Abstract
Glaucoma is one of the most common causes of treatable visual impairment in the developed world, affecting approximately 64 million people worldwide, some of whom will be bilaterally blind from irreversible optic nerve damage. The optic nerve head is a key site of damage in glaucoma where there is fibrosis of the connective tissue in the lamina cribrosa (LC) extracellular matrix. As a ubiquitous second messenger, calcium (Ca2+) can interact with various cellular proteins to regulate multiple physiological processes and contribute to a wide range of diseases, including cancer, fibrosis, and glaucoma. Our research has shown evidence of oxidative stress, mitochondrial dysfunction, an elevated expression of Ca2+ entry channels, Ca2+-dependent pumps and exchangers, and an abnormal rise in cytosolic Ca2+ in human glaucomatous LC fibroblast cells. We have evidence that this increase is dependent on Ca2+ entry channels located in the plasma membrane, and its release is from internal stores in the endoplasmic reticulum (ER), as well as from the mitochondria. Here, we summarize some of the molecular Ca2+-dependent mechanisms related to this abnormal Ca2+-signalling in human glaucoma LC cells, with a view toward identifying potential therapeutic targets for ongoing optic neuropathy.
1. Introduction
1.1. Glaucoma, Optic Nerve Fibrosis, Lamina Cribrosa Fibroblasts, and Calcium
Glaucoma is the one of most common causes of treatable visual impairment in the developed world [1], affecting approximately 64.3 million people worldwide, and these numbers were estimated to increase to 76.0 million in 2020 and 111.8 million by 2040 [2]. Optic disc cupping is a characteristic clinical feature of the glaucomatous optic nerve head (ONH), and it occurs due to a loss of neural tissue (axons of the retinal ganglion cells [RGC]) and remodeling of the connective tissue (CT) in the ONH, leading to progressive and irreversible visual field loss. It manifests clinically as an increase in the cup-to-disc ratio, a deepening of the cup, and a greater visibility of the lamina cribrosa (LC). Although there may be other factors in the retina and brain that contribute to RGC axonal damage and loss, the preponderance of evidence suggests that the laminar region of the ONH is the principal site of damage.
The LC’s topography in glaucoma includes shearing and a collapse of its beams, resulting in a thinning and backward bowing of the LC. Histologically, there are significant changes in the extra-cellular matrix (ECM) in human and monkey glaucoma optic nerve head specimens [3], with increased deposition of collagen, elastin, and proteoglycan and increased expression of the major pro-fibrotic growth factor transforming growth factor beta (TGFβ) [4].
The lamina cribrosa cells of the ONH were first characterized by the Hernandez group in 1988 [5]. Furthermore, Lambert et al. continued to study LC characterization by testing whether human LC cells and tissue express neurotrophin and tyrosine kinase receptor [6]. In a further study, the lamina cribrosa cells were identified and localized in the beams of the LC [7]. Figure 1 illustrates an example of phase contrast microscopy images of cultured LC cells from non-glaucomatous and glaucomatous human donors.
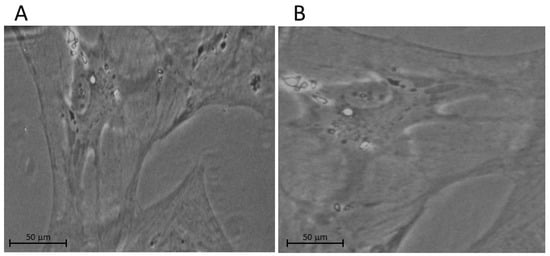
Figure 1.
Examples of phase contrast microscopy images of cultured LC cells from (A) non-glaucomatous and (B) glaucomatous human donors. We note that the glaucomatous LC cell is larger than the non-glaucomatous LC cell.
These LC cells stain positively for α-smooth muscle actin (α-SMA), fibronectin, collagen1A1, and vitronectin, and they appear to occur in humans (and likely in other primates) but have not been identified in rodents such as mice or rats. These latter animals undergo a gliosis at the optic nerve head (and not the typical 3-D fibrotic ECM remodelling, as seen in human glaucoma) [8].
The cells that play a role in this CT remodeling include ONH astrocytes and LC cells. Hernandez has shown a significant number of alterations in LC astrocytes, including an increase in the synthesis of ECM macromolecules, cell adhesion molecules and growth factors, and cytokines [9].
Fibrosis is attributed to excessive ECM accumulation which ultimately damages the connective tissues [10]. External stimuli such as oxidative stress, mechanical stretch, growth factors, and increased substrate stiffness cause fibroblasts to change their phenotype and differentiate into myofibroblasts to drive fibrosis [11,12]. The main features of myofibroblast differentiation are the disproportionate increases in the expression of structural ECM proteins, matricellular proteins, smooth muscle α-actin (αSMA), and transforming growth factor beta (TGF-β) [13]. Moreover, TGF-β plays a critical role in fibrosis [14], is an effective inducer of myofibroblasts, and stimulates the expression of important genes in fibrosis through several downstream pathways, especially Smad signalling [15,16].
It has been found that the CT of the LC and the trabecular meshwork (TM) show substantial ECM fibrosis in glaucoma [17,18]. Our previous work has focused on decoding the fibrotic signature of LC cells in response to glaucomatous change. We have shown that human glaucomatous LC cells [19] have many characteristics of myofibroblasts, including the expression of α-SMA and fibrotic genes (e.g., collagen 1A1, periostin, and fibronectin) in response to TGFβ stimulation [20], cyclic stretch [21], and hypoxia [22]. Furthermore, we found that LC cells grown on stiff substrates show the enhanced expression of αSMA, F-Actin, and vinculin [23]. In Table 1, we summarize our laboratory results on ECM gene expression, abnormal Ca2+ signalling, and mitochondrial dysfunction in glaucoma LC cells.

Table 1.
Summary of dysfunctional intracellular Ca2+ concentration, Ca2+ entry and exit ion channels, Ca2+-related ECM genes and proteins, disturbed Ca2+-signalling pathways, and dysfunctional cell proliferation and autophagy in glaucomatous LC myofibroblasts obtained by the authors.
In addition, we previously used oxidative stress to model glaucoma in LC cells, and we found that both basal- and oxidative-stress-induced levels of cytosolic calcium (Ca2+) were abnormally elevated in glaucoma LC cells [24]. It is well known that Ca2+ is a key driver/player of fibrosis [33]. In addition, we demonstrated an increase in L-type Ca2+ channels in glaucoma LC cells [32]. In the same study, we showed that L-type Ca2+ channel blockade with verapamil reduces the mechanical-strain-induced ECM gene response in human LC cells. Furthermore, we showed that Ca2+-dependent potassium channel Maxi-K+ expression and activity are significantly elevated in glaucoma LC cells [27]. More recently, by reducing the oxidative-stress-induced production of ECM genes and LC cell proliferation trough a signalling pathway mechanism involving nuclear factor of activated T-cells (NFATc3), we found that the voltage non-dependent, stretch-activated cation channels, transient receptor potential canonical TRPC1 and TRPC6, are highly expressed in glaucoma LC cells and are also involved in the aberrantly elevated intracellular [Ca2+]i levels found in glaucoma LC cells [26].
In order to clarify the molecular mechanisms underpinning fibrosis in glaucoma, we investigated intracellular Ca2+-related signalling pathways by exploring the protein kinases expression and activity of PKCα and RAS-RAF-MAPK in human normal and glaucoma LC myofibroblasts using hypo-osmotic-induced cell membrane stretch to model glaucoma. We found significant increases in both the expression (in resting conditions) and the activity (phosphorylation in a hypo-osmotic-induced cellular swelling condition) of the protein kinases PKCα, p38, and p42/44 in glaucoma LC cells [29]. Taken together, these data may suggest a possible coordinating effect of these protein kinases and Ca2+ in the development of fibrosis in glaucoma, and they may also provide the molecular bases for the therapeutic outcome of targeting the PKCα, p38MAPK, and p42/44 MAPK kinases. Hence, a strategy to inhibit their signalling pathways may be central for an efficient treatment of fibrosis in glaucoma. On the other hand, we also explored the bioenergetics of glaucoma LC cells, and we observed that glaucoma LC cells exhibit dysfunctional mitochondria [30], mitochondrial fission [30], and an increase in glycolysis, with a decrease in OXPHOS [31].
