
Assessments of Alpha-Amylase Inhibitory Potential of Tagetes Flavonoids through In Vitro, Molecular Docking, and Molecular Dynamics Simulation Studies

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. In Vitro AAI Activity
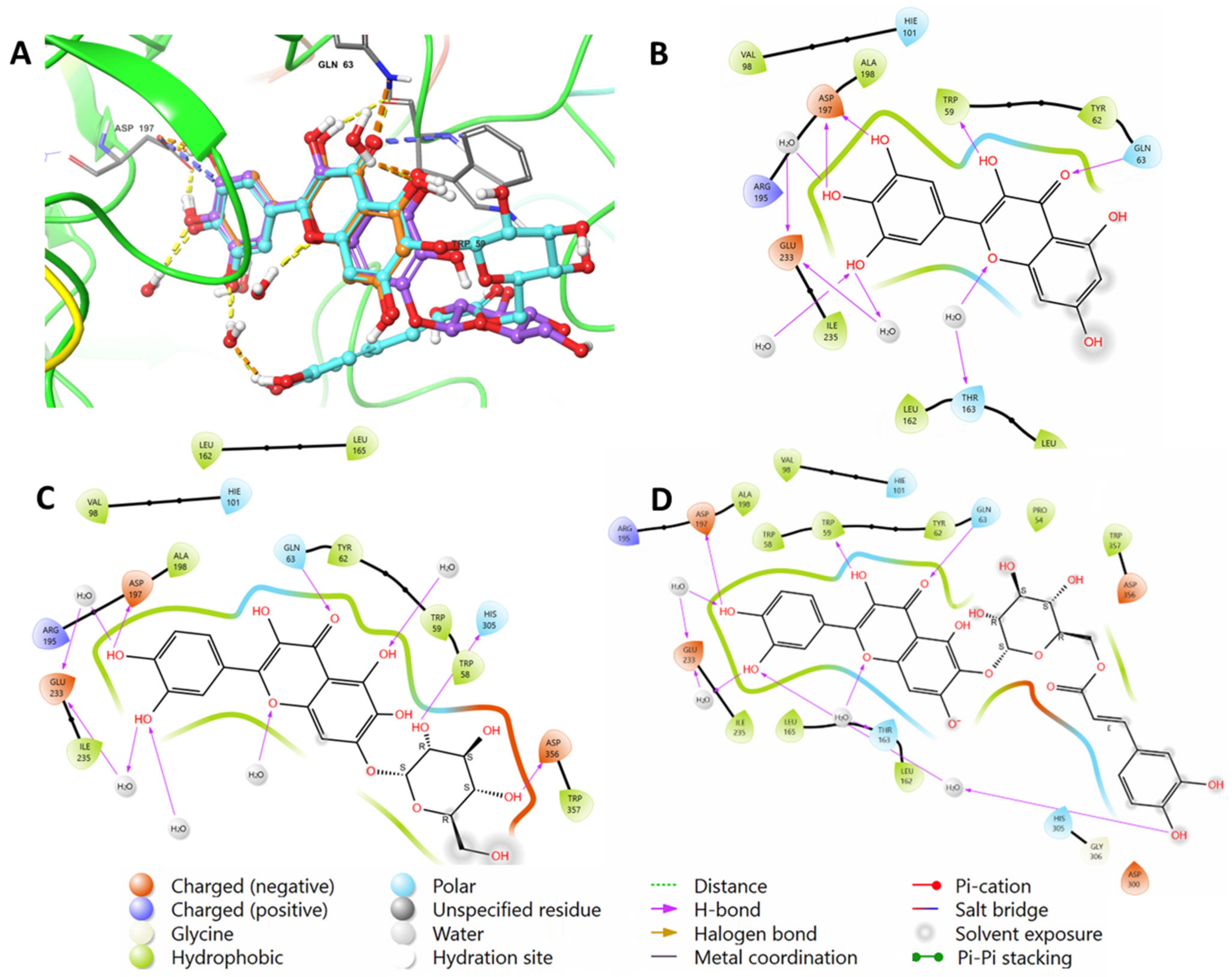
2.2. Molecular Docking Studies
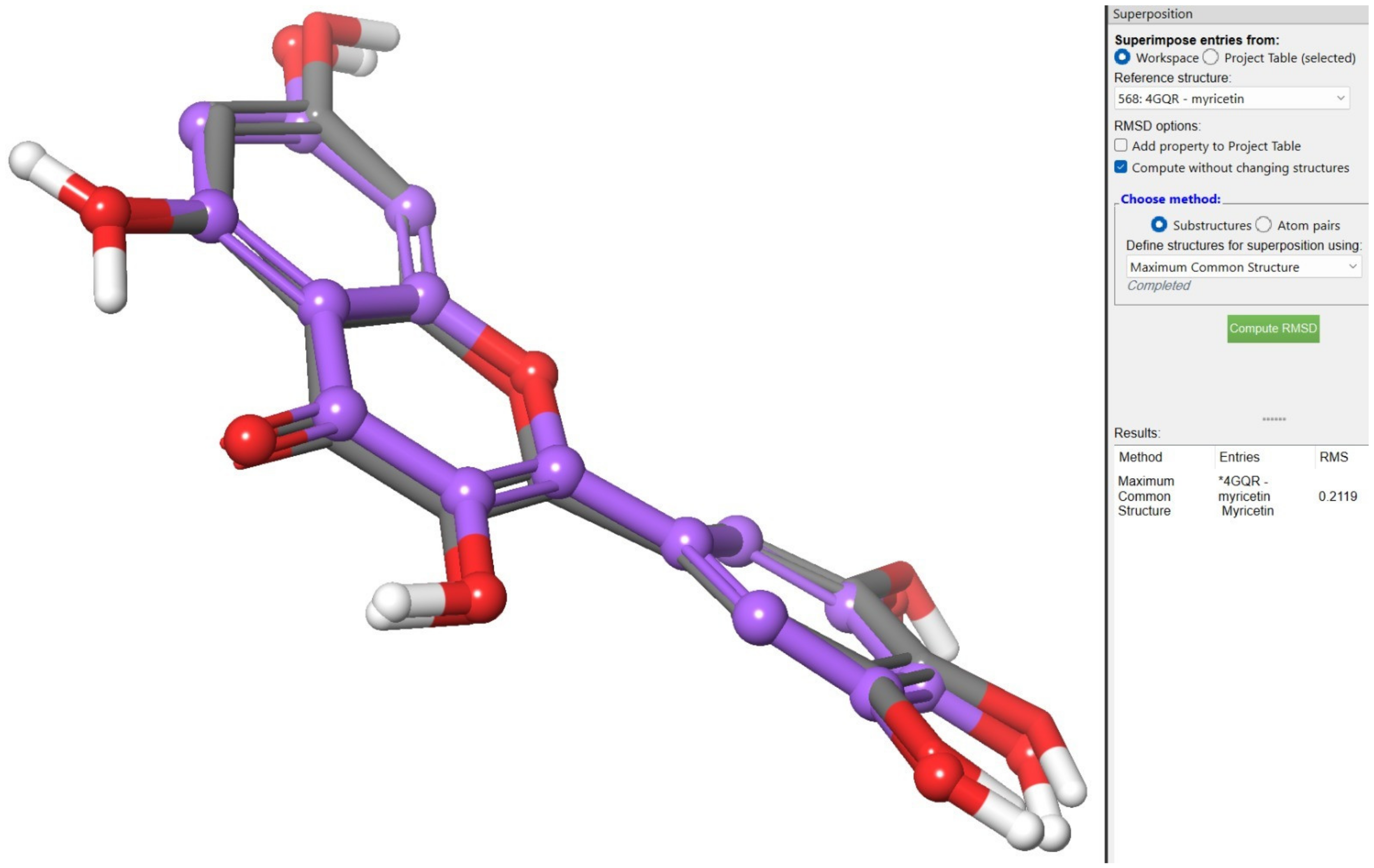
