bethesda, MD, USA), which allows selecting and calculating the area of pixels with similar colors. Results are shown as a percentage of positively stained area versus total quantified area from 10 fields per kidney (×200 magnification) [56]. Samples were examined in a blinded manner.
4.4. Data Mining
Information on the expression of genes encoding enzymes in the Gb3 pathway in human CKD transcriptomics datasets was obtained from Nephroseq v5 (http://v5.nephroseq.org/) accessed on 19 September 2022. A high-sensitivity approach was used, in which statistically significant differences (p < 0.05) in gene expression or correlation with analytical values were selected when representing a fold change >1.25 or an r value > 0.25 in DKD samples and their controls. Additionally, the Kidney Interactive Transcriptomics (KIT) webpage (http://humphreyslab.com/SingleCell/displaycharts.php; accessed on 19 May 2023) was used to identify individual cell types that may show differential expression of genes encoding Gb3 pathway enzymes in human DKD [57,58].
4.5. Statistics
Statistical analysis was performed using GraphPad Prism Software 8 (GraphPad Software, San Diego, CA, USA). Results are expressed as mean ± SEM. Significance (p < 0.05) was assessed by Student’s t-test for two groups of data and ANOVA for three or more groups with Bonferroni post-hoc correction. Pearson correlation was used to assess relationships between two continuous variables.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms242115853/s1. Reference [59] is cited in the supplementary materials.
Author Contributions
Conceptualization, A.O. and M.D.S.-N.; methodology, A.O. and M.D.S.-N.; validation, A.O. and M.D.S.-N.; formal analysis, A.O. and M.D.S.-N.; investigation, M.I.C.; resources, M.I.C.; data curation, S.C. and A.P.-C.; writing—original draft preparation, A.O. and M.D.S.-N. Writing—review & editing, A.B.S. and M.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Sanofi-Genzyme (GZ-2017-11724) and grants from the Instituto de Salud Carlos III (ISCIII)–Fondo de Investigacion Sanitaria (FIS)–Fondo Europeo de Desarrollo Regional (FEDER) grants PI21/00251 and PI22/00050. M.D.S-N. and A.B.S. were supported by MICINN Ramon y Cajal program RYC2018-024461-I and RYC2019-026916-I, respectively.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of IIS-FJD (PROEX 036/16).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used and/or analysed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
A.O. has received grants from Sanofi and consultancy or speaker fees or travel support from Adviccene, Astellas, Astrazeneca, Amicus, Amgen, Fresenius Medical Care, GSK, Bayer, Sanofi-Genzyme, Menarini, Kyowa Kirin, Alexion, Idorsia, Chiesi, Otsuka, Novo-Nordisk and Vifor Fresenius Medical Care Renal Pharma and is Director of the Catedra UAM-Mundipharma of diabetic kidney disease and the Catedra UAM-Astrazeneca of chronic kidney disease and electrolytes.
References
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Cammarata, G.; Fatuzzo, P.; Rodolico, M.S.; Colomba, P.; Sicurella, L.; Iemolo, F.; Zizzo, C.; Alessandro, R.; Bartolotta, C.; Duro, G.; et al. High variability of Fabry disease manifestations in an extended Italian family. Biomed Res. Int. 2015, 2015, 504784. [Google Scholar] [CrossRef] [PubMed]
- Rigoldi, M.; Concolino, D.; Morrone, A.; Pieruzzi, F.; Ravaglia, R.; Furlan, F.; Santus, F.; Strisciuglio, P.; Torti, G.; Parini, R. Intrafamilial phenotypic variability in four families with Anderson-Fabry disease. Clin. Genet. 2014, 86, 258–263. [Google Scholar] [CrossRef]
- Gilchrist, M.; Casanova, F.; Tyrrell, J.S.; Cannon, S.; Wood, A.R.; Fife, N.; Young, K.; Oram, R.A.; Weedon, M.N. Prevalence of Fabry disease-causing variants in the UK Biobank. J. Med. Genet. 2022, 60, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Brand, E.; Burlina, A.; Cecchi, F.; Garman, S.C.; Kempf, J.; Laney, D.A.; Linhart, A.; Maródi, L.; Nicholls, K.; et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol. Genet. Genom. Med. 2018, 6, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Boenink, R.; Astley, M.E.; Huijben, J.A.; Stel, V.S.; Kerschbaum, J.; Ots-Rosenberg, M.; Åsberg, A.A.; Lopot, F.; Golan, E.; Castro de la Nuez, P.; et al. The ERA Registry Annual Report 2019: Summary and age comparisons. Clin. Kidney J. 2021, 15, 452–472. [Google Scholar] [CrossRef]