In a recent study, we found that glaucomatous LC cells proliferate at a higher rate, and we showed that yes-associated protein (YAP) expression levels were relatively enhanced in glaucoma LC cells (Table 1). Furthermore, the enhanced cell proliferation in glaucoma LC cells was reduced following treatment with the known YAP inhibitor verteporfin [28].
1.2. General Concept of Ca2+-Signalling Homeostasis (Figure 2)
Ca2+ enters into a cell and interacts with different Ca2+-binding proteins that function either as Ca2+ effectors or buffers. Ca2+ ions are key signalling ions for regulating numerous physiological processes [34,35]. It is, therefore, not surprising that the disruption of Ca2+ homeostasis and its downstream signalling is responsible for many pathological states including apoptosis, excessive proliferation, angiogenesis, fibrosis, and cancer. The increase in intracellular Ca2+ concentration ([Ca2+]i) results from either the influx of extracellular Ca2+ through the plasma membrane Ca2+ entry channels or its release from internal stores such as the endo/sarcoplasmic reticulum, primarily through 1,4,5-trphosphate receptor (IP3R) and ryanodine receptors (RyR). In most cells, external stimuli bind to ligand-engaged G protein-coupled receptors (GPCRs), leading to the subsequent synthesis of IP3 and the activation of the IP3 receptor at the ER membrane, resulting in the release of Ca2+ from the ER [36,37]. In resting cells, the cytosolic Ca2+ concentration is maintained at very low levels (~100 nM) by two ATP-dependent systems: plasma membrane Ca2+ ATPases (PMCAs), which hydrolyze ATP to provide the needed energy to extrude Ca2+ to the extracellular space, and sarco-endoplasmic reticulum Ca2+ ATPases (SERCAs) pumps, which provide sufficient energy to re-uptake the Ca2+ into the ER lumen and the mitochondria [38,39]. The Na+/Ca2+ exchanger (NCX) also uses the energy contained within the Na+ concentration gradient (Na+/K+-ATPase pump) to extrude Ca2+ out of the cell [40]. Thus, cells provide most of their energy to maintain [Ca2+]i at the lower levels so that small increases in plasma membrane Ca2+ influx or efflux from the internal store can trigger rapid, transient, and marked increases in [Ca2+]i. It is these increases in [Ca2+]i that are a key signal in gene transcription regulation.

Figure 2.
General concept of Ca2+-signalling homeostasis. Stimuli induce both the entry of external Ca2+ and the release Ca2+ from the internal stores of the ER/SR via IP3R and RYR. In activated cells, Ca2+ enters cells through different types of Ca2+ channels, including voltage-operated channels (VOC), second messenger-operated channels (SMOC), store-operated channels (SOC), and receptor-operated channels (ROC). We note that VOCs are activated by membrane depolarization and SMOCs are activated by small messenger molecules, such as InsP3. In resting cells, Ca2+ is removed from the cell by exchangers and pumps. The NCX and PMCA extrude Ca2+ from the cytosol to the extracellular milieu, whereas the ER/SR Ca2+ -ATPase (SERCA) pumps pump Ca2+ back into the ER. Mitochondria also have an active function during the recovery process in that they sequester Ca2+ rapidly through a uniporter, which is then released more slowly back into the cytosol.
1.3. Ca2+ Signalling (Figure 2)
Intracellular Ca2+ plays a key role in multiple signal transduction pathways in a wide variety of cell types by modulating critical cellular functions such as cell death and gene transcription. For example, the pro-hypertrophic ECM gene expression in cardiac myocytes is seen in pathological cardiac growth and is characterized by the elevation of cytosolic Ca2+, acting via numerous signalling cascades, including the protein kinase c α (PKCα) pathways [41]. Cells throughout the body have a vast array of mechanisms that tightly regulate intracellular Ca2+ levels to maintain low cytosolic levels relative to higher levels outside of a cell. These mechanisms include Ca2+ entry and exit pathways and Ca2+ stores, buffers, and transporters.
The chronic elevation of intracellular Ca2+ levels activates numerous downstream Ca2+-dependent signalling pathways that can mediate maladaptive ECM remodeling, resulting in connective tissue fibrosis [42]. This includes the increased expression of PKC alpha, IP3R, calcineurin, and calmodulin (CaM), which results in the activation of nuclear transcription factors (NFAT) and many other Ca2+-binding proteins, leading, for example, to pathological cardiac hypertrophy, pulmonary fibrosis, and other forms of fibrosis [43].
The most studied Ca2+-dependent signalling protein is CaM. A rise in cytosolic Ca2+ levels activates CaM, which can activate several Ca2+-dependent kinases, including Ca2+-CaM dependent kinase (CamK) [44]. The transcription factor NF-κB is normally kept in the cytosol by IκB; however, IkB phosphorylation by CamK leads to its degradation, allowing NF-κB to translocate to the nucleus and promote Ca2+-dependent gene transcription [45]. A large number of studies have shown that T-type and L-type Ca2+ channel blockers are useful in animal models of fibrosis in several tissues, including the kidneys [46], liver [47,48,49], heart [50,51], and skin [52,53]. For example, the calcineurin inhibitor cyclosporin A, an immuno-suppressive and anti-fibrotic agent, inhibits TGF-β-induced ECM deposition in cardiac-activated fibroblasts through the calcineurin–NFAT pathway, thus preventing cardiac cell hypertrophy [51,54]. Other studies have shown alterations of calcium homeostasis in models of glaucoma [55,56,57].
We previously reported elevated cytosolic Ca2+ in human glaucoma LC fibroblasts [24]. Moreover, we found that cyclosporin A blocked NFATc3 nuclear translocation, which reduced the ECM fibrosis gene expression in glaucoma LC fibroblasts [25]. Similar results (of elevated intracellular Ca2+) have been shown in TM cells from human glaucoma donors [58]. More recently, we showed that a rise in [Ca2+]i also induced a sequential phosphorylation of PKCα and the downstream series of phosphorylation of p38MAPK and p42/44 MAPK, resulting in Ca2+-dependent genes, such as profibrotic ECM genes, and, ultimately, the proliferation of glaucoma LC cells [29].
2. Ca2+ Entry (Figure 2)
Calcium homeostasis is regulated by a number of Ca2+ channels. Ca2+ entry channels are integral membrane proteins that allow the passage of Ca2+ ions across the cell membrane either under their electrochemical gradient (‘passive’ passage) or in response to specific activating external stimuli (‘active’ passage). Cells use this external source of signal Ca2+ by stimulating various Ca2+ entry channels. Among these Ca2+ channels, voltage-operated channels (VOCs), the best known Ca2+ entry channels, are found in excitable cells and generate the rapid Ca2+ fluxes that control rapid cellular processes such as muscle contraction or exocytosis at synaptic transmission. T-type Ca2+ currents serve as pacemakers of rhythmic activity in a diverse array of cell types [59,60]. These channels are activated by relatively small membrane depolarization [61].
L-type Ca2+ channels play a key role in many cell types where they mediate large changes in [Ca2+]i in response to changes in membrane potential [34,61,62]. The membrane depolarization and accumulation of Ca2+ in these channels in turn causes a delayed inactivation of the channels, providing a negative feedback control loop for this Ca2+ influx pathway [63].
