
Nuclear Magnetic Resonance Relaxation Pathways in Electrolytes for Energy Storage



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Electrolytes
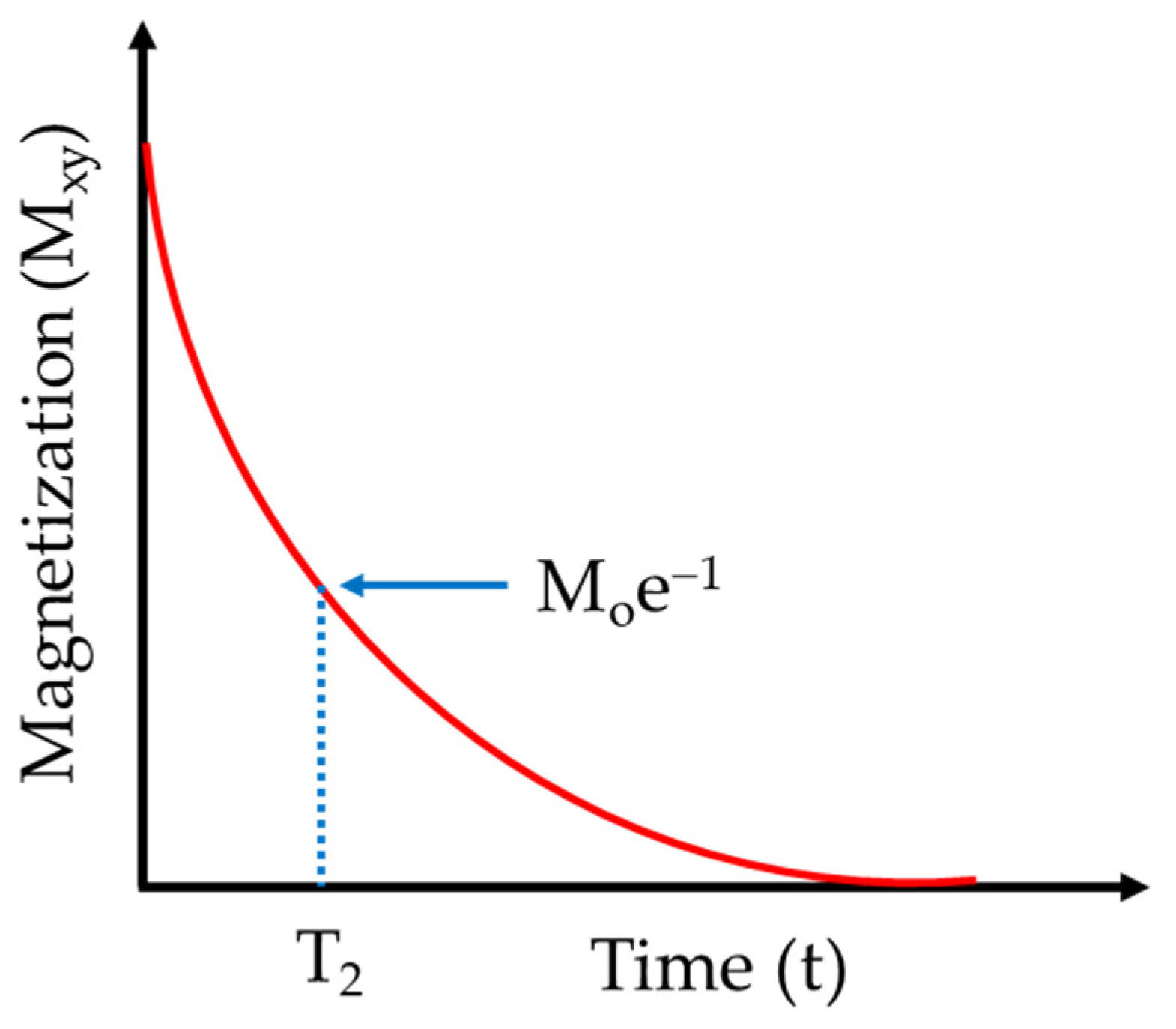
3. NMR Relaxation
4. Translational and Rotational Dynamics in Liquid and Confined Electrolytes
4.1. Ionic Liquids Dynamics
4.2. Deep Eutectic Solvents (DESs)
4.3. Organic Solvent Electrolytes
4.4. Confined Liquid Electrolytes
4.4.1. Ionogels (IGs)
4.4.2. Gel Polymers Electrolytes (GPEs)
5. Solid Electrolytes
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BF4 | Tetrafluoroborate |
| BMS | Battery Management System |
| BPP | Bloemburgen, Purcell, and Pound |
| C2C1im | 1-ethyl-3-methylimidazolium (also called EMIM) |
| C4C1im | 1-butyl-3-methylimidazolium (also called BMIM) |
| C8C1im | 1-methyl-3-octylimidazolium |
| CD | Cole-Davidson |
| (CF3SO2)2N | Bis(trifluoromethylsulfonyl)amide (also called TFSI, TFSA) |
| CIP | Contact-ion-pair |
| DEC | Diethyl carbonate |
| DMC | Dimethyl carbonate |
| DES | Deep eutectic solvents |
| EC | Ethylene carbonate |
| EDLC | Electric double layer capacitor |
| EFG | Electric field gradient |
| FEC | Fluoroethylene carbonate |
| FFC | Fast field cycling |
| FFHS | Force-free-hard-sphere |
| FSI | Bis(fluorosulfonyl)imide (also called FSA, FSI) |
| GPE | Gel polymer electrolytes |
| IL | Ionic Liquid |
| IG | Ionogel |
| LEDC | Lithium ethylene decarbonates |
| LIB | Lithium-ion battery |
| LiBF4 | Lithium tetrafluoroborate |
| LiClO4 | Lithium perchlorate |
| LiCF3SO3 | Lithium trifluoromethanesulfonate |
| MDS | Molecular dynamics simulations |
| NTf2 | bis(trifluoromethanesulfonyl)imide (also called TFSA, TFSI) |
| PC | Propylene carbonate |
| PF6 | Hexafluorophosphate |
| PEFC | Polymer electrolyte fuel cell |
| PEO | Poly (ethylene oxide) |
| PVDF | Poly (vinylidene fluoride) |
| QCC | Quadrupolar coupling constant |
| RMTD | Reorientations mediated by translational displacements |
| SEI | Solid electrolyte interface |
| SED | Stokes-Einstein-Debye |
| SOFC | Solid oxide fuel cell |
| SPE | Solid polymer electrolytes |
| TEP | Triethyl phosphate |
| TMP | Trimethyl phosphate |
| VC | Vinylene carbonate |
References
- Vangari, M.; Pryor, T.; Jiang, L. Supercapacitors: Review of materials and fabrication methods. J. Energy Eng. 2013, 139, 72–79. [Google Scholar] [CrossRef]
- Zhong, C.; Deng, Y.; Hu, W.; Qiao, J.; Zhang, L.; Zhang, J. A review of electrolyte materials and compositions for electrochemical supercapacitors. Chem. Soc. Rev. 2015, 44, 7484–7539. [Google Scholar] [CrossRef]
- Zang, X.; Shen, C.; Sanghadasa, M.; Lin, L. High-voltage supercapacitors based on aqueous electrolytes. ChemElectroChem 2019, 6, 976–988. [Google Scholar] [CrossRef]
- Conti, J.; Holtberg, P.; Diefenderfer, J.; LaRose, A.; Turnure, J.T.; Westfall, L. International Energy Outlook 2016 with Projections to 2040; USDOE Energy Information Administration (EIA): Washington, DC, USA, 2016. [Google Scholar]
- Hansen, K.; Breyer, C.; Lund, H. Status and perspectives on 100% renewable energy systems. Energy 2019, 175, 471–480. [Google Scholar] [CrossRef]
- Cantarero, M.M.V. Of renewable energy, energy democracy, and sustainable development: A roadmap to accelerate the energy transition in developing countries. Energy Res. Soc. Sci. 2020, 70, 101716. [Google Scholar] [CrossRef]
- Iqbal, M.Z.; Siddique, S.; Hussain, G.; Iqbal, M.W. Room temperature spin valve effect in the NiFe/Gr–hBN/Co magnetic tunnel junction. J. Mater. Chem. C 2016, 4, 8711–8715. [Google Scholar] [CrossRef]
- Blomgren, G.E. The development and future of lithium ion batteries. J. Electrochem. Soc. 2016, 164, A5019. [Google Scholar] [CrossRef]
- Placke, T.; Kloepsch, R.; Dühnen, S.; Winter, M. Lithium ion, lithium metal, and alternative rechargeable battery technologies: The odyssey for high energy density. J. Solid State Electrochem. 2017, 21, 1939–1964. [Google Scholar] [CrossRef]
- Zubi, G.; Dufo-López, R.; Carvalho, M.; Pasaoglu, G. The lithium-ion battery: State of the art and future perspectives. Renew. Sustain. Energy Rev. 2018, 89, 292–308. [Google Scholar] [CrossRef]
- Ding, Y.; Cano, Z.P.; Yu, A.; Lu, J.; Chen, Z. Automotive Li-ion batteries: Current status and future perspectives. Electrochem. Energy Rev. 2019, 2, 1–28. [Google Scholar] [CrossRef]
- Miller, J.R.; Simon, P. Electrochemical capacitors for energy management. Science 2008, 321, 651–652. [Google Scholar] [CrossRef]
- Stoller, M.D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R.S. Graphene-based ultracapacitors. Nano Lett. 2008, 8, 3498–3502. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Z.; Huang, Y.; Ma, Y.; Wang, C.; Chen, M.; Chen, Y. Supercapacitor devices based on graphene materials. J. Phys. Chem. C 2009, 113, 13103–13107. [Google Scholar] [CrossRef]
- Wu, H.; He, D.; Wang, Y.; Fu, M.; Liu, Z.; Wang, J.; Wang, H. Graphene as the electrode material in supercapacitors. In Proceedings of the 2010 8th International Vacuum Electron Sources Conference and Nanocarbon, Nanjing, China, 14–16 October 2010; pp. 465–466. [Google Scholar]
- Ervin, M.H.; Miller, B.S.; Hanrahan, B.; Mailly, B.; Palacios, T. A comparison of single-wall carbon nanotube electrochemical capacitor electrode fabrication methods. Electrochim. Acta 2012, 65, 37–43. [Google Scholar] [CrossRef]
- Pope, M.A.; Korkut, S.; Punckt, C.; Aksay, I.A. Supercapacitor electrodes produced through evaporative consolidation of graphene oxide-water-ionic liquid gels. J. Electrochem. Soc. 2013, 160, A1653. [Google Scholar] [CrossRef]
- Feng, X.; Ouyang, M.; Liu, X.; Lu, L.; Xia, Y.; He, X. Thermal runaway mechanism of lithium ion battery for electric vehicles: A review. Energy Storage Mater. 2018, 10, 246–267. [Google Scholar] [CrossRef]