2.3. QM/MM (Quantum Mechanics/Molecular Mechanics) Analysis
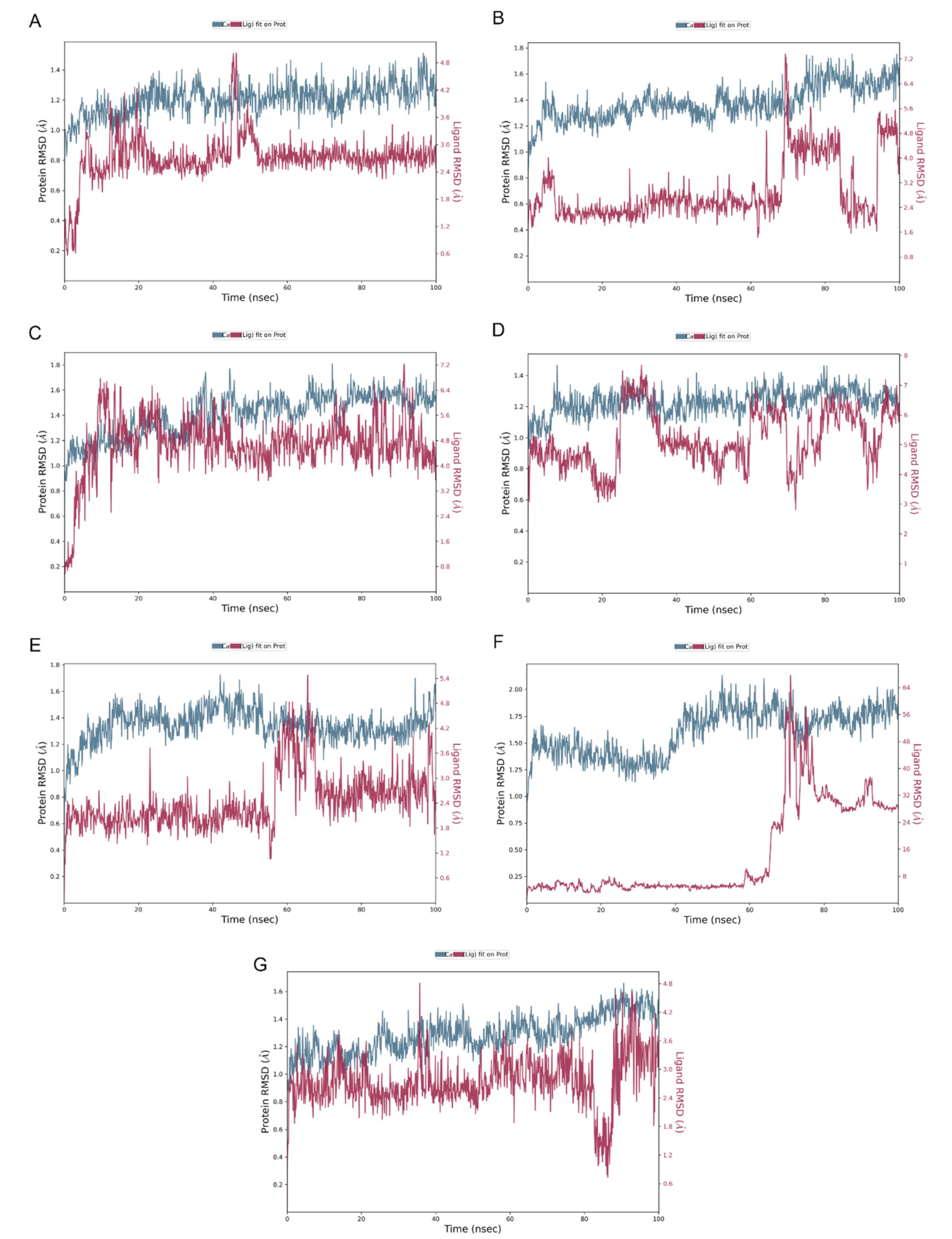
2.4. Molecular Dynamics Simulation
2.5. ADMET Properties
3. Materials and Methods
3.1. AAI Evaluation
3.2. Preparation of Proteins
3.3. Ligand Preparation
3.4. Grid Generation and Molecular Docking
3.5. Induced-Fit Docking
3.6. Molecular Dynamics Simulation (MD)
3.7. QM/MM (Quantum Mechanics/Molecular Mechanics) Calculations
3.8. Prediction of ADMET
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mecha-nisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Diabetes: Key Facts—World Health Organization. Available online: http://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 23 April 2022).