Receptor-operated channels (ROCs) are the other class of Ca2+ permeable channels that open in response to external signals, such as the NMDA (N-methyl-D-aspartate) receptors that respond to glutamate. In addition to these channel-opening mechanisms, there are many other channel types that are sensitive to a diverse array of stimuli, such as store-operated channels (SOCs), thermo-sensors and stretch-activated channels (SACs), and Ca2+-release-activated Ca2+ channels (CRACs), which mediate the store-operated Ca2+ channel entry (SOCE). The SOCE refills the stores after depletion, and they involve Ca2+ influx via ORAI1 Ca2+ channels after activation by the ER Ca2+ store sensor stromal interaction molecule 1(STIM1). Many of these channels belong to the large transient receptor protein (TRP) ion-channel family [64,65,66,67], and they are encoded by up to 30 different genes. TRP channels are a family of voltage independent Ca2+ channels which can respond to a diverse selection of stimuli, including internal Ca2+ store depletion, cyclic stretch, and other types of stresses [68]. This family is formed by three major groups: the canonical TRPC family, the vanilloid TRPV family, and the melastatin TRPM family. Members of the TRP family are particularly important in controlling slow cellular processes such as cell differentiation and proliferation.
We found elevated voltage-gated channels (L-type Ca2+ channels) [32] and elevated voltage-independent ion channels (TRPC1/6) in glaucomatous LC cells compared to normal non-glaucomatous LC cells [26].
3. Calcium Release from Internal Stores (Figure 3)
Calcium and the Endoplasmic Reticulum
The ER is an essential central cellular organelle in each eukaryotic cell, and it plays a critical role in Ca2+ handling, protein synthesis, and protein processing [69]. The ER ensures proper protein synthesis and folding by regulating many post-translational modifications [70,71,72]. Several factors, including ATP and Ca2+ signals, regulate protein folding through disulfide-bond formation [73]. The ER process directs the transit of folded proteins in membrane vesicles to different intracellular organelles and to the extracellular space of the cell [74,75]. Ca2+ concentration in the ER is a key regulator of protein folding. With prolonged stress conditions, damaged cells are eliminated by the activation of programmed cell death signalling. Therefore, disruptions in Ca2+ concentrations lead to reductions in the protein folding capacity of the ER, leading to the excessive accumulation and aggregation of unfolded proteins (UPR) and an increase in protein secretion and/or protein misfolding [69,76,77], resulting in ER stress. Prolonged UPR activation can promote a pro-survival response to a pro-apoptotic signalling, especially in a pathological condition [78]. We recently discussed this in the context of LC fibrosis in glaucoma [79].

Figure 3.
Endoplasmic reticulum (ER) stress and unfolded protein response (UPR). The ER’s functions include proper protein synthesis and folding to maintain cellular homeostasis. The disturbance of cellular ATP production or Ca2+ concentration affects ER functioning, leading to the excessive accumulation and aggregation of unfolded proteins and generating ER stress, which further activates the UPR. The UPR plays key roles in adaptive responses, feedback control, and cell fates. In an adaptive response, the UPR reduces ER stress and restores ER homeostasis. UPR signalling is inhibited through a negative feedback mechanism.
4. Calcium and Mitochondria
4.1. Mitochondrial Function Regulation
Mitochondria are involved in numerous cellular biological functions through their roles in adenosine-triphosphate (ATP) production through the tricarboxylic acid (TCA) cycle and electron transport chain and through the electrochemical gradients across the mitochondrial membrane to drive the oxidative phosphorylation (OXPHOS) process [80,81,82]. The metabolic pathways of mitochondria, including glycolysis and respiration, are major sources of ATP production in living cells. As a result of anaerobic respiration, glycolysis produces the lactate that is necessary for cell growth and proliferation [83]. It has been demonstrated that the inhibition of mitochondrial respiration induces a switch to glycolysis, stimulates cell differentiation, and reduces cell proliferation [84]. Dysregulation of these regulatory mechanisms has been identified in different fibrotic diseases [85].
Beyond energy production, the mitochondrion has many other functions, including the generation of redox molecules, reactive oxygen species (ROS) production, intracellular Ca2+ regulation, cell proliferation, and apoptosis [86,87,88]. For normal metabolism, cells must produce ROS. However, excessive ROS production leads to oxidative damage of the structure and function of mitochondria and also to the excessive release of the Ca2+ from mitochondria via the mitochondrial permeability transition pore (mPTP) (Figure 2) [89]. It has been found that the dysregulation of Ca2+-signalling homeostasis also increases ROS generation and over-activates mitophagy, resulting in mitochondrial damage and impaired respiratory function, and it also promotes apoptosis [90,91].
Apoptosis plays a vital role in the elimination of cells, which is important for the processes of embryogenesis, development, and tissue homeostasis [92]. Ca2+ is a major player in the regulation of cell death [93], and severe Ca2+ dysregulation can induce ER stress-mediated apoptosis in response to various pathological conditions [94]. The B-cell lymphoma 2 (Bcl-2) protein family is a key part of the protein complexes that curb the response to ER stress, with apoptosis and autophagy as the possible end-results [95]. Bcl-2 has been defined as a rheostat that belongs to a large family of proteins that includes pro-apoptotic and anti-apoptotic molecules [95]. The pro-apoptotic members of the Bcl-2 family trigger mitochondrial outer membrane permeabilization (MOMP), leading to the release of cytochrome c and to the assembly of the apoptosome [96].
Mitochondrial Dysfunction Regulation in Glaucoma
Numerous publications have shown mitochondrial dysfunction and altered cell bioenergetics in diverse forms of organ fibrosis, including in cardiac-, pulmonary-, renal-, and cancer-associated fibroblasts [85].
It is well known that mitochondrial dysfunction plays a key role in the development of glaucoma, and it has also been investigated as a potential drug target for glaucoma treatment [97,98,99,100,101]. For example, the rapamycin (mTOR) signalling pathway and nicotinamide treatment were used in clinical therapies to observe glaucoma-related mitochondria dysfunction [102,103]. While mitochondrial function is regulated by several signalling pathways, Ca2+ signalling is considered to play a key role in the regulation of mitochondria [104]. Reports have shown that Ca2+ entry channel inhibitors reduce acute axonal degeneration and improve regeneration after optic nerve damage [105]. A combination of Ca2+ entry channel inhibitors [106] indicates that ROS generation and downstream Ca2+ signalling are crucial during the progression of glaucoma.
Under the physiological conditions of cytosolic Ca2+ buffering, mitochondria play a key role in the “gating” of store-operated channels (SOC). Mitochondria actively coordinate spatiotemporal cytosolic Ca2+ under both physiological and pathological conditions [107,108]. By retaking the Ca2+ that has been released from the ER, mitochondrial buffering results in larger store depletion and, hence, the activation of Ca2+-release-activated channels (CRAC). Studies of mitochondria-dependent Ca2+ handling have revealed the molecular identities of the Ca2+- control components, including the mitochondrial Ca2+ uniporter (MCU) [109].
We previously examined the mitochondrial function and bioenergetics of glaucoma LC cells, and we observed evidence of reduced mitochondrial membrane potential in glaucoma LC cells [24], mitochondrial fission [30], and an increase in glycolysis, with a decrease in OXPHOS [31].
5. Ca2+ and Oxidative Stress
Oxidative stress can arise from Ca2+ dysregulation through several mechanisms, including increasing metabolic rate [110] and the activation of ROS-producing enzymes such as nitric oxide synthase and nicotinamide adenine dinucleotide phosphate oxidase [97,98,99,111]. ROS formation can damage proteins, lipids, and nucleic acids. Oxidative stress also creates a positive feedback loop with Ca2+ dysregulation. ROS depolarize the mitochondrial membrane and impair its energy metabolism, leading to a decrease in the ability of mitochondria to buffer Ca2+ [112]. In addition, the excessive production of ROS promotes Ca2+ release from internal stores via RYR and IP3R. Ca2+ ATPase pumps and the Na+-Ca2+ exchangers are responsible of maintaining the Ca2+ gradient across the plasma membrane [35].
Several anti-oxidative markers are elevated in the aqueous humor of glaucoma patients, including catalase, glutathione peroxidase, superoxide dismutase, and malondialdehyde [113]. In human glaucomatous retinas and optic nerve heads, glial-related oxidative stress pathways are upregulated [114]. Numerous studies have shown that oxidative stress is primarily involved in glaucoma at multiple levels and contributes to pro-fibrotic remodeling, IOP dysregulation, and impeded RGC axoplasmic transport [115].