- Deng, K.; Zeng, Q.; Wang, D.; Liu, Z.; Qiu, Z.; Zhang, Y.; Xiao, M.; Meng, Y. Single-ion conducting gel polymer electrolytes: Design, preparation and application. J. Mater. Chem. A 2020, 8, 1557–1577. [Google Scholar] [CrossRef]
- Aurbach, D.; Zaban, A.; Schechter, A.; Ein-Eli, Y.; Zinigrad, E.; Markovsky, B. The study of electrolyte solutions based on ethylene and diethyl carbonates for rechargeable Li batteries: I. Li metal anodes. J. Electrochem. Soc. 1995, 142, 2873. [Google Scholar] [CrossRef]
- Xu, K.; Lam, Y.; Zhang, S.S.; Jow, T.R.; Curtis, T.B. Solvation sheath of Li+ in nonaqueous electrolytes and its implication of graphite/electrolyte interface chemistry. J. Phys. Chem. C 2007, 111, 7411–7421. [Google Scholar] [CrossRef]
- Shi, S.; Lu, P.; Liu, Z.; Qi, Y.; Hector, L.G., Jr.; Li, H.; Harris, S.J. Direct calculation of Li-ion transport in the solid electrolyte interphase. J. Am. Chem. Soc. 2012, 134, 15476–15487. [Google Scholar] [CrossRef]
- Shi, S.; Qi, Y.; Li, H.; Hector, L.G., Jr. Defect thermodynamics and diffusion mechanisms in Li2CO3 and implications for the solid electrolyte interphase in Li-ion batteries. J. Phys. Chem. C 2013, 117, 8579–8593. [Google Scholar] [CrossRef]
- Philippe, B.; Dedryvère, R.m.; Gorgoi, M.; Rensmo, H.k.; Gonbeau, D.; Edström, K. Role of the LiPF6 salt for the long-term stability of silicon electrodes in Li-ion batteries—A photoelectron spectroscopy study. Chem. Mater. 2013, 25, 394–404. [Google Scholar]
- Seo, D.M.; Chalasani, D.; Parimalam, B.S.; Kadam, R.; Nie, M.; Lucht, B.L. Reduction reactions of carbonate solvents for lithium ion batteries. ECS Electrochem. Lett. 2014, 3, A91. [Google Scholar]
- Yoon, T.; Milien, M.S.; Parimalam, B.S.; Lucht, B.L. Thermal decomposition of the solid electrolyte interphase (SEI) on silicon electrodes for lithium ion batteries. Chem. Mater. 2017, 29, 3237–3245. [Google Scholar] [CrossRef]
- Parimalam, B.S.; MacIntosh, A.D.; Kadam, R.; Lucht, B.L. Decomposition reactions of anode solid electrolyte interphase (SEI) components with LiPF6. J. Phys. Chem. C 2017, 121, 22733–22738. [Google Scholar]
- Rezqita, A.; Kathribail, A.-R.; Kahr, J.; Jahn, M. Analysis of degradation of Si/Carbon||LiNi0.5Mn0.3Co0.2O2 full cells: Effect of prelithiation. J. Electrochem. Soc. 2019, 166, A5483. [Google Scholar]
- Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D. Challenges in the development of advanced Li-ion batteries: A review. Energy Environ. Sci. 2011, 4, 3243–3262. [Google Scholar]
- Ponrouch, A.; Marchante, E.; Courty, M.; Tarascon, J.-M.; Palacin, M.R. In search of an optimized electrolyte for Na-ion batteries. Energy Environ. Sci. 2012, 5, 8572–8583. [Google Scholar]
- Bhide, A.; Hofmann, J.; Dürr, A.K.; Janek, J.; Adelhelm, P. Electrochemical stability of non-aqueous electrolytes for sodium-ion batteries and their compatibility with Na0.7CoO2. Phys. Chem. Chem. Phys. 2014, 16, 1987–1998. [Google Scholar] [CrossRef]
- Shakourian-Fard, M.; Kamath, G.; Smith, K.; Xiong, H.; Sankaranarayanan, S.K. Trends in Na-ion solvation with alkyl-carbonate electrolytes for sodium-ion batteries: Insights from first-principles calculations. J. Phys. Chem. C 2015, 119, 22747–22759. [Google Scholar]
- Cresce, A.V.; Russell, S.M.; Borodin, O.; Allen, J.A.; Schroeder, M.A.; Dai, M.; Peng, J.; Gobet, M.P.; Greenbaum, S.G.; Rogers, R.E. Solvation behavior of carbonate-based electrolytes in sodium ion batteries. Phys. Chem. Chem. Phys. 2017, 19, 574–586. [Google Scholar]
- Xing, L.; Zheng, X.; Schroeder, M.; Alvarado, J.; von Wald Cresce, A.; Xu, K.; Li, Q.; Li, W. Deciphering the ethylene carbonate–propylene carbonate mystery in Li-ion batteries. Acc. Chem. Res. 2018, 51, 282–289. [Google Scholar]
- Li, K.; Zhang, J.; Lin, D.; Wang, D.-W.; Li, B.; Lv, W.; Sun, S.; He, Y.-B.; Kang, F.; Yang, Q.-H. Evolution of the electrochemical interface in sodium ion batteries with ether electrolytes. Nat. Commun. 2019, 10, 725. [Google Scholar]
- Paterno, D.; Rock, E.; Forbes, A.; Iqbal, R.; Mohammad, N.; Suarez, S. Aluminum ions speciation and transport in acidic deep eutectic AlCl3 amide electrolytes. J. Mol. Liq. 2020, 319, 114118. [Google Scholar]
- Paterno, D.; Suarez, S. Aluminum Ion Species Transport in Pure and Additive Modulated Deep Eutectic Solvents (DES) Electrolytes. ECS Trans. 2020, 98, 401. [Google Scholar]
- Rumble, C.A.; Kaintz, A.; Yadav, S.K.; Conway, B.; Araque, J.C.; Baker, G.A.; Margulis, C.; Maroncelli, M. Rotational dynamics in ionic liquids from NMR relaxation experiments and simulations: Benzene and 1-ethyl-3-methylimidazolium. J. Phys. Chem. B 2016, 120, 9450–9467. [Google Scholar] [PubMed]
- Lo Celso, F.; Appetecchi, G.B.; Simonetti, E.; Zhao, M.; Castner, E.W., Jr.; Keiderling, U.; Gontrani, L.; Triolo, A.; Russina, O. Microscopic structural and dynamic features in triphilic room temperature ionic liquids. Front. Chem. 2019, 7, 285. [Google Scholar] [PubMed]
- Zhao, M.; Wu, B.; Lall-Ramnarine, S.I.; Ramdihal, J.D.; Papacostas, K.A.; Fernandez, E.D.; Sumner, R.A.; Margulis, C.J.; Wishart, J.F.; Castner, E.W., Jr. Structural analysis of ionic liquids with symmetric and asymmetric fluorinated anions. J. Chem. Phys. 2019, 151, 074504. [Google Scholar]
- Alam, T.M.; Dreyer, D.R.; Bielwaski, C.W.; Ruoff, R.S. Measuring molecular dynamics and activation energies for quaternary acyclic ammonium and cyclic pyrrolidinium ionic liquids using 14N NMR spectroscopy. J. Phys. Chem. A 2011, 115, 4307–4316. [Google Scholar] [PubMed]
- Alam, T.M.; Dreyer, D.R.; Bielawski, C.W.; Ruoff, R.S. Combined measurement of translational and rotational diffusion in quaternary acyclic ammonium and cyclic pyrrolidinium ionic liquids. J. Phys. Chem. B 2013, 117, 1967–1977. [Google Scholar]
- Suarez, S.N.; Rúa, A.; Cuffari, D.; Pilar, K.; Hatcher, J.L.; Ramati, S.; Wishart, J.F. Do TFSA anions slither? Pressure exposes the role of TFSA conformational exchange in self-diffusion. J. Phys. Chem. B 2015, 119, 14756–14765. [Google Scholar]