- Germain, D.P.; Altarescu, G.; Barriales-Villa, R.; Mignani, R.; Pawlaczyk, K.; Pieruzzi, F.; Terryn, W.; Vujkovac, B.; Ortiz, A. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol. Genet. Metab. 2022, 137, 49–61. [Google Scholar] [CrossRef]
- Najafian, B.; Svarstad, E.; Bostad, L.; Gubler, M.C.; Tøndel, C.; Whitley, C.; Mauer, M. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011, 79, 663–670. [Google Scholar] [CrossRef]
- Tøndel, C.; Bostad, L.; Larsen, K.K.; Hirth, A.; Vikse, B.E.; Houge, G.; Svarstad, E. Agalsidase benefits renal histology in young patients with Fabry disease. J. Am. Soc. Nephrol. 2013, 24, 137–148. [Google Scholar] [CrossRef]
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Sanz, A.B.; Carrasco, S.; Saleem, M.A.; Mathieson, P.W.; Valdivielso, J.M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Globotriaosylsphingosine actions on human glomerular podocytes: Implications for Fabry nephropathy. Nephrol. Dial. Transpl. 2011, 26, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Park, A.S.D.; Mohtat, D.; Thomas, D.B.; Pullman, J.M.; Susztak, K. Transcriptome analysis of human diabetic kidney disease. Diabetes 2011, 60, 2354–2369. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Nair, V.; Smith, S.; Zhu, L.; Shedden, K.; Song, P.X.K.; Mariani, L.H.; Eichinger, F.H.; Berthier, C.C.; Randolph, A.; et al. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci. Transl. Med. 2015, 7, 316ra193. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.; Boucherot, A.; Yasuda, Y.; Henger, A.; Brunner, B.; Eichinger, F.; Nitsche, A.; Kiss, E.; Bleich, M.; Gröne, H.-J.; et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes 2006, 55, 2993–3003. [Google Scholar] [CrossRef]
- Hodgin, J.B.; Nair, V.; Zhang, H.; Randolph, A.; Harris, R.C.; Nelson, R.G.; Weil, E.J.; Cavalcoli, J.D.; Patel, J.M.; Brosius, F.C.; et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 2013, 62, 299–308. [Google Scholar] [CrossRef]
- Shayman, J.A. Targeting Glucosylceramide Synthesis in the Treatment of Rare and Common Renal Disease. Semin. Nephrol. 2018, 38, 183–192. [Google Scholar] [CrossRef]
- Van Eijk, M.; Ferra, M.J.; Boot, R.G.; Aerts, J.M.F.G. Lyso-glycosphingolipids: Presence and consequences. Essays Biochem. 2020, 64, 565–578. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Marques, A.R.A.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef]
- Deegan, P.B.; Goker-Alpan, O.; Geberhiwot, T.; Hopkin, R.J.; Lukina, E.; Tylki-Szymanska, A.; Zaher, A.; Sensinger, C.; Gaemers, S.J.M.; Modur, V.; et al. Venglustat, an orally administered glucosylceramide synthase inhibitor: Assessment over 3 years in adult males with classic Fabry disease in an open-label phase 2 study and its extension study. Mol. Genet. Metab. 2022, 138, 106963. [Google Scholar] [CrossRef]
- Valbuena, C.; Oliveira, J.P.; Carneiro, F.; Relvas, S.; Ganhão, M.; Sá-Miranda, M.C.; Rodrigues, L.G. Kidney histologic alterations in α-Galactosidase-deficient mice. Virchows Arch. 2011, 458, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Sanchez-Niño, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Cuarental, L.; Ribagorda, M.; Ceballos, M.I.; Pintor-Chocano, A.; Carriazo, S.M.; Dopazo, A.; Vazquez, E.; Suarez-Alvarez, B.; Cannata-Ortiz, P.; Sanz, A.B.; et al. The transcription factor Fosl1 preserves Klotho expression and protects from acute kidney injury. Kidney Int. 2022, 103, 686–701. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sánchez-Ramos, C.; Monsalve, M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int. 2016, 89, 399–410. [Google Scholar] [CrossRef]
- Mora-Fernández, C.; Sánchez-Niño, M.D.; Donate-Correa, J.; Martín-Núñez, E.; Pérez-Delgado, N.; Valiño-Rivas, L.; Fernández-Fernández, B.; Ortiz, A.; Navarro-González, J.F. Sodium-glucose co-transporter-2 inhibitors increase Klotho in patients with diabetic kidney disease: A clinical and experimental study. Biomed. Pharmacother. 2022, 154, 113677. [Google Scholar] [CrossRef]