We previously examined the level of oxidative stress in LC cells obtained from normal and glaucomatous human donor eyes, and our data showed evidence of oxidative stress in primary cultured glaucomatous fibroblast LC cells [24]. We found that glaucoma LC cells exhibit a significant increase in malondialdehyde (MDA) and reduced antioxidants such as aldo-keto reductase family 1 member C1 (AKR1C1) and glutamate—cysteine ligase catalytic subunit (GCLC). The same study showed evidence of mitochondrial dysfunction and abnormal elevated intracellular Ca2+ levels in glaucoma fibroblast LC cells [24].
Calcium and Cell Proliferation
It is well-known that cell proliferation is dependent on the cell cycle, which consists of four primary phases: G1, the first phase; S phase, in which nucleic acids occurs; G2, the second phase; and M phase, or mitosis. The switches between these phases are tightly controlled, and checkpoints during the cell cycle determine if the cell proceeds to the next phase [116]. These checkpoints have been shown to be dependent on Ca2+. Variations in [Ca2+]i are known to play a pivotal role throughout the cell cycle [35]. Several studies have established that cell proliferation is dependent on extracellular Ca2+ [117,118]. Ca2+ is required early in G1, as cells re-enter the cell cycle, to promote the activation of AP1 (FOS and JUN) transcription factors, c-AMP-responsive element binding (CREB) protein, and NFAT. Ca2+ plays a key role through these factors in coordinating the expression of cell cycle regulators such as the D-type cyclins, which are required for the activation of cyclin-dependent kinase 4 complexes. The initiation of the centrosomal duplication at the G1/S phase is also dependent on Ca2+ and on calmodulin (CaM), CaM kinase II (CaMK), and the centrosomal protein CP110. The Ca2+/CaM/CaMK pathways were shown to be necessary for cell cycle progression [117,118]. Calcineurin, a Ca2+-dependent phosphatase, is known to activate the nuclear factor of the activated T-cell transcription factor NFAT, and a demonstrated link with MYC [119] regulating gene transcription of cyclins E provides a connection between Ca2+-dependent pathways and proliferation.
The Ca2+/calcineurin/NFAT pathway is one of the major routes that can be activated by the entry of Ca2+ through plasma membrane Ca2+ channels. The use of various Ca2+ channel blockers has supported the idea that Ca2+ influx plays a role in cell proliferation. These observations suggest that a decrease in Ca2+ channel expression will lead to cell cycle arrest [120]. However, fibrosis and cancer are characterized by alterations in the Ca2+ signalling involved in cell proliferation. It has been found that the enhanced TRPC3-dependent Ca2+ influx led to increased proliferation in ovarian cancer [121].
In human LC cells, we previously found that the Ca2+ entry channels TRPC1/C6 contribute to oxidative stress-induced ECM gene transcription and cell proliferation [26]. The TRPC1/C6 channels may constitute important therapeutic targets for preventing ECM remodeling and fibrosis progression in glaucoma optic neuropathy [26]. Furthermore, we found that glaucomatous LC cells proliferate at higher rate and we showed that yes-associated protein (YAP) expression levels were relatively enhanced in glaucoma LC cells [28] (Table 1). Furthermore, the enhanced cell proliferation in glaucoma LC cells was reduced following treatment with the known YAP inhibitor verteporfin [28].
6. Calcium and Autophagy
Autophagy is another metabolic pathway that regulates the degradation of unfolded proteins and cellular components [122]. During the cellular autophagy process, some soluble proteins and cell organelles (mitochondria and endoplasmic reticulum) which have been dysfunctional in the cytoplasm are surrounded by autophagosomes. The autophagosome and lysosome fuse to form an autolysosome. Hypoxia [123], oxidative stress [124], and ER stress [125] induce cell autophagy. When ER stress is prolonged and unfolded proteins go beyond the capacity of the proteasome degradation system, autophagy may be triggered. The activation of PERK leads to the phosphorylation of the eukaryotic initiation factor (eIF2), which inhibits protein synthesis [126,127]. The activation of IRE1 and ATF6 promotes the transcription of UPR target genes. ER stress also leads to Ca2+ release from the ER to the cytosol, leading to the activation of numerous kinases and proteases involved in autophagy, including CaMKKβ [126], which, in turn, stimulates the disruption of the Beclin 1 inhibitory complexes (Beclin 1-IP3-R or Beclin 1-Bcl-2). In addition, CaMKKβ is also an upstream activator of AMPK, which inhibits mTORC1 [126].
It is known that ER stress-induced autophagy depends on Ca2+ homeostasis. Several studies have shown that imbalanced Ca2+ homeostasis can induce apoptosis in cancer cells. There is evidence that β-lapachone induces μ-calpain-mediated activity and is independent of caspase activity cell death in MCF-7 cells [128]. Other studies have reported the proapoptotic effect of EGTA and EDTA (Ca2+ ion chelators) in adenocarcinoma cells [129]. Furthermore, it has been found that factors increasing intracellular Ca2+ concentration, such as vitamin D3, ATP, ionomycin, and thapsigargin (an inhibitor of the ER Ca2+-ATPase pump), induced autophagic cell death in MCF-7 breast cancer cells [130]. In the Ca2+-dependent induction of autophagy, Ca2+ released from intracellular stores or fluxed from extracellular space via distinct Ca2+ channels activate CaMKKβ, which mediates the AMPK-dependent inhibition of mTORC1 [131]. Studies carried out on thapsigargin have revealed that the IRE1-JNK pathway is required for autophagy activation.
Studies on human primary cultures of TM cells have also shown that during glaucoma, the autophagic mechanism in TM cells is dysregulated. TM cells isolated from glaucomatous patients show dysregulation in the autophagic signalling pathway and a reduction in the autophagic response to oxidative stress [132]. The same study found that glaucomatous TM cells exhibited an overall reduction in LC3 and an increase in lipofuscin (or non-degradable lysosomal content) [132]. Other proteins associated with autophagy that were found to be down regulated during glaucoma are sequestosome-1 (p62) (a scaffold that targets ubiquitinated proteins for autophagic degradation), scCTSB (a lysosomal protein), and LC3B-II [131]. Furthermore, the same group, using transcriptome analysis, showed that autophagy regulated TGFβ/Smad-induced fibrogenesis in trabecular meshwork cells [133].
In human LC cells, we found that glaucoma LC cells exhibit a significant increased number of peri-nuclear lysosomes and an increase in autophagy in glaucoma LC myofibroblasts [24]. Glaucomatous LC cells contain significantly higher expression levels of cathepsin K mRNA and Atg5. Enhanced levels of LC3-II were found in both LC fibroblast cells and optic nerve head sections from glaucoma donors, indicating that intracellular lipofuscin accumulation may have important effects on autophagy [24]. Taken together, these data show that autophagic systems are dysfunctional in the TM and LC cells of glaucoma patients. Thus, targeting these signalling pathways could be beneficial in treating glaucoma.
7. Concluding Remarks
Ca2+ homeostasis is a crucial determinant of cellular function and survival. Intracellular Ca2+ ions are dynamically regulated by the plasma membrane, endoplasmic reticulum, and mitochondria. Ca2+ is also a ubiquitous and versatile intracellular second messenger contributing to several critical signalling pathways that participate in the regulation of numerous physiological processes [35,40]. In response to different stressors, Ca2+ enters into a cell and interacts with different Ca2+-binding proteins, of which there are over 200 encoded by the human genome that function either as Ca2+ effectors or buffers. Disruption of cytosolic Ca2+ homeostasis/signalling is responsible for many pathological states including proliferation, apoptosis, autophagy, angiogenesis, fibrosis, neurodegeneration, and cancer diseases. This review highlights the multiple abnormalities that we have identified in Ca2+ homeostasis in glaucomatous lamina cribrosa cells associated with increased fibrosis in the optic nerve head (summarized in Table 1 and Figure 4). Recent genome-wide association studies have described mutations in a number of calcium genes in glaucoma, requiring further studies [134,135]. Therapeutic targeting based on these abnormalities may help to reduce the global burden of visual impairment associated with glaucoma.