- Yoshimura, Y.; Shigemi, M.; Takaku, M.; Yamamura, M.; Takekiyo, T.; Abe, H.; Hamaya, N.; Wakabayashi, D.; Nishida, K.; Funamori, N. Stability of the liquid state of imidazolium-based ionic liquids under high pressure at room temperature. J. Phys. Chem. B 2015, 119, 8146–8153. [Google Scholar]
- Harris, K.R.; Kanakubo, M. Self-diffusion, velocity cross-correlation, distinct diffusion and resistance coefficients of the ionic liquid [BMIM][Tf2N] at high pressure. Phys. Chem. Chem. Phys. 2015, 17, 23977–23993. [Google Scholar]
- Nazet, A.; Sokolov, S.; Sonnleitner, T.; Makino, T.; Kanakubo, M.; Buchner, R. Densities, viscosities, and conductivities of the imidazolium ionic liquids [Emim][Ac], [Emim][FAP], [Bmim][BETI], [Bmim][FSI], [Hmim][TFSI], and [Omim][TFSI]. J. Chem. Eng. Data 2015, 60, 2400–2411. [Google Scholar]
- Pilar, K.; Rua, A.; Suarez, S.N.; Mallia, C.; Lai, S.; Jayakody, J.; Hatcher, J.L.; Wishart, J.F.; Greenbaum, S. Investigation of dynamics in BMIM TFSA ionic liquid through variable temperature and pressure NMR relaxometry and diffusometry. J. Electrochem. Soc. 2017, 164, H5189. [Google Scholar] [CrossRef]
- Shimizu, K.; Freitas, A.A.; Lopes, J.N.C. Structural characterization of the [CnC1im][C4F9SO3] ionic liquid series: Alkyl versus perfluoroalkyl side chains. J. Mol. Liq. 2017, 226, 28–34. [Google Scholar]
- Brooks, N.J.; Castiglione, F.; Doherty, C.M.; Dolan, A.; Hill, A.J.; Hunt, P.A.; Matthews, R.P.; Mauri, M.; Mele, A.; Simonutti, R. Linking the structures, free volumes, and properties of ionic liquid mixtures. Chem. Sci. 2017, 8, 6359–6374. [Google Scholar]
- Harris, K.R.; Kanakubo, M.; Kodama, D.; Makino, T.; Mizuguchi, Y.; Watanabe, M.; Watanabe, T. Temperature and density dependence of the transport properties of the ionic liquid triethylpentylphosphonium bis(trifluoromethanesulfonyl)amide, [P222, 5][Tf2N]. J. Chem. Eng. Data 2018, 63, 2015–2027. [Google Scholar]
- Bagh, F.S.G.; Shahbaz, K.; Mjalli, F.S.; Hashim, M.A.; AlNashef, I.M. Zinc (II) chloride-based deep eutectic solvents for application as electrolytes: Preparation and characterization. J. Mol. Liq. 2015, 204, 76–83. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, R.; Meng, X.; Liu, H.; Xu, C.; Liu, Z. Structural and spectroscopic characterizations of amide–AlCl3-based ionic liquid analogues. Inorg. Chem. 2016, 55, 2374–2380. [Google Scholar]
- Zhang, Y.; Han, J.; Liao, C. Insights into the properties of deep eutectic solvent based on reline for Ga-controllable CIGS solar cell in one-step electrodeposition. J. Electrochem. Soc. 2016, 163, D689. [Google Scholar]
- Angell, M.; Pan, C.-J.; Rong, Y.; Yuan, C.; Lin, M.-C.; Hwang, B.-J.; Dai, H. High Coulombic efficiency aluminum-ion battery using an AlCl3-urea ionic liquid analog electrolyte. Proc. Natl. Acad. Sci. USA 2017, 114, 834–839. [Google Scholar]
- Liu, C.; Chen, W.; Wu, Z.; Gao, B.; Hu, X.; Shi, Z.; Wang, Z. Density, viscosity and electrical conductivity of AlCl3-amide ionic liquid analogues. J. Mol. Liq. 2017, 247, 57–63. [Google Scholar]
- Tome, L.I.; Baiao, V.; da Silva, W.; Brett, C.M. Deep eutectic solvents for the production and application of new materials. Appl. Mater. Today 2018, 10, 30–50. [Google Scholar]
- Garaga, M.N.; Persson, M.; Yaghini, N.; Martinelli, A. Local coordination and dynamics of a protic ammonium based ionic liquid immobilized in nano-porous silica micro-particles probed by Raman and NMR spectroscopy. Soft Matter 2016, 12, 2583–2592. [Google Scholar]
- Guyomard-Lack, A.; Said, B.; Dupré, N.; Galarneau, A.; Le Bideau, J. Enhancement of lithium transport by controlling the mesoporosity of silica monoliths filled by ionic liquids. New J. Chem. 2016, 40, 4269–4276. [Google Scholar]
- Garaga, M.N.; Aguilera, L.; Yaghini, N.; Matic, A.; Persson, M.; Martinelli, A. Achieving enhanced ionic mobility in nanoporous silica by controlled surface interactions. Phys. Chem. Chem. Phys. 2017, 19, 5727–5736. [Google Scholar] [PubMed]
- Ashby, D.S.; DeBlock, R.H.; Lai, C.-H.; Choi, C.S.; Dunn, B.S. Patternable, solution-processed ionogels for thin-film lithium-ion electrolytes. Joule 2017, 1, 344–358. [Google Scholar]
- Chen, N.; Zhang, H.; Li, L.; Chen, R.; Guo, S. Ionogel electrolytes for high-performance lithium batteries: A review. Adv. Energy Mater. 2018, 8, 1702675. [Google Scholar]
- Jayakody, N.K.; Fraenza, C.C.; Greenbaum, S.G.; Ashby, D.; Dunn, B.S. NMR relaxometry and diffusometry analysis of dynamics in ionic liquids and ionogels for use in lithium-ion batteries. J. Phys. Chem. B 2020, 124, 6843–6856. [Google Scholar]
- Liang, S.; Yan, W.; Wu, X.; Zhang, Y.; Zhu, Y.; Wang, H.; Wu, Y. Gel polymer electrolytes for lithium ion batteries: Fabrication, characterization and performance. Solid State Ion. 2018, 318, 2–18. [Google Scholar]
- Cheng, X.; Pan, J.; Zhao, Y.; Liao, M.; Peng, H. Gel polymer electrolytes for electrochemical energy storage. Adv. Energy Mater. 2018, 8, 1702184. [Google Scholar]
- Richardson, P.; Voice, A.; Ward, I. NMR self diffusion and relaxation time measurements for poly (vinylidene fluoride)(PVDF) based polymer gel electrolytes containing LiBF4 and propylene carbonate. Polymer 2016, 97, 69–79. [Google Scholar]
- Woo, H.-S.; Son, H.; Min, J.-Y.; Rhee, J.; Lee, H.-T.; Kim, D.-W. Ionic liquid-based gel polymer electrolyte containing zwitterion for lithium-oxygen batteries. Electrochim. Acta 2020, 345, 136248. [Google Scholar]
- Tikekar, M.D.; Archer, L.A.; Koch, D.L. Stabilizing electrodeposition in elastic solid electrolytes containing immobilized anions. Sci. Adv. 2016, 2, e1600320. [Google Scholar]
- Yue, L.; Ma, J.; Zhang, J.; Zhao, J.; Dong, S.; Liu, Z.; Cui, G.; Chen, L. All solid-state polymer electrolytes for high-performance lithium ion batteries. Energy Storage Mater. 2016, 5, 139–164. [Google Scholar]