- Sugiura, H.; Yoshida, T.; Tsuchiya, K.; Mitobe, M.; Nishimura, S.; Shirota, S.; Akiba, T.; Nihei, H. Klotho reduces apoptosis in experimental ischaemic acute renal failure. Nephrol. Dial. Transpl. 2005, 20, 2636–2645. [Google Scholar] [CrossRef]
- Sugiura, H.; Yoshida, T.; Mitobe, M.; Yoshida, S.; Shiohira, S.; Nitta, K.; Tsuchiya, K. Klotho reduces apoptosis in experimental ischaemic acute kidney injury via HSP-70. Nephrol. Dial. Transpl. 2010, 25, 60–68. [Google Scholar] [CrossRef]
- Shi, M.; Flores, B.; Gillings, N.; Bian, A.; Cho, H.J.; Yan, S.; Liu, Y.; Levine, B.; Moe, O.W.; Hu, M.C. αKlotho Mitigates Progression of AKI to CKD through Activation of Autophagy. J. Am. Soc. Nephrol. 2016, 27, 2331–2345. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Gillings, N.; Flores, B.; Takahashi, M.; Kuro-o, M.; Moe, O.W. Recombinant α-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 2017, 91, 1104–1114. [Google Scholar] [CrossRef]
- Jou-Valenci, D.; Molema, G.; Popa, E.; Aslan, A.; Van Dijk, F.; Mencke, R.; Hillebrands, J.L.; Heeringa, P.; Hoenderop, J.G.; Zijlstra, J.G.; et al. Renal Klotho is Reduced in Septic Patients and Pretreatment With Recombinant Klotho Attenuates Organ Injury in Lipopolysaccharide-Challenged Mice. Crit. Care Med. 2018, 46, E1196–E1203. [Google Scholar] [CrossRef]
- Shin, Y.J.; Luo, K.; Quan, Y.; Ko, E.J.; Chung, B.H.; Lim, S.W.; Yang, C.W. Therapeutic Challenge of Minicircle Vector Encoding Klotho in Animal Model. Am. J. Nephrol. 2019, 49, 413–423. [Google Scholar] [CrossRef]
- Li, P.; Shi, M.; Maique, J.; Shaffer, J.; Yan, S.; Moe, O.W.; Hu, M.C. Beclin 1/Bcl-2 complex-dependent autophagy activity modulates renal susceptibility to ischemia-reperfusion injury and mediates renoprotection by Klotho. Am. J. Physiol. Renal Physiol. 2020, 318, F772–F792. [Google Scholar] [CrossRef]
- Zhu, X.; Li, S.; Lin, Q.; Shao, X.; Wu, J.; Zhang, W.; Cai, H.; Zhou, W.; Jiang, N.; Zhang, Z.; et al. αKlotho protein has therapeutic activity in contrast-induced acute kidney injury by limiting NLRP3 inflammasome-mediated pyroptosis and promoting autophagy. Pharmacol. Res. 2021, 167, 105531. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Cao, J.; Wei, X.; Ge, Y.; Su, Z.; Yu, D. Klotho alleviates contrast-induced acute kidney injury by suppressing oxidative stress, inflammation, and NF-KappaB/NLRP3-mediated pyroptosis. Int. Immunopharmacol. 2023, 118, 110105. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Weinberg, J.M. The tubule pathology of septic acute kidney injury: A neglected area of research comes of age. Kidney Int. 2012, 81, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Stallons, L.J.; Whitaker, R.M.; Schnellmann, R.G. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol. Lett. 2014, 224, 326–332. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tran, M.T.; Parikh, S.M. PGC1α in the kidney. Am. J. Physiol. Renal Physiol. 2018, 314, F1–F8. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tran, M.T.; Ralto, K.M.; Zsengeller, Z.K.; Raman, V.; Bhasin, S.S.; Sun, N.; Chen, X.; Brown, D.; Rovira, I.I.; et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight 2019, 4, e126749. [Google Scholar] [CrossRef]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martín-Sánchez, D.; Martinez-Moreno, J.M.; Carrasco, S.; Ruiz-Andrés, O.; Monsalve, M.; Sanchez-Ramos, C.; Gómez, M.J.; Ruiz-Ortega, M.; Sánchez-Niño, M.D.; et al. PGC-1α deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J. Pathol. 2019, 249, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Takla, M.; Keshri, S.; Rubinsztein, D.C. The post-translational regulation of transcription factor EB (TFEB) in health and disease. EMBO Rep. 2023, e57574. [Google Scholar] [CrossRef]