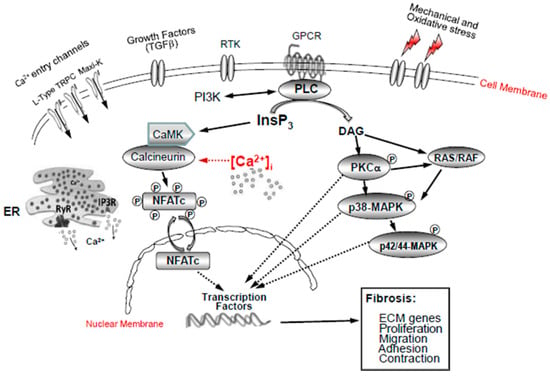
Figure 4.
Ca2+ signalling pathways in activated lamina cribrosa fibroblasts in glaucoma. Mechanical and oxidative stress and growth factors (TGFβ) stimulate Ca2+ ion channels (L-type, TRPC, Maxi-K+) and intracellular Ca2+ release from internal stores (ER and mitochondria). This stimulates PLC, which, in turn, activates a variety of signalling pathways, such as RAS/RAF and p38MAPK, as well as PKC p42/44-MAPK, CamK-calcineurin-NFATc, the SERCA pumps, and the PI3K signalling pathways, leading to the activation of Ca2+-dependent gene transcription factors (NFATc3 and YAP).
Author Contributions
M.I.: writing—original draft preparation; writing—review and editing. C.J.O.: review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable. As there was no use of animals or humans use in this study.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. No animals or humans were used in this study.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this article.
Abbreviations
| AMPK | AMP-activated protein kinase |
| ATF 4 | Activating transcription factor 4 |
| ATF 6 | Activating transcription factor 6 |
| Atg | Autophagy-related gene |
| Bad | Bcl-2-associated death |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| Bip | Immunoglobulin binding protein |
| CaMK | Ca2+/calmodulin-dependent kinase |
| CHOP | C/EBP-homologous protein |
| CRAC | Calcium release-activated channel |
| elF2α | Eukaryotic initiation factor |
| ER | Endoplasmic reticulum |
| IP3-R | Inositol triphosphate receptor |
| IRE1 | Inositol-requiring enzyme 1 |
| JNK | c-Jun NH2-terminal kinase |
| mTOR | Mammalian target of rapamycin kinase |
| mTORC1: | Mammalian target of rapamycin complex 1 |
| PERK | RNA-dependent protein ER kinase |
| PI3-K | Phosphoinositide 3-kinase |
| PMCA | Plasma membrane calcium ATPase |
| Raf-1 | Ras protooncogene serine/threonine protein kinase |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| SERCA | Smooth ER Ca2+-ATPase |
| SOCE | Store-operated calcium channel entry |
| UPR | Unfolded protein response |
| AMPK | AMP activated protein kinase |
| XBP-1 | X-box binding protein 1 |
| [Ca2+] | Intracellular Ca2+ concentration |
| PMCA | Plasma membrane Ca2+-ATPase |
| ECM | Extracellular matrix |
| ERAD | ER-associated degradation |
| GRP78 | Glucose-regulated protein 78 |
| IOP | Intraocular pressure |
| ISR | Integrated stress response |
| PKC | Protein kinase C |
| POAG | Primary open-angle glaucoma |
| TM | Trabecular meshwork |
| CREB | c-AMP-responsive element binding |
| mPTP | Mitochondrial permeability transition pore |
References
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.R.; Ye, H. Changes in the extracellular matrix of the human optic nerve head in primary open-angle glaucoma. Am. J. Ophthalmol. 1990, 109, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Pena, J.D.; Taylor, A.W.; Ricard, C.S.; Vidal, I.; Hernandez, M.R. Transforming growth factor beta isoforms in human optic nerve heads. Br. J. Ophthalmol. 1999, 83, 209–218. [Google Scholar] [CrossRef]
- Hernandez, M.R.; Igoe, F.; Neufeld, A.H. Cell culture of the human lamina cribrosa. Investig. Ophthalmol. Vis. Sci. 1988, 29, 78–89. [Google Scholar]
- Lambert, W.; Agarwal, R.; Howe, W.; Clark, A.F.; Wordinger, R.J. Neurotrophin and Neurotrophin Receptor Expression by Cells of the Human Lamina Cribrosa. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2315–2323. [Google Scholar]
- Tovar-Vidales, T.; Wordinger, R.J.; Clark, A.F. Identification and localization of lamina cribrosa cells in the human optic nerve head. Exp. Eye Res. 2016, 147, 94–97. [Google Scholar] [CrossRef]
- Dillinger, A.E.; Weber, G.R.; Mayer, M.; Schneider, M.; Göppner, C.; Ohlmann, A.; Shamonin, M.; Monkman, G.J.; Fuchshofer, R. CCN2/CTGF-A Modulator of the Optic Nerve Head Astrocyte. Front. Cell Dev. Biol. 2022, 10, 864433. [Google Scholar] [CrossRef]
- Hernandez, M.R. The optic nerve head in glaucoma: Role of astrocytes in tissue remodeling. Prog. Retin. Eye Res. 2000, 19, 297–321. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Borne, S.W.M.V.D.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2009, 7, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Chai, B.; Duan, R.; Zhou, Y.; Huang, X.; Li, Q. Inhibition of FKBP10 Attenuates Hypertrophic Scarring through Suppressing Fibroblast Activity and Extracellular Matrix Deposition. J. Investig. Dermatol. 2017, 137, 2326–2335. [Google Scholar] [CrossRef]
- Tomcik, M.; Palumbo-Zerr, K.; Zerr, P.; Sumova, B.; Avouac, J.; Dees, C.; Distler, A.; Becvar, R.; Distler, O.; Schett, G.; et al. Tribbles homologue 3 stimulates canonical TGF-β signalling to regulate fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 2016, 75, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Tamm, E.R.; Braunger, B.M.; Fuchshofer, R. Intraocular Pressure and the Mechanisms Involved in Resistance of the Aqueous Humor Flow in the Trabecular Meshwork Outflow Pathways. Prog. Mol. Biol. Transl. Sci. 2015, 134, 301–314. [Google Scholar]
- Hopkins, A.; Murphy, R.; Irnaten, M.; Wallace, D.M.; Quill, B.; O’Brien, C. The role of lamina cribrosa tissue stiffness and fibrosis as fundamental biomechanical drivers of pathological glaucoma cupping. Am. J. Physiol. Cell. Physiol. 2020, 319, C611–C623. [Google Scholar] [CrossRef]
- Kirwan, R.P.; Wordinger, R.J.; Clark, A.F.; O’Brien, C.J. Differential global and extra-cellular matrix focused gene expression patterns between normal and glaucomatous human lamina cribrosa cells. Mol. Vis. 2009, 15, 76–88. [Google Scholar] [PubMed]
- Kirwan, R.P.; Leonard, M.O.; Murphy, M.; Clark, A.F.; O’Brien, C.J. Transforming growth factor-beta-regulated gene transcription and protein expression in human GFAP-negative lamina cribrosa cells. Glia 2005, 52, 309–324. [Google Scholar] [CrossRef]
- Kirwan, R.P.; Crean, J.K.; Fenerty, C.H.; Clark, A.F.; O’Brien, C.J. Effect of cyclical mechanical stretch and exogenous transforming growth factor-beta1 on matrix metalloproteinase-2 activity in lamina cribrosa cells from the human optic nerve head. J. Glaucoma 2004, 13, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, R.P.; Felice, L.; Clark, A.F.; O’Brien, C.J.; Leonard, M.O. Hypoxia regulated gene transcription in human optic nerve lamina cribrosa cells in culture. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kilpatrick, J.I.; Lukasz, B.; Jarvis, S.P.; McDonnell, F.; Wallace, D.M.; Clark, A.F.; O’Brien, C.J. Increased Substrate Stiffness Elicits a Myofibroblastic Phenotype in Human Lamina Cribrosa Cells. Investig. Ophthalmol. Vis. Sci. 2018, 59, 803–814. [Google Scholar] [CrossRef] [PubMed]
- McElnea, E.M.; Quill, B.; Docherty, N.G.; Irnaten, M.; Siah, W.F.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol. Vis. 2011, 17, 1182–1191. [Google Scholar] [PubMed]