- Mauger, A.; Armand, M.; Julien, C.; Zaghib, K. Challenges and issues facing lithium metal for solid-state rechargeable batteries. J. Power Source 2017, 353, 333–342. [Google Scholar]
- Famprikis, T.; Canepa, P.; Dawson, J.A.; Islam, M.S.; Masquelier, C. Fundamentals of inorganic solid-state electrolytes for batteries. Nat. Mater. 2019, 18, 1278–1291. [Google Scholar]
- Kato, Y.; Hori, S.; Saito, T.; Suzuki, K.; Hirayama, M.; Mitsui, A.; Yonemura, M.; Iba, H.; Kanno, R. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 2016, 1, 16030. [Google Scholar]
- Wang, W.; Fang, Z.; Zhao, M.; Peng, Y.; Zhang, J.; Guan, S. Solid polymer electrolytes based on the composite of PEO–LiFSI and organic ionic plastic crystal. Chem. Phys. Lett. 2020, 747, 137335. [Google Scholar]
- Cai, D.; Wang, D.; Chen, Y.; Zhang, S.; Wang, X.; Xia, X.; Tu, J. A highly ion-conductive three-dimensional LLZAO-PEO/LiTFSI solid electrolyte for high-performance solid-state batteries. Chem. Eng. J. 2020, 394, 124993. [Google Scholar]
- Sengwa, R.; Dhatarwal, P. Predominantly chain segmental relaxation dependent ionic conductivity of multiphase semicrystalline PVDF/PEO/LiClO4 solid polymer electrolytes. Electrochim. Acta 2020, 338, 135890. [Google Scholar]
- Zhou, Q.; Li, Q.; Liu, S.; Yin, X.; Huang, B.; Sheng, M. High Li-ion conductive composite polymer electrolytes for all-solid-state Li-metal batteries. J. Power Source 2021, 482, 228929. [Google Scholar]
- Polu, A.R.; Singh, P.K. Improved ion dissociation and amorphous region of PEO based solid polymer electrolyte by incorporating tetracyanoethylene. Mater. Today Proc. 2022, 49, 3093–3097. [Google Scholar] [CrossRef]
- Ngai, K.S.; Ramesh, S.; Ramesh, K.; Juan, J.C. A review of polymer electrolytes: Fundamental, approaches and applications. Ionics 2016, 22, 1259–1279. [Google Scholar]
- Mindemark, J.; Lacey, M.J.; Bowden, T.; Brandell, D. Beyond PEO—Alternative host materials for Li+-conducting solid polymer electrolytes. Prog. Polym. Sci. 2018, 81, 114–143. [Google Scholar]
- Xue, Z.; He, D.; Xie, X. Poly (ethylene oxide)-based electrolytes for lithium-ion batteries. J. Mater. Chem. A 2015, 3, 19218–19253. [Google Scholar]
- Brooks, D.J.; Merinov, B.V.; Goddard, W.A., III; Kozinsky, B.; Mailoa, J. Atomistic description of ionic diffusion in PEO–LiTFSI: Effect of temperature, molecular weight, and ionic concentration. Macromolecules 2018, 51, 8987–8995. [Google Scholar] [CrossRef]
- Mongcopa, K.I.S.; Tyagi, M.; Mailoa, J.P.; Samsonidze, G.; Kozinsky, B.; Mullin, S.A.; Gribble, D.A.; Watanabe, H.; Balsara, N.P. Relationship between segmental dynamics measured by quasi-elastic neutron scattering and conductivity in polymer electrolytes. ACS Macro Lett. 2018, 7, 504–508. [Google Scholar]
- Chen, F.; Pringle, J.M.; Forsyth, M. Insights into the transport of alkali metal ions doped into a plastic crystal electrolyte. Chem. Mater. 2015, 27, 2666–2672. [Google Scholar]
- Lin, D.; Liu, W.; Liu, Y.; Lee, H.R.; Hsu, P.-C.; Liu, K.; Cui, Y. High ionic conductivity of composite solid polymer electrolyte via in situ synthesis of monodispersed SiO2 nanospheres in poly (ethylene oxide). Nano Lett. 2016, 16, 459–465. [Google Scholar] [CrossRef]
- Fu, X.; Liu, Y.; Wang, W.; Han, L.; Yang, J.; Ge, M.; Yao, Y.; Liu, H. Probing the fast lithium-ion transport in small-molecule solid polymer electrolytes by solid-state NMR. Macromolecules 2020, 53, 10078–10085. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Cui, Z.; Shangguan, X.; Zhang, H.; Zhang, J.; Tang, K.; Li, L.; Zhou, X.; Cui, G. A fluorinated polycarbonate based all solid state polymer electrolyte for lithium metal batteries. Electrochim. Acta 2020, 337, 135843. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Oxford University Press: Oxford, UK, 1961. [Google Scholar]
- Mason, J. Multinuclear NMR; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Slichter, C.P. Principles of Magnetic Resonance; Springer: Berlin/Heidelberg, Germany, 1996; Volume 1. [Google Scholar]
- Kimmich, R. NMR: Tomography, Diffusometry, Relaxometry; Springer: Berlin/Heidelberg, Germany, 1997. [Google Scholar]
- Kowalewski, J.; Maler, L. Nuclear Spin Relaxation in Liquids: Theory, Experiments, and Applications; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Levitt, M.H. Spin Dynamics: Basics of Nuclear Magnetic Resonance; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Price, W.S. NMR Studies of Translational Motion: Principles and Applications; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Chiappe, C.; Sanzone, A.; Mendola, D.; Castiglione, F.; Famulari, A.; Raos, G.; Mele, A. Pyrazolium-versus imidazolium-based ionic liquids: Structure, dynamics and physicochemical properties. J. Phys. Chem. B 2013, 117, 668–676. [Google Scholar] [CrossRef]
- Castiglione, F.; Famulari, A.; Raos, G.; Meille, S.V.; Mele, A.; Appetecchi, G.B.; Passerini, S. Pyrrolidinium-based ionic liquids doped with lithium salts: How does Li+ coordination affect its diffusivity? J. Phys. Chem. B 2014, 118, 13679–13688. [Google Scholar] [CrossRef]
- Khatun, S.; Castner, E.W., Jr. Ionic liquid–solute interactions studied by 2D NOE NMR spectroscopy. J. Phys. Chem. B 2015, 119, 9225–9235. [Google Scholar] [CrossRef]
- Di Pietro, M.E.; Castiglione, F.; Mele, A. Anions as dynamic probes for ionic liquid mixtures. J. Phys. Chem. B 2020, 124, 2879–2891. [Google Scholar] [CrossRef] [PubMed]
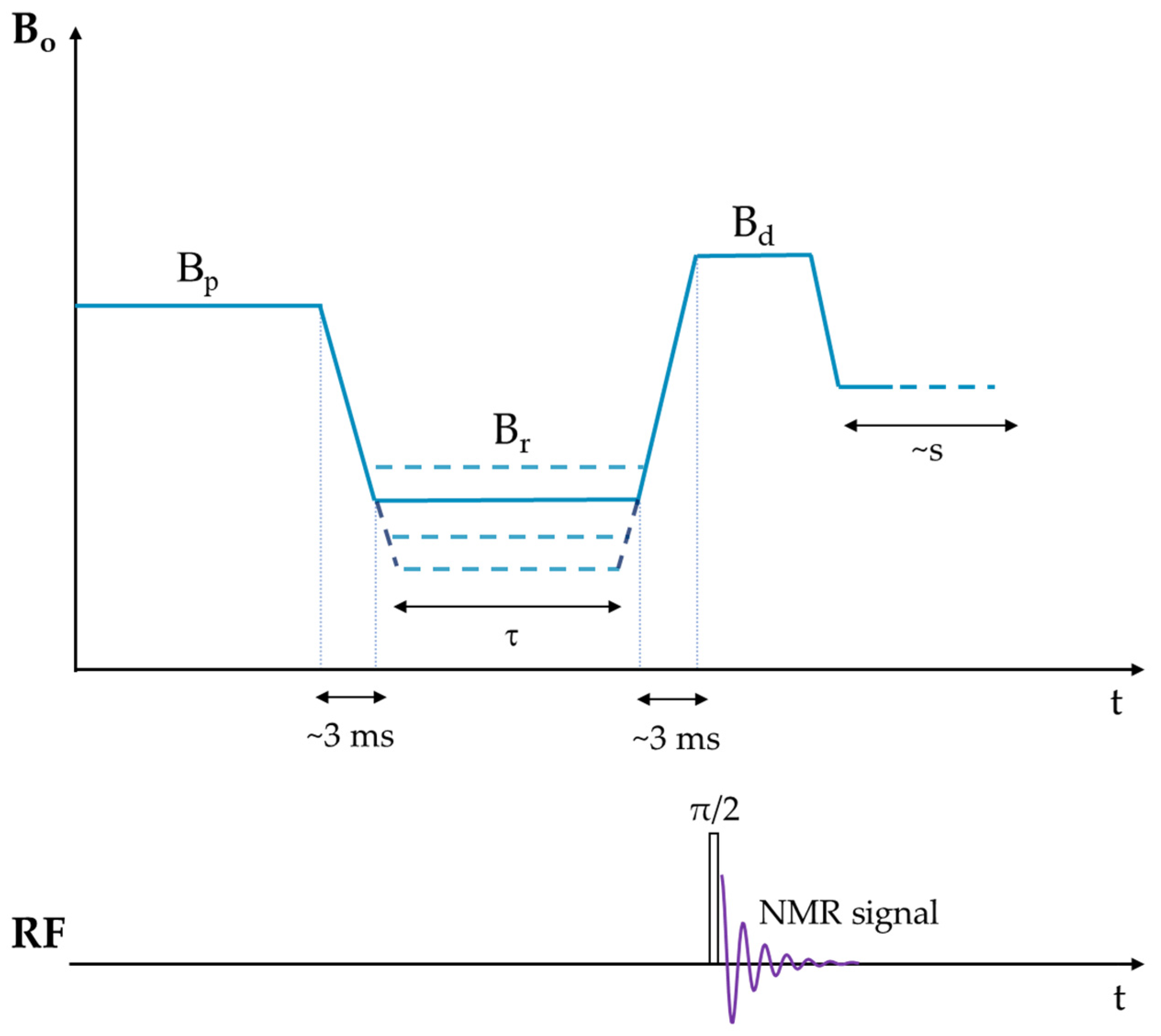
- Kimmich, R.; Anoardo, E. Field-cycling NMR relaxometry. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 257–320. [Google Scholar] [CrossRef]