- Wen, W.; Zheng, H.; Li, W.; Huang, G.; Chen, P.; Zhu, X.; Cao, Y.; Li, J.; Huang, X.; Huang, Y. Transcription factor EB: A potential integrated network regulator in metabolic-associated cardiac injury. Metabolism 2023, 147, 155662. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Zhang, L.; Qin, Y.; Yu, D. Dissecting the multifaced function of transcription factor EB (TFEB) in human diseases: From molecular mechanism to pharmacological modulation. Biochem. Pharmacol. 2023, 215, 115698. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Perera, R.M. Emerging roles of the MiT/TFE factors in cancer. Trends Cancer 2023, 9, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-barriuso, M.; Martin-sanchez, D.; Martinez-moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-niño, M.D.; Ruiz-ortega, M.; Ortiz, A.; Sanz, A.B. The role of PGC-1α and mitochondrial biogenesis in kidney diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Moreno, J.A.; Ruiz-Ortega, M.; Ramos, A.M.; Sanz, A.B.; Ortiz, A. Downregulation of kidney protective factors by inflammation: Role of transcription factors and epigenetic mechanisms. Am. J. Physiol. Renal Physiol. 2016, 311, F1329–F1340. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Guerrero-Mauvecin, J.; Miguel, V.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Tubular Mitochondrial Dysfunction, Oxidative Stress, and Progression of Chronic Kidney Disease. Antioxidants 2022, 11, 1356. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Zhu, X.L.; Wang, Z.Z.; Bai, H.; Zhang, J.J.; Hao, C.Y.; Duan, H. Bin A Novel Immune-Related Prognostic Biomarker and Target Associated With Malignant Progression of Glioma. Front. Oncol. 2021, 11, 643159. [Google Scholar] [CrossRef]
- Marquez-Exposito, L.; Tejedor-Santamaria, L.; Santos-Sanchez, L.; Valentijn, F.A.; Cantero-Navarro, E.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marchant, V.; Sanz, A.B.; et al. Acute Kidney Injury is Aggravated in Aged Mice by the Exacerbation of Proinflammatory Processes. Front. Pharmacol. 2021, 12, 662020. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; Van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Cianciaruso, B.; Cizmarik, M.; Germain, D.P.; Mignani, R.; Oliveira, J.P.; Villalobos, J.; Vujkovac, B.; Waldek, S.; Wanner, C.; et al. End-stage renal disease in patients with Fabry disease: Natural history data from the Fabry Registry. Nephrol. Dial. Transpl. 2010, 25, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Taguchi, A.; Nishikawa, Y.; Guili, C.; Mikame, M.; Nameta, M.; Yamaguchi, Y.; Ueno, M.; Imai, N.; Ito, Y.; et al. Medullary thick ascending limb impairment in the GlatmTg(CAG-A4GALT) Fabry model mice. FASEB J. 2018, 32, 4544–4559. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Sarafidis, P.; Kanbay, M.; Navarro-González, J.F.; Soler, M.J.; Górriz, J.L.; Ortiz, A. SGLT2 inhibitors for non-diabetic kidney disease: Drugs to treat CKD that also improve glycaemia. Clin. Kidney J. 2020, 13, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Sanz, A.B.; Lorz, C.; Gnirke, A.; Rastaldi, M.P.; Nair, V.; Egido, J.; Ruiz-Ortega, M.; Kretzler, M.; Ortiz, A. BASP1 promotes apoptosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 610–621. [Google Scholar] [CrossRef]
- Valiño-Rivas, L.; Cuarental, L.; Ceballos, M.I.; Pintor-Chocano, A.; Perez-Gomez, M.V.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Growth differentiation factor-15 preserves Klotho expression in acute kidney injury and kidney fibrosis. Kidney Int. 2022, 101, 1200–1215. [Google Scholar] [CrossRef]
- Wu, H.; Malone, A.F.; Donnelly, E.L.; Kirita, Y.; Uchimura, K.; Ramakrishnan, S.M.; Gaut, J.P.; Humphreys, B.D. Single-Cell Transcriptomics of a Human Kidney Allograft Biopsy Specimen Defines a Diverse Inflammatory Response. J. Am. Soc. Nephrol. 2018, 29, 2069–2080. [Google Scholar] [CrossRef]
- Wilson, P.C.; Wu, H.; Kirita, Y.; Uchimura, K.; Ledru, N.; Rennke, H.G.; Welling, P.A.; Waikar, S.S.; Humphreys, B.D. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 19619–19625. [Google Scholar] [CrossRef]
- Sanchez-Nin, M.D.; Sanz, A.B.; Ihalmo, P.; Lassila, M.; Holthofer, H.; Mezzano, S.; Aros, C.; Groop, P.H.; Saleem, M.A.; Mathieson, P.W.; et al. The MIF receptor CD74 in diabetic podocyte injury. J. Am. Soc. Nephrol. 2009, 20, 353–362. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).