- Irnaten, M.; Zhdanov, A.; Brennan, D.; Crotty, T.; Clark, A.; Papkovsky, D.; O’Brien, C. Activation of the NFAT-Calcium Signaling Pathway in Human Lamina Cribrosa Cells in Glaucoma. Investig. Ophthalmol. Vis. Sci. 2018, 59, 831–842. [Google Scholar] [CrossRef]
- Irnaten, M.; O’Malley, G.; Clark, A.; O’Brien, C. Transient receptor potential channels TRPC1/TRPC6 regulate lamina cribrosa cell extracellular matrix gene transcription and proliferation. Exp. Eye Res. 2020, 193, 107980. [Google Scholar] [CrossRef]
- Irnaten, M.; Barry, R.C.; Wallace, D.M.; Docherty, N.G.; Quill, B.; Clark, A.F.; O’Brien, C.J. Elevated maxi-K(+) ion channel current in glaucomatous lamina cribrosa cells. Exp. Eye Res. 2013, 115, 224–229. [Google Scholar] [CrossRef]
- Murphy, R.; Irnaten, M.; Hopkins, A.; O’Callaghan, J.; Stamer, W.D.; Clark, A.F.; Wallace, D.; O’Brien, C.J. Matrix Mechanotransduction via Yes-Associated Protein in Human Lamina Cribrosa Cells in Glaucoma. Investig. Ophthalmol. Vis. Sci. 2022, 63, 16. [Google Scholar] [CrossRef]
- Irnaten, M.; Duff, A.; Clark, A.; O’Brien, C.J. Intra-Cellular Calcium Signaling Pathways (PKC, RAS/RAF/MAPK, PI3K) in Lamina Cribrosa Cells in Glaucoma. Clin Med. 2020, 10, 62. [Google Scholar] [CrossRef]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial dysfunction in ocular disease: Focus on glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar] [CrossRef]
- Kamel, K.; O’Brien, C.J.; Zhdanov, A.V.; Papkovsky, D.B.; Clark, A.F.; Stamer, W.D.; Irnaten, M. Reduced Oxidative Phosphorylation and Increased Glycolysis in Human Glaucoma Lamina Cribrosa Cells. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4. [Google Scholar] [CrossRef] [PubMed]
- Quill, B.; Irnaten, M.; Docherty, N.G.; McElnea, E.M.; Wallace, D.M.; Clark, A.F.; O’Brien, C.J. Calcium channel blockade reduces mechanical strain-induced extracellular matrix gene response in lamina cribrosa cells. Br. J. Ophthalmol. 2015, 99, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Xie, J.; Nattel, S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 2011, 89, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell. Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis, and remodelling. Nat. Rev. Mol. Cell. Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Jain, R.; Watson, U.; Vasudevan, L.; Saini, D.K. ERK Activation Pathways Downstream of GPCRs. Int. Rev. Cell. Mol. Biol. 2018, 338, 79–109. [Google Scholar]
- Prole, D.L.; Taylor, C.W. Structure and Function of IP3 Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035063. [Google Scholar] [CrossRef]
- Capiod, T.; Mauger, J.P.; Binet, A.; Claret, M. Regulation of calcium in non-excitable cells. Curr. Opin. Cell Biol. 1989, 1, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell. 2020, 78, 1055–1069. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Liu, Q.; Molkentin, J.D. Protein kinase Cα as a heart failure therapeutic target. J. Mol. Cell Cardiol. 2011, 51, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; McKinsey, T.A.; Olson, E.N. Decoding calcium signals involved in cardiac growth and function. Nat. Med. 2000, 6, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004, 94, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Smedler, E.; Uhlén, P. Frequency decoding of calcium oscillations. Biochim. Biophys. Acta 2014, 1840, 964–969. [Google Scholar] [CrossRef]
- Oruganti, S.R.; Edin, S.; Grundström, C.; Grundström, T. CaMKII targets Bcl10 in T-cell receptor induced activation of NF-κB. Mol. Immunol. 2011, 48, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Mori, T.; Kurumazuka, D.; Kitada, K.; Hayashi, T.; Nagatoya, K.; Inoue, T.; Ukimura, A.; Matsumura, Y.; Ishizaka, N.; et al. Inhibitory effects of T/L-type calcium channel blockers on tubulointerstitial fibrosis in obstructed kidneys in rats. Urology 2011, 77, 249.e9–249.e15. [Google Scholar] [CrossRef]
- Shafik, A.N.; Khodeir, M.M.; Gouda, N.A.; Mahmoud, M.E. Improved antifibrotic effect of a combination of verapamil and silymarin in rat-induced liver fibrosis. Arab J. Gastroenterol. 2011, 12, 143–149. [Google Scholar] [CrossRef]
- Nakagami, H.; Shimamura, M.; Miyake, T.; Shimosato, T.; Minobe, N.; Moritani, T.; Kiomy Osako, M.; Nakagami, F.; Koriyama, H.; Kyutoku, M.; et al. Nifedipine prevents hepatic fibrosis in a non-alcoholic steatohepatitis model induced by an L-methionine-and choline-deficient diet. Mol. Med. Rep. 2012, 5, 37–40. [Google Scholar]
- Ohyama, T.; Sato, K.; Kishimoto, K.; Yamazaki, Y.; Horiguchi, N.; Ichikawa, T.; Kakizaki, S.; Takagi, H.; Izumi, T.; Mori, M. Azelnidipine is a calcium blocker that attenuates liver fibrosis and may increase antioxidant defence. Br. J. Pharmacol. 2012, 165, 1173–1187. [Google Scholar] [CrossRef]
- Orenes-Piñero, E.; Hernández-Romero, D.; Jover, E.; de la Morena, G.; Valdés, M.; Marín, F. An insight of novel pharmacological therapies in hypertrophic cardiomyopathy. Med. Chem. 2011, 7, 275–285. [Google Scholar] [CrossRef]
- Horiba, M.; Muto, T.; Ueda, N.; Opthof, T.; Miwa, K.; Hojo, M.; Lee, J.K.; Kamiya, K.; Kodama, I.; Yasui, K. T-type Ca2+ channel blockers prevent cardiac cell hypertrophy through an inhibition of calcineurin-NFAT3 activation as well as L-type Ca2+ channel blockers. Life Sci. 2008, 82, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Zunwen, L.; Shizhen, Z.; Dewu, L.; Yungui, M.; Pu, N. Effect of tetrandrine on the TGF-β–induced Smad signal transduction pathway in human hypertrophic scar fibroblasts in vitro. Burns 2012, 38, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Huang, C.Y. The effect of combined steroid and calcium channel blocker injection on human hypertrophic scars in animal model: A new strategy for the treatment of hypertrophic scars. Dermatol. Surg. 2010, 36, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Gooch, J.L.; Gorin, Y.; Zhang, B.X.; Abboud, H.E. Involvement of calcineurin in transforming growth factor-beta–mediated regulation of extracellular matrix accumulation. J. Biol. Chem. 2004, 279, 15561–15570. [Google Scholar] [CrossRef] [PubMed]
- Niittykoski, M.; Kalesnykas, G.; Larsson, K.; Kaarniranta, K.; Akerman, K.; Uusitalo, H. Altered calcium signaling in an experimental model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6387–6393. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, J.; Starr, C.; Mohns, E.; Li, Y.; Chen, E.; Yoon, Y.; Kellner, C.; Tanaka, K.; Wang, H.; et al. Preservation of vision after CaMKII-mediated protection of retinal ganglion cells. Cell 2021, 184, 4299–4314.e12. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Frye, A.M.; Phuong, T.T.T.; Yarishkin, O.; Jo, A.O.; Xu, Y.; Lakk, M.; Iuso, A.; Redmon, S.N.; Ambati, B.; et al. TRPV4 regulates calcium homeostasis, cytoskeletal remodeling, conventional outflow and intraocular pressure in the mammalian eye. Sci. Rep. 2016, 6, 30583. [Google Scholar] [CrossRef]