- Kimmich, R. Field-Cycling NMR Relaxometry: Instrumentation, Model Theories and Applications; Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Yasaka, Y.; Kimura, Y. Polarity and nonpolarity of ionic liquids viewed from the rotational dynamics of carbon monoxide. J. Phys. Chem. B 2015, 119, 15493–15501. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Sumida, H.; Fujii, K.; Takahashi, K.; Kimura, Y. Heterogeneous Structures of Ionic Liquids as Probed by CO Rotation with Nuclear Magnetic Resonance Relaxation Analysis and Molecular Dynamics Simulations. J. Phys. Chem. B 2020, 124, 10465–10476. [Google Scholar] [CrossRef] [PubMed]
- Strate, A.; Neumann, J.; Overbeck, V.; Bonsa, A.-M.; Michalik, D.; Paschek, D.; Ludwig, R. Rotational and translational dynamics and their relation to hydrogen bond lifetimes in an ionic liquid by means of NMR relaxation time experiments and molecular dynamics simulation. J. Chem. Phys. 2018, 148, 193843. [Google Scholar] [CrossRef]
- Kruk, D.; Meier, R.; Rachocki, A.; Korpała, A.; Singh, R.; Rössler, E. Determining diffusion coefficients of ionic liquids by means of field cycling nuclear magnetic resonance relaxometry. J. Chem. Phys. 2014, 140, 244509. [Google Scholar] [CrossRef]
- Seyedlar, A.O.; Stapf, S.; Mattea, C. Dynamics of the ionic liquid 1-butyl-3-methylimidazolium bis (trifluoromethylsulphonyl) imide studied by nuclear magnetic resonance dispersion and diffusion. Phys. Chem. Chem. Phys. 2015, 17, 1653–1659. [Google Scholar] [CrossRef]
- Kruk, D.; Wojciechowski, M.; Brym, S.; Singh, R.K. Dynamics of ionic liquids in bulk and in confinement by means of 1 H NMR relaxometry–BMIM-OcSO4 in an SiO2 matrix as an example. Phys. Chem. Chem. Phys. 2016, 18, 23184–23194. [Google Scholar] [CrossRef] [PubMed]
- Wencka, M.; Apih, T.; Korošec, R.C.; Jenczyk, J.; Jarek, M.; Szutkowski, K.; Jurga, S.; Dolinšek, J. Molecular dynamics of 1-ethyl-3-methylimidazolium triflate ionic liquid studied by 1 H and 19 F nuclear magnetic resonances. Phys. Chem. Chem. Phys. 2017, 19, 15368–15376. [Google Scholar] [CrossRef] [PubMed]
- Beira, M.; Daniel, C.I.; Almeida, P.L.; Corvo, M.C.; Rosatella, A.A.; Afonso, C.A.; Sebastião, P.J. 1H NMR Relaxometry and Diffusometry Study of Magnetic and Nonmagnetic Ionic Liquid-Based Solutions: Cosolvent and Temperature Effects. J. Phys. Chem. B 2017, 121, 11472–11484. [Google Scholar] [CrossRef]
- Kruk, D.; Wojciechowski, M.; Florek-Wojciechowska, M.; Singh, R.K. Dynamics of Ionic Liquids in Confinement by Means of NMR Relaxometry—EMIM-FSI in a Silica Matrix as an Example. Materials 2020, 13, 4351. [Google Scholar] [CrossRef]
- Overbeck, V.; Schröder, H.; Bonsa, A.-M.; Neymeyr, K.; Ludwig, R. Insights into the translational and rotational dynamics of cations and anions in protic ionic liquids by means of NMR fast-field-cycling relaxometry. Phys. Chem. Chem. Phys. 2021, 23, 2663–2675. [Google Scholar] [CrossRef]
- Garaga, M.N.; Jayakody, N.; Fraenza, C.C.; Itin, B.; Greenbaum, S. Molecular-level insights into structure and dynamics in ionic liquids and polymer gel electrolytes. J. Mol. Liq. 2021, 329, 115454. [Google Scholar] [CrossRef]
- Overbeck, V.; Appelhagen, A.; Rößler, R.; Niemann, T.; Ludwig, R. Rotational correlation times, diffusion coefficients and quadrupolar peaks of the protic ionic liquid ethylammonium nitrate by means of 1H fast field cycling NMR relaxometry. J. Mol. Liq. 2021, 322, 114983. [Google Scholar] [CrossRef]
- Kruk, D.; Masiewicz, E.; Lotarska, S.; Markiewicz, R.; Jurga, S. Relationship between Translational and Rotational Dynamics of Alkyltriethylammonium-Based Ionic Liquids. Int. J. Mol. Sci. 2022, 23, 1688. [Google Scholar] [CrossRef]
- Silva, G.M.; Beira, M.J.; Morgado, P.; Branco, L.C.; Sebastião, P.J.; Lopes, J.N.C.; Filipe, E.J. Ionic liquids with hydrogenated and perfluorinated chains: Structural study of the [P6, 6, 6, 14][FnCOO] n = 7, 9, 11. Checking the existence of polar–hydrogenated–perfluorinated triphilic continuity. J. Mol. Liq. 2022, 367, 120506. [Google Scholar] [CrossRef]
- Hwang, L.P.; Freed, J.H. Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. J. Chem. Phys. 1975, 63, 4017–4025. [Google Scholar] [CrossRef]
- Ayant, Y.; Belorizky, E.; Aluzon, J.; Gallice, J. Calcul des densités spectrales résultant d’un mouvement aléatoire de translation en relaxation par interaction dipolaire magnétique dans les liquides. J. Phys. 1975, 36, 991–1004. [Google Scholar] [CrossRef]
- Sholl, C. Nuclear spin relaxation by translational diffusion in liquids and solids: High-and low-frequency limits. J. Phys. C Solid State Phys. 1981, 14, 447. [Google Scholar] [CrossRef]
- Honegger, P.; Overbeck, V.; Strate, A.; Appelhagen, A.; Sappl, M.; Heid, E.; Schröder, C.; Ludwig, R.; Steinhauser, O. Understanding the nature of nuclear magnetic resonance relaxation by means of fast-field-cycling relaxometry and molecular dynamics simulations—The validity of relaxation models. J. Phys. Chem. Lett. 2020, 11, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Overbeck, V.; Golub, B.; Schroeder, H.; Appelhagen, A.; Paschek, D.; Neymeyr, K.; Ludwig, R. Probing relaxation models by means of Fast Field-Cycling relaxometry, NMR spectroscopy and molecular dynamics simulations: Detailed insight into the translational and rotational dynamics of a protic ionic liquid. J. Mol. Liq. 2020, 319, 114207. [Google Scholar] [CrossRef]
- Torrey, H.C. Nuclear spin relaxation by translational diffusion. Phys. Rev. 1953, 92, 962. [Google Scholar] [CrossRef]
- Freed, J.H. Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. II. Finite jumps and independent T 1 processes. J. Chem. Phys. 1978, 68, 4034–4037. [Google Scholar] [CrossRef]
- Kaszyńska, J.; Rachocki, A.; Bielejewski, M.; Tritt-Goc, J. Influence of cellulose gel matrix on BMIMCl ionic liquid dynamics and conductivity. Cellulose 2017, 24, 1641–1655. [Google Scholar] [CrossRef]
- Woessner, D. Spin relaxation processes in a two-proton system undergoing anisotropic reorientation. J. Chem. Phys. 1962, 36, 1–4. [Google Scholar] [CrossRef]
- Rachocki, A.; Andrzejewska, E.; Dembna, A.; Tritt-Goc, J. Translational dynamics of ionic liquid imidazolium cations at solid/liquid interface in gel polymer electrolyte. Eur. Polym. J. 2015, 71, 210–220. [Google Scholar] [CrossRef]
- Alfurayj, I.; Fraenza, C.C.; Zhang, Y.; Pandian, R.; Spittle, S.; Hansen, B.; Dean, W.; Gurkan, B.; Savinell, R.; Greenbaum, S. Solvation Dynamics of Wet Ethaline: Water is the Magic Component. J. Phys. Chem. B 2021, 125, 8888–8901. [Google Scholar] [CrossRef] [PubMed]
- Triolo, A.; Di Pietro, M.E.; Mele, A.; Lo Celso, F.; Brehm, M.; Di Lisio, V.; Martinelli, A.; Chater, P.; Russina, O. Liquid structure and dynamics in the choline acetate: Urea 1: 2 deep eutectic solvent. J. Chem. Phys. 2021, 154, 244501. [Google Scholar] [CrossRef] [PubMed]
- Fraenza, C.C.; Elgammal, R.A.; Garaga, M.N.; Bhattacharyya, S.; Zawodzinski, T.A.; Greenbaum, S.G. Dynamics of glyceline and interactions of constituents: A multitechnique NMR study. J. Phys. Chem. B 2022, 126, 890–905. [Google Scholar] [CrossRef]
- de Araujo Lima e Souza, G.; Di Pietro, M.E.; Castiglione, F.; Vanoli, V.; Mele, A. Insights into the Effect of Lithium Doping on the Deep Eutectic Solvent Choline Chloride: Urea. Materials 2022, 15, 7459. [Google Scholar] [CrossRef]
- Di Pietro, M.E.; Goloviznina, K.; van den Bruinhorst, A.; de Araujo Lima e Souza, G.; Costa Gomes, M.; Padua, A.A.; Mele, A. Lithium Salt Effects on the Liquid Structure of Choline Chloride–Urea Deep Eutectic Solvent. ACS Sustain. Chem. Eng. 2022, 10, 11835–11845. [Google Scholar] [CrossRef]
- Peng, J.; Gobet, M.; Devany, M.; Xu, K.; von Wald Cresce, A.; Borodin, O.; Greenbaum, S. Multinuclear magnetic resonance investigation of cation-anion and anion-solvent interactions in carbonate electrolytes. J. Power Source 2018, 399, 215–222. [Google Scholar] [CrossRef]
- Chen, Y.; Jaegers, N.R.; Wang, H.; Han, K.S.; Hu, J.Z.; Mueller, K.T.; Murugesan, V. Role of Solvent Rearrangement on Mg2+ Solvation Structures in Dimethoxyethane Solutions using Multimodal NMR Analysis. J. Phys. Chem. Lett. 2020, 11, 6443–6449. [Google Scholar] [CrossRef]
- Salama, M.; Shterenberg, I.; Gizbar, H.; Eliaz, N.N.; Kosa, M.; Keinan-Adamsky, K.; Afri, M.; Shimon, L.J.; Gottlieb, H.E.; Major, D.T. Unique behavior of dimethoxyethane (DME)/Mg(N(SO2CF3)2)2 solutions. J. Phys. Chem. C 2016, 120, 19586–19594. [Google Scholar] [CrossRef]
- Kubisiak, P.; Eilmes, A. Solvation of Mg2+ Ions in Mg(TFSI)2–dimethoxyethane electrolytes—A view from molecular dynamics simulations. J. Phys. Chem. C 2018, 122, 12615–12622. [Google Scholar] [CrossRef]
- Self, J.; Hahn, N.T.; Fong, K.D.; McClary, S.A.; Zavadil, K.R.; Persson, K.A. Ion pairing and redissociaton in low-permittivity electrolytes for multivalent battery applications. J. Phys. Chem. Lett. 2020, 11, 2046–2052. [Google Scholar] [CrossRef]
- Patel, M.U.; Demir-Cakan, R.; Morcrette, M.; Tarascon, J.M.; Gaberscek, M.; Dominko, R. Li-S Battery Analyzed by UV/Vis in Operando Mode. ChemSusChem 2013, 6, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Lowe, M.A.; Gao, J.; Abruña, H.D. Mechanistic insights into operational lithium–sulfur batteries by in situ X-ray diffraction and absorption spectroscopy. RSC Adv. 2014, 4, 18347–18353. [Google Scholar] [CrossRef]
- See, K.A.; Leskes, M.; Griffin, J.M.; Britto, S.; Matthews, P.D.; Emly, A.; Van der Ven, A.; Wright, D.S.; Morris, A.J.; Grey, C.P. Ab initio structure search and in situ 7Li NMR studies of discharge products in the Li–S battery system. J. Am. Chem. Soc. 2014, 136, 16368–16377. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-S.; Fu, Y.; Cochell, T.; Manthiram, A. A strategic approach to recharging lithium-sulphur batteries for long cycle life. Nat. Commun. 2013, 4, 2985. [Google Scholar] [CrossRef]
- Cuisinier, M.; Cabelguen, P.-E.; Adams, B.; Garsuch, A.; Balasubramanian, M.; Nazar, L. Unique behaviour of nonsolvents for polysulphides in lithium–sulphur batteries. Energy Environ. Sci. 2014, 7, 2697–2705. [Google Scholar] [CrossRef]
- Yamada, Y.; Furukawa, K.; Sodeyama, K.; Kikuchi, K.; Yaegashi, M.; Tateyama, Y.; Yamada, A. Unusual stability of acetonitrile-based superconcentrated electrolytes for fast-charging lithium-ion batteries. J. Am. Chem. Soc. 2014, 136, 5039–5046. [Google Scholar] [CrossRef]
- See, K.A.; Wu, H.-L.; Lau, K.C.; Shin, M.; Cheng, L.; Balasubramanian, M.; Gallagher, K.G.; Curtiss, L.A.; Gewirth, A.A. Effect of hydrofluoroether cosolvent addition on Li solvation in acetonitrile-based solvate electrolytes and its influence on S reduction in a Li–S battery. ACS Appl. Mater. Interfaces 2016, 8, 34360–34371. [Google Scholar] [CrossRef]
- Cheng, L.; Curtiss, L.A.; Zavadil, K.R.; Gewirth, A.A.; Shao, Y.; Gallagher, K.G. Sparingly solvating electrolytes for high energy density lithium–sulfur batteries. ACS Energy Lett. 2016, 1, 503–509. [Google Scholar] [CrossRef]
- Lee, C.-W.; Pang, Q.; Ha, S.; Cheng, L.; Han, S.-D.; Zavadil, K.R.; Gallagher, K.G.; Nazar, L.F.; Balasubramanian, M. Directing the lithium–sulfur reaction pathway via sparingly solvating electrolytes for high energy density batteries. ACS Cent. Sci. 2017, 3, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.; Wu, H.-L.; Narayanan, B.; See, K.A.; Assary, R.S.; Zhu, L.; Haasch, R.T.; Zhang, S.; Zhang, Z.; Curtiss, L.A. Effect of the hydrofluoroether cosolvent structure in acetonitrile-based solvate electrolytes on the Li+ solvation structure and Li–S battery performance. ACS Appl. Mater. Interfaces 2017, 9, 39357–39370. [Google Scholar] [CrossRef] [PubMed]
- Bielejewski, M.; Puszkarska, A.; Tritt-Goc, J. Thermal properties, conductivity, and spin-lattice relaxation of gel electrolyte based on low molecular weight gelator and solution of high temperature ionic liquid. Electrochim. Acta 2015, 165, 122–129. [Google Scholar] [CrossRef]