- He, Y.; Ge, J.; Tombran-Tink, J. Mitochondrial defects and dysfunction in calcium regulation in glaucomatous trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4912–4922. [Google Scholar] [CrossRef]
- Fry, C.H.; Sui, G.; Wu, C. T-type Ca21 channels in non-vascular smooth muscles. Cell Calcium. 2006, 40, 231–239. [Google Scholar] [CrossRef]
- Tanaka, H.; Komikado, C.; Namekata, I.; Nakamura, H.; Suzuki, M.; Tsuneoka, Y.; Shigenobu, K.; Takahara, A. Species difference in the contribution of T-type calcium current to cardiac pacemaking as revealed by r(-)-efonidipine. J. Pharmacol. Sci. 2008, 107, 99–102. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Simms, B.A.; Zamponi, G.W. Neuronal voltage-gated calcium channels: Structure, function, and dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Ben-Johny, M.; Yue, D.T. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol. 2014, 143, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Hughes, F.M.J.; Huang, Y.; Cidlowski, J.A.; Putney, J.W., Jr. Roles of cytoplasmic Ca2+ and intracellular Ca2+ stores in induction and suppression of apoptosis in S49 cells. Am. J. Physiol. Cell Physiol. 1997, 272, C1241–C1249. [Google Scholar] [CrossRef]
- Bird, G.S.J.; Bian, X.; Putney, J.W. Calcium entry signal? Nature 1995, 373, 481–482. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.S.J.; Putney, J.W., Jr. Effect of inositol 1,3,4,5-tetrakisphosphate on inositol trisphosphate-activated Ca[IMAGE]. Signaling in mouse lacrimal acinar cells. J. Biol. Chem. 1996, 271, 6766–6770. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.S.J.; Putney, J.W.J. Inhibition of thapsigargin-induced calcium entry by microinjected guanine nucleotide analogues. Evidence for the involvement of a small G-protein in capacitative calcium entry. J. Biol. Chem. 1993, 268, 21486–21488. [Google Scholar] [CrossRef]
- Kaneko, Y.; Szallasi, A. Transient receptor potential (TRP) channels: A clinical perspective. Br. J. Pharmacol. 2014, 171, 2474–2507. [Google Scholar]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Reid, D.W.; Nicchitta, C.V. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2015, 16, 221–231. [Google Scholar] [CrossRef]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Gaut, J.R.; Hendershot, L.M. The modification and assembly of proteins in the endoplasmic reticulum. Curr. Opin. Cell Biol. 1993, 5, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Canino, L.S.; McCammon, J.A. Unfolding proteins under external forces: A solvable model under the self-consistent pair contact probability approximation. Phys. Rev. Lett. 2002, 89, 068103. [Google Scholar] [CrossRef]
- Scull, C.M.; Tabas, I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arter. Thromb. Vasc. Biol. 2011, 31, 2792–2797. [Google Scholar] [CrossRef]
- Jaronen, M.; Goldsteins, G.; Koistinaho, J. ER stress and unfolded protein response in amyotrophic lateral sclerosis-a controversial role of protein disulphide isomerase. Front. Cell Neurosci. 2014, 8, 402. [Google Scholar] [CrossRef]
- Nguyen, P.; Calderon, R.; Rodriguez-Ledezma, Y.; Araujo, K.; Bhandari, D. GIV/Girdin promotes cell survival during endoplasmic reticulum stress. Mol. Cell Biochem. 2019, 453, 79–88. [Google Scholar] [CrossRef]
- Hurley, D.J.; Normile, C.; Irnaten, M.; O’Brien, C. The Intertwined Roles of Oxidative Stress and Endoplasmic Reticulum Stress in Glaucoma. Antioxidants 2022, 11, 886. [Google Scholar] [CrossRef]
- Ricci, J.E.; Muñoz-Pinedo, C.; Fitzgerald, P.; Bailly-Maitre, B.; Perkins, G.A.; Yadava, N.; Scheffler, I.E.; Ellisman, M.H.; Green, D.R. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 2004, 117, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef]
- Hüttemann, M.; Helling, S.; Sanderson, T.H.; Hüttemann, M.; Helling, S.; Sanderson, T.H.; Sinkler, C.; Samavati, L.; Mahapatra, G.; Varughese, A.; et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: Cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371.e359. [Google Scholar] [CrossRef]
- Coutelle, O.; Hornig-Do, H.T.; Witt, A.; Andree, M.; Schiffmann, L.M.; Piekarek, M.; Brinkmann, K.; Seeger, J.M.; Liwschitz, M.; Miwa, S.; et al. Embelin inhibits endothelial mitochondrial respiration and impairs neoangiogenesis during tumor growth and wound healing. EMBO Mol. Med. 2014, 6, 624–639. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [PubMed]
- Mela, L. Inhibition and activation of calcium transport in mitochondria. Effet of lanthanides and local anesthetic drugs. Biochemistry 1969, 8, 2481–2486. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Simpson, A.W.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef]
- Valero, R.A.; Senovilla, L.; Núñez, L.; Villalobos, C. The role of mitochondrial potential in control of calcium signals involved in cell proliferation. Cell Calcium. 2008, 44, 259–269. [Google Scholar] [CrossRef]
- Luis-García, E.; Becerril, C.; Salgado-Aguayo, A.; Aparicio-Trejo, O.; Romero, Y.; Flores-Soto, E.; Mendoza-Milla, C.; Montaño, M.; Chagoya, V.; Pedraza-Chaverri, J.; et al. Mitochondrial Dysfunction and Alterations in Mitochondrial Permeability Transition Pore (mPTP) Contribute to Apoptosis Resistance in Idiopathic Pulmonary Fibrosis Fibroblasts. Int. J. Mol. Sci. 2021, 22, 7870. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, H.; Wang, X.; Wang, Q.C.; Zhang, C.; Wang, J.Q.; Wang, Y.-H.; An, C.-Q.; Yang, K.-Y.; Wang, Y.; et al. TMCO1 is essential for ovarian follicle development by regulating ER ca (2+) store of granulosa cells. Cell Death Differ. 2018, 25, 1686–1701. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Q.C.; Sun, Z.; Li, T.; Yang, K.; An, C.; Guo, C.; Tang, T.-S. ER stress mediated degradation of diacylglycerol acyltransferase impairs mitochondrial functions in TMCO1 deficient cells. Biochem. Biophys. Res. Commun. 2019, 512, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Twomey, C.; McCarthy, J.V. Pathways of apoptosis and importance in development. J. Cell Mol. Med. 2005, 9, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Romagnoli, A.; Pinton, P.; Rizzuto, R. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 2008, 8, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef] [PubMed]
- García-Murria, M.J.; Duart, G.; Grau, B.; Diaz-Beneitez, E.; Rodríguez, D.; Mingarro, I.; Martínez-Gil, L. Viral Bcl2s’ transmembrane domain interact with host Bcl2 proteins to control cellular apoptosis. Nat. Commun. 2020, 11, 6056. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Li, T.; Yang, Y.; Zhang, J.-T.; Antonsson, B.; Brustovetsky, N. BAX insertion, oligomerization, and outer membrane permeabilization in brain mitochondria: Role of permeability transition and SH-redox regulation. Biochim. Biophys. Acta 2010, 1797, 1795–1806. [Google Scholar] [CrossRef]
- Van Bergen, N.J.; Chakrabarti, R.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial disorders and the eye. Eye Brain 2011, 3, 29–47. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Trounce, I.A.; Crowston, J.G. Mechanisms of retinal ganglion cell injury in aging and glaucoma. Ophthalmic Res. 2010, 44, 173–178. [Google Scholar] [CrossRef]