- Kruk, D.; Wojciechowski, M.; Verma, Y.L.; Chaurasia, S.K.; Singh, R.K. Dynamical properties of EMIM-SCN confined in a SiO2 matrix by means of 1 H NMR relaxometry. Phys. Chem. Chem. Phys. 2017, 19, 32605–32616. [Google Scholar] [CrossRef]
- Ordikhani Seyedlar, A.; Stapf, S.; Mattea, C. Nuclear magnetic relaxation and diffusion study of the ionic liquids 1-ethyl-and 1-butyl-3-methylimidazolium bis (trifluoromethylsulfonyl) imide confined in porous glass. Magn. Reson. Chem. 2019, 57, 818–828. [Google Scholar] [CrossRef]
- Kowalczuk, J.; Bielejewski, M.; Tritt-Goc, J. Ionic liquid dynamics and electrical conductivity under confinement within micro and nanocellulose ionogels. Cellulose 2023, 30, 3551–3567. [Google Scholar] [CrossRef]
- Bielejewski, M.; Rachocki, A.; Kaszyńska, J.; Tritt-Goc, J. The gelation influence on diffusion and conductivity enhancement effect in renewable ionic gels based on a LMWG. Phys. Chem. Chem. Phys. 2018, 20, 5803–5817. [Google Scholar] [CrossRef]
- Zavada, T.; Kimmich, R. The anomalous adsorbate dynamics at surfaces in porous media studied by nuclear magnetic resonance methods. The orientational structure factor and Lévy walks. J. Chem. Phys. 1998, 109, 6929–6939. [Google Scholar] [CrossRef]
- Kim, G.-T.; Appetecchi, G.B.; Alessandrini, F.; Passerini, S. Solvent-free, PYR1ATFSI ionic liquid-based ternary polymer electrolyte systems: I. Electrochemical characterization. J. Power Source 2007, 171, 861–869. [Google Scholar] [CrossRef]
- Joost, M.; Kunze, M.; Jeong, S.; Schönhoff, M.; Winter, M.; Passerini, S. Ionic mobility in ternary polymer electrolytes for lithium-ion batteries. Electrochim. Acta 2012, 86, 330–338. [Google Scholar] [CrossRef]
- Li, M.; Yang, B.; Wang, L.; Zhang, Y.; Zhang, Z.; Fang, S.; Zhang, Z. New polymerized ionic liquid (PIL) gel electrolyte membranes based on tetraalkylammonium cations for lithium ion batteries. J. Membr. Sci. 2013, 447, 222–227. [Google Scholar] [CrossRef]
- Gouverneur, M.; Jeremias, S.; Schönhoff, M. 7Li nuclear magnetic resonance studies of dynamics in a ternary gel polymer electrolyte based on polymeric ionic liquids. Electrochim. Acta 2015, 175, 35–41. [Google Scholar] [CrossRef]
- Shaplov, A.S.; Marcilla, R.; Mecerreyes, D. Recent advances in innovative polymer electrolytes based on poly (ionic liquid) s. Electrochim. Acta 2015, 175, 18–34. [Google Scholar] [CrossRef]
- Bhandary, R.; Schoenhoff, M. Polymer effect on lithium ion dynamics in gel polymer electrolytes: Cationic versus acrylate polymer. Electrochim. Acta 2015, 174, 753–761. [Google Scholar] [CrossRef]
- Shaplov, A.; Ponkratov, D.; Vygodskii, Y.S. Poly (ionic liquid)s: Synthesis, properties, and application. Polym. Sci. Ser. B 2016, 58, 73–142. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, H.; Girard, G.M.; Yunis, R.; MacFarlane, D.R.; Mecerreyes, D.; Bhattacharyya, A.J.; Howlett, P.C.; Forsyth, M. Preparation and characterization of gel polymer electrolytes using poly (ionic liquids) and high lithium salt concentration ionic liquids. J. Mater. Chem. A 2017, 5, 23844–23852. [Google Scholar] [CrossRef]
- Kerner, M.; Johansson, P. Pyrrolidinium FSI and TFSI-based polymerized ionic liquids as electrolytes for high-temperature lithium-ion batteries. Batteries 2018, 4, 10. [Google Scholar] [CrossRef]
- Brinkkötter, M.; Gouverneur, M.; Sebastião, P.; Chávez, F.V.; Schönhoff, M. Spin relaxation studies of Li+ ion dynamics in polymer gel electrolytes. Phys. Chem. Chem. Phys. 2017, 19, 7390–7398. [Google Scholar] [CrossRef]
- Brinkkötter, M.; Lozinskaya, E.I.; Ponkratov, D.O.; Vygodskii, Y.; Schmidt, D.F.; Shaplov, A.S.; Schönhoff, M. Influence of cationic poly (ionic liquid) architecture on the ion dynamics in polymer gel electrolytes. J. Phys. Chem. C 2019, 123, 13225–13235. [Google Scholar] [CrossRef]
- Tominaga, Y.; Yamazaki, K. Fast Li-ion conduction in poly (ethylene carbonate)-based electrolytes and composites filled with TiO2 nanoparticles. Chem. Commun. 2014, 50, 4448–4450. [Google Scholar] [CrossRef]
- Lago, N.; Garcia-Calvo, O.; Lopez del Amo, J.M.; Rojo, T.; Armand, M. All-solid-state lithium-ion batteries with grafted ceramic nanoparticles dispersed in solid polymer electrolytes. ChemSusChem 2015, 8, 3039–3043. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Ren, C.; Luo, F.; Ma, Q.; Hu, Y.-S.; Zhou, Z.; Li, H.; Huang, X.; Chen, L. A ceramic/polymer composite solid electrolyte for sodium batteries. J. Mater. Chem. A 2016, 4, 15823–15828. [Google Scholar] [CrossRef]
- Xuefu, S.; Nemori, H.; Mitsuoka, S.; Xu, P.; Matsui, M.; Takeda, Y.; Yamamoto, O.; Imanishi, N. High Lithium-Ion-Conducting NASICON-Type Li1+xAlxGeyTi2−x−y (PO4)3 Solid Electrolyte. Front. Energy Res. 2016, 4, 12. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z.; Feng, Y.; Tan, R.; Zuo, Y.; Gao, R.; Zhao, Y.; Han, L.; Wang, Z.; Pan, F. Flexible composite solid electrolyte facilitating highly stable “soft contacting” Li–electrolyte interface for solid state lithium-ion batteries. Adv. Energy Mater. 2017, 7, 1701437. [Google Scholar] [CrossRef]
- Peng, J.; Xiao, Y.; Clarkson, D.A.; Greenbaum, S.G.; Zawodzinski, T.A.; Chen, X.C. A Nuclear magnetic resonance study of cation and anion dynamics in polymer–ceramic composite solid electrolytes. ACS Appl. Polym. Mater. 2020, 2, 1180–1189. [Google Scholar] [CrossRef]
- Ding, C.; Nohira, T.; Kuroda, K.; Hagiwara, R.; Fukunaga, A.; Sakai, S.; Nitta, K.; Inazawa, S. NaFSA–C1C3pyrFSA ionic liquids for sodium secondary battery operating over a wide temperature range. J. Power Source 2013, 238, 296–300. [Google Scholar] [CrossRef]
- Noor, S.A.M.; Howlett, P.C.; MacFarlane, D.R.; Forsyth, M. Properties of sodium-based ionic liquid electrolytes for sodium secondary battery applications. Electrochim. Acta 2013, 114, 766–771. [Google Scholar] [CrossRef]
- Monti, D.; Jónsson, E.; Palacín, M.R.; Johansson, P. Ionic liquid based electrolytes for sodium-ion batteries: Na+ solvation and ionic conductivity. J. Power Source 2014, 245, 630–636. [Google Scholar] [CrossRef]
- Ding, C.; Nohira, T.; Hagiwara, R.; Matsumoto, K.; Okamoto, Y.; Fukunaga, A.; Sakai, S.; Nitta, K.; Inazawa, S. Na [FSA]-[C3C1pyrr][FSA] ionic liquids as electrolytes for sodium secondary batteries: Effects of Na ion concentration and operation temperature. J. Power Source 2014, 269, 124–128. [Google Scholar] [CrossRef]
- Pope, C.R.; Romanenko, K.; MacFarlane, D.R.; Forsyth, M.; O’Dell, L.A. Sodium ion dynamics in a sulfonate based ionomer system studied by 23Na solid-state nuclear magnetic resonance and impedance spectroscopy. Electrochim. Acta 2015, 175, 62–67. [Google Scholar] [CrossRef]
- Guin, M.; Tietz, F. Survey of the transport properties of sodium superionic conductor materials for use in sodium batteries. J. Power Source 2015, 273, 1056–1064. [Google Scholar] [CrossRef]
- Alam, T.M.; Bell, N.; Wheeler, J.; Spoerke, E.D.; Cygan, R.T.; Ingersoll, D. Exploring the role of phosphate structural distortions on the sodium jump dynamics in NaSICON phases. MRS Online Proc. Libr. (OPL) 2015, 1773, 1–6. [Google Scholar] [CrossRef]
- Ma, Q.; Guin, M.; Naqash, S.; Tsai, C.-L.; Tietz, F.; Guillon, O. Scandium-substituted Na3Zr2(SiO4)2(PO4) prepared by a solution-assisted solid-state reaction method as sodium-ion conductors. Chem. Mater. 2016, 28, 4821–4828. [Google Scholar] [CrossRef]
- Naqash, S.; Ma, Q.; Tietz, F.; Guillon, O. Na3Zr2(SiO4)2(PO4) prepared by a solution-assisted solid state reaction. Solid State Ion. 2017, 302, 83–91. [Google Scholar] [CrossRef]
- Zinkevich, T.; Fiedler, A.; Guin, M.; Tietz, F.; Guillon, O.; Ehrenberg, H.; Indris, S. Na+ ion mobility in Na3+xSc2(SiO4)x(PO4)3−x (0.1< x< 0.8) observed by 23Na NMR spectroscopy. Solid State Ion. 2020, 348, 115277. [Google Scholar]
- Guin, M.; Tietz, F.; Guillon, O. New promising NASICON material as solid electrolyte for sodium-ion batteries: Correlation between composition, crystal structure and ionic conductivity of Na3+xSc2SixP3−xO12. Solid State Ion. 2016, 293, 18–26. [Google Scholar] [CrossRef]
- Gabriel, J.; Petrov, O.V.; Kim, Y.; Martin, S.W.; Vogel, M. Lithium ion dynamics in Li2S+GeS2+GeO2 glasses studied using 7Li NMR field-cycling relaxometry and line-shape analysis. Solid State Nucl. Magn. Reson. 2015, 70, 53–62. [Google Scholar] [CrossRef]
- Martin, S.W.; Bischoff, C.; Schuller, K. Composition Dependence of the Na+ Ion Conductivity in 0.5Na2S+ 0.5[x GeS2+(1–x)PS5/2] Mixed Glass Former Glasses: A Structural Interpretation of a Negative Mixed Glass Former Effect. J. Phys. Chem. B 2015, 119, 15738–15751. [Google Scholar] [CrossRef]
- Storek, M.; Adjei-Acheamfour, M.; Christensen, R.; Martin, S.W.; Böhmer, R. Positive and negative mixed glass former effects in sodium borosilicate and borophosphate glasses studied by 23Na NMR. J. Phys. Chem. B 2016, 120, 4482–4495. [Google Scholar] [CrossRef]
- Watson, D.E.; Martin, S.W. Short range order characterization of the Na2S+ SiS2 glass system using Raman, infrared and 29Si magic angle spinning nuclear magnetic resonance spectroscopies. J. Non-Cryst. Solids 2017, 471, 39–50. [Google Scholar] [CrossRef]
- Shastri, A.; Watson, D.; Ding, Q.-P.; Furukawa, Y.; Martin, S.W. 23Na nuclear magnetic resonance study of yNa2S+(1 − y)[xSiS2+(1 − x)PS5/2] glassy solid electrolytes. Solid State Ion. 2019, 340, 115013. [Google Scholar] [CrossRef]
- Anderson, O.; Stuart, D. Calculation of activation energy of ionic conductivity in silica glasses by classical methods. J. Am. Ceram. Soc. 1954, 37, 573–580. [Google Scholar] [CrossRef]
- Haaks, M.; Martin, S.W.; Vogel, M. Relation of short-range and long-range lithium ion dynamics in glass-ceramics: Insights from Li 7 NMR field-cycling and field-gradient studies. Phys. Rev. B 2017, 96, 104301. [Google Scholar] [CrossRef]
- Becher, M.; Becker, S.; Hecht, L.; Vogel, M. From local to diffusive dynamics in polymer electrolytes: NMR studies on coupling of polymer and ion dynamics across length and time scales. Macromolecules 2019, 52, 9128–9139. [Google Scholar] [CrossRef]
- Sinai, Y.G. The limiting behavior of a one-dimensional random walk in a random medium. Theory Probab. Its Appl. 1983, 27, 256–268. [Google Scholar] [CrossRef]
- Oshanin, G.; Burlatsky, S.; Moreau, M.; Gaveau, B. Behavior of transport characteristics in several one-dimensional disordered systems. Chem. Phys. 1993, 177, 803–819. [Google Scholar] [CrossRef]
- Kariyo, S.; Brodin, A.; Gainaru, C.; Herrmann, A.; Hintermeyer, J.; Schick, H.; Novikov, V.; Rössler, E. From simple liquid to polymer melt. Glassy and polymer dynamics studied by fast field cycling NMR relaxometry: Rouse regime. Macromolecules 2008, 41, 5322–5332. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraenza, C.C.; Greenbaum, S.G.; Suarez, S.N. Nuclear Magnetic Resonance Relaxation Pathways in Electrolytes for Energy Storage. Int. J. Mol. Sci. 2023, 24, 10373. https://doi.org/10.3390/ijms241210373
Fraenza CC, Greenbaum SG, Suarez SN. Nuclear Magnetic Resonance Relaxation Pathways in Electrolytes for Energy Storage. International Journal of Molecular Sciences. 2023; 24(12):10373. https://doi.org/10.3390/ijms241210373
Chicago/Turabian StyleFraenza, Carla C., Steve G. Greenbaum, and Sophia N. Suarez. 2023. "Nuclear Magnetic Resonance Relaxation Pathways in Electrolytes for Energy Storage" International Journal of Molecular Sciences 24, no. 12: 10373. https://doi.org/10.3390/ijms241210373
APA StyleFraenza, C. C., Greenbaum, S. G., & Suarez, S. N. (2023). Nuclear Magnetic Resonance Relaxation Pathways in Electrolytes for Energy Storage. International Journal of Molecular Sciences, 24(12), 10373. https://doi.org/10.3390/ijms241210373