- Kong, G.Y.X.; Van Bergen, N.J.; Trounce, I.; Crowston, J.G. Mitochondrial Dysfunction and Glaucoma. Eur. J. Gastroenterol. Hepatol. 2009, 18, 93–100. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress, and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef]
- Zhou, D.B.; Castanos, M.V.; Geyman, L.; Rich, C.A.; Tantraworasin, A.; Ritch, R.; Rosen, R.B. Mitochondrial Dysfunction in Primary Open-Angle Glaucoma Characterized by Flavoprotein Fluorescence at the Optic Nerve Head. Ophthalmol. Glaucoma 2022, 5, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Núñez-Álvarez, C.; Joglar, B.; Del Olmo-Aguado, S. Glaucoma: Focus on mitochondria in relation to pathogenesis and neuroprotection. Eur. J. Pharmacol. 2016, 787, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Vosler, P.S.; Brennan, C.S.; Chen, J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 2008, 38, 78–100. [Google Scholar] [CrossRef]
- Ribas, V.T.; Lingor, P. Calcium channel inhibition-mediated axonal stabilization improves axonal regeneration after optic nerve crush. Neural Regen Res. 2016, 11, 1245–1246. [Google Scholar] [CrossRef]
- Savigni, D.L.; O’Hare Doig, R.L.; Szymanski, C.R.; Bartlett, C.A.; Lozić, I.; Smith, N.M.; Fitzgerald, M. Three Ca2+ channel inhibitors in combination limit chronic secondary degeneration following neurotrauma. Neuropharmacology 2013, 75, 380–390. [Google Scholar] [CrossRef]
- Gunter, T.E.; Buntinas, L.; Sparagna, G.; Eliseev, R.; Gunter, K. Mitochondrial calcium transport: Mechanisms and functions. Cell Calcium. 2000, 28, 285–296. [Google Scholar] [CrossRef]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.I.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102. [Google Scholar] [CrossRef]
- Mammucari, C.; Gherardi, G.; Rizzuto, R. Structure, Activity Regulation, and Role of the Mitochondrial Calcium Uniporter in Health and Disease. Front. Oncol. 2017, 7, 139. [Google Scholar] [CrossRef]
- Zündorf, G.; Reiser, G.J. The phosphorylation status of extracellular-regulated kinase 1/2 in astrocytes and neurons from rat hippocampus determines the thrombin-induced calcium release and ROS generation. J. Neurochem. 2011, 119, 1194–1204. [Google Scholar] [CrossRef]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Murchison, D.; Griffith, W.H. Calcium buffering systems and calcium signaling in aged rat basal forebrain neurons. Aging Cell. 2007, 6, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.A.; Arafa, L.F.; El-Baz, A. Oxidative stress markers in patients with primary open-angle glaucoma. Curr. Eye Res. 2010, 35, 295–301. [Google Scholar] [CrossRef]
- Tezel, G.; Luo, C.; Yang, X. Accelerated aging in glaucoma: Immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1201–1211. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Umen, J.G.; Goodenough, U.W. Control of cell division by a retinoblastoma protein homolog in Chlamydomonas. Genes Dev. 2001, 15, 1652–1661. [Google Scholar] [CrossRef]
- Kahl, C.R.; Means, A.R. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef]
- Capiod, T. Cell proliferation, calcium influx and calcium channels. Biochimie 2011, 93, 2075–2079. [Google Scholar] [CrossRef]
- Buchholz, M.; Schatz, A.; Wagner, M.; Michl, P.; Linhart, T.; Adler, G.; Gress, T.M.; Ellenrieder, V. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1.and the Ca2+/calcineurin signaling pathway. EMBO J. 2006, 25, 3714–3724. [Google Scholar] [CrossRef]
- Borowiec, A.S.; Bidaux, G.; Pigat, N.; Goffin, V.; Bernichtein, S.; Capiod, T. Calcium channels, external calcium concentration and cell proliferation. Eur. J. Pharmacol. 2014, 739, 19–25. [Google Scholar] [CrossRef]
- Yang, S.L.; Cao, Q.; Zhou, K.C.; Feng, Y.J.; Wang, Y.Z. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene 2009, 28, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Elżbieta, K.; Pająk, B.; Orzechowski, A. Calcium Homeostasis and ER Stress in Control of Autophagy in Cancer Cells. Biomed. Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, C.; Shen, M.; Yang, M.; Jin, Z.; Ding, L.; Jiang, W.; Yang, J.; Chen, H.; Cao, F.; et al. Autophagy mediates the beneficial effect of hypoxic preconditioning on bone marrow mesenchymal stem cells for the therapy of myocardial infarction. Stem Cell Res. Ther. 2017, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Jacobi, A.; Vater, C.; Zou, L.; Zou, X.; Stiehler, M. Icariin Promotes Angiogenic Differentiation and Prevents Oxidative Stress-Induced Autophagy in Endothelial Progenitor Cells. Stem Cells 2015, 33, 1863–1877. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Sung, M.S.; Lee, E.G.; Yoo, H.G.; Cheon, Y.H.; Chae, H.J.; Yoo, W.H. A pathogenic role for ER stress-induced autophagy and ER chaperone GRP78/BiP in T lymphocyte systemic lupus erythematosus. J. Leukoc. Biol. 2015, 97, 425–433. [Google Scholar] [CrossRef]
- Jiang, C.; Li, Z.; Wang, X.-B.; Li, J.; Wang, B.; Lv, G.-H.; Liu, F.-B. Ca2+ Regulates Autophagy Through CaMKKβ/AMPK/mTOR Signaling Pathway in Mechanical Spinal cord Injury: An in vitro Study. Neurochem. Res. 2022. [Google Scholar] [CrossRef]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell. 2011, 22, 4390–4405. [Google Scholar] [CrossRef]
- Tagliarino, C.; Pink, J.J.; Reinicke, K.E.; Simmers, S.M.; Wuerzberger-Davis, S.M.; Boothman, D.A. μ-calpain activation in β-lapachone-mediated apoptosis. Cancer Biol. Ther. 2003, 2, 141–152. [Google Scholar] [CrossRef]
- Pajak, B.; Orzechowski, A. Ethylenediaminetetraacetic acid affects subcellular expression of clusterin protein in human colon adenocarcinoma COLO 205 cell line. Anti-Cancer Drugs 2007, 18, 55–63. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Jäättelä, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase Kinase-β, and Bcl-2. Mol. Cell. 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Porter, K.; Hirt, J.; Stamer, W.D.; Liton, P.B. Autophagic dysregulation in glaucomatous trabecular meshwork cells. Biochim. Biophys. Acta 2015, 1852, 379–385. [Google Scholar] [CrossRef]
- Nettesheim, A.; Shim, M.S.; Hirt, J.; Liton, P.B. Transcriptome analysis reveals autophagy as regulator of TGFβ/Smad-induced fibrogenesis in trabecular meshwork cells. Sci. Rep. 2019, 9, 16092. [Google Scholar] [CrossRef]
- Chen, F.; Klein, A.P.; Klein, B.E.K.; Lee, K.E.; Truitt, B.; Klein, R.; Iyengar, S.K.; Duggal, P. Exome Array Analysis Identifies CAV1/CAV2 as a Susceptibility Locus for Intraocular Pressure. Investig. Ophthalmol. Vis. Sci. Jan. 2015, 56, 544–551. [Google Scholar] [CrossRef]
- Burdon, K.P.; Macgregor, S.; Hewitt, A.W.; Sharma, S.; Chidlow, G.; Mills, R.A.; Danoy, P.; Casson, R.; Viswanathan, A.C.; Liu, J.Z.; et al. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat. Genet. 2011, 43, 574–578. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).