- Darenskaya, M.A.; Kolesnikova, L.I.; Kolesnikov, S.I. Oxidative Stress: Pathogenetic Role in Diabetes Mellitus and Its Complications and Therapeutic Approaches to Correction. Bull. Exp. Biol. Med. 2021, 171, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.-A. Role of Reactive Oxygen Species in the Progression of Type 2 Diabetes and Atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: Facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Mohamed, G.A.; Abdel-Latif, M.M.M.; El-Messery, S.M.; Al Musayeib, N.M.; Shehata, I.A. Minutaside A, new α-amylase inhibitor flavonol glucoside from Tagetes minuta: Antidiabetic, Antioxidant, and Molecular Modeling Studies. Starch/Stärke 2015, 67, 976–984. [Google Scholar] [CrossRef]
- Dal, S.; Sigrist, S. The Protective Effect of Antioxidants Consumption on Diabetes and Vascular Complications. Diseases 2016, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; Bhutani, J.; O’Keefe, J.H. Acarbose: Safe and effective for lowering postprandial hyperglycaemia and improving cardiovascular outcomes. Open Hear. 2015, 2, e000327. [Google Scholar] [CrossRef]
- Hyun, T.K.; Kim, H.-C.; Kim, J.-S. Antioxidant and antidiabetic activity of Thymus quinquecostatus Celak. Ind. Crop. Prod. 2014, 52, 611–616. [Google Scholar] [CrossRef]
- Jugran, A.K.; Rawat, S.; Devkota, H.P.; Bhatt, I.D.; Rawal, R.S. Diabetes and plant-derived natural products: From ethnopharmacological approaches to their potential for modern drug discovery and development. Phytother. Res. 2021, 35, 223–245. [Google Scholar] [CrossRef]
- Gong, X.; Ji, M.; Xu, J.; Zhang, C.; Li, M. Hypoglycemic effects of bioactive ingredients from medicine food homology and medicinal health food species used in China. Crit. Rev. Food Sci. Nutr. 2020, 60, 2303–2326. [Google Scholar] [CrossRef]
- Yeung, A.W.K.; Tzvetkov, N.T.; Durazzo, A.; Lucarini, M.; Souto, E.B.; Santini, A.; Gan, R.-Y.; Jozwik, A.; Grzybek, W.; Horbańczuk, J.O.; et al. Natural products in diabetes research: Quantitative literature analysis. Nat. Prod. Res. 2021, 35, 5813–5827. [Google Scholar] [CrossRef]
- Shahwan, M.; Alhumaydhi, F.; Ashraf, G.M.; Hasan, P.M.; Shamsi, A. Role of polyphenols in combating Type 2 Diabetes and insulin resistance. Int. J. Biol. Macromol. 2022, 206, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Soule, J.A. Medicinal and Beverage Uses of Tagetes (Tageteae: Compositae). Am. J. Bot. 1993, 80, 177. [Google Scholar]
- Salehi, B.; Valussi, M.; Morais-Braga, M.F.B.; Carneiro, J.N.P.; Leal, A.L.A.B.; Coutinho, H.D.M.; Vitalini, S.; Kręgiel, D.; Antolak, H.; Sharifi-Rad, M.; et al. Tagetes spp. Essential Oils and Other Extracts: Chemical Characterization and Biological Activity. Molecules 2018, 23, 2847. [Google Scholar] [CrossRef]
- Wanzala, W.; Wagacha, J.M.; Dossaji, S.F.; Gakuubi, M.M. Bioactive properties of Tagetes minuta L. (Asteraceae) essential oils: A Review. Am. J. Essent. Oils Nat. Prod. 2016, 4, 27–36. [Google Scholar]
- Singh, P.; Krishna, A.; Kumar, V.; Krishna, S.; Singh, K.; Gupta, M.; Singh, S. Chemistry and biology of industrial crop Tagetes species: A review. J. Essent. Oil Res. 2016, 28, 1–14. [Google Scholar] [CrossRef]
- Shirazi, M.T.; Gholami, H.; Kavoosi, G.; Rowshan, V.; Tafsiry, A. Chemical Composition, Antioxidant, Antimicrobial and Cytotoxic Activities of T Agetes Minuta and O Cimum Basilicum Essential Oils. Food Sci. Nutr. 2014, 2, 146–155. [Google Scholar] [CrossRef]
- Paniagua-Zambrana, N.Y.; Bussmann, R.W.; Echeverría, J.; Romero, C. Tagetes elliptica Sm. Tagetes erecta L. Tagetes filifolia Lag. Tagetes minuta L. Tagetes multiflora Kunth ASTERACEAE. In Ethnobotany of the Andes. Ethnobotany of Mountain Regions; Paniagua-Zambrana, N., Bussmann, R., Eds.; Springer: Cham, Switzerland, 2020; pp. 1–12. [Google Scholar] [CrossRef]
- Walia, S.; Kumar, R. Wild marigold (Tagetes minuta L.) an important industrial aromatic crop: Liquid gold from the Himalaya. J. Essent. Oil Res. 2020, 32, 373–393. [Google Scholar] [CrossRef]
- Ibrahim, S.R.; Mohamed, G.A.; Al Haidari, R.A.; El-Kholy, A.A.; Zayed, M.F.; Khayat, M.T. Tagetnoic acid, a new lipoxygenase inhibitor peroxy fatty acid from Tagetes minuta growing in Saudi Arabia. Nat. Prod. Res. 2020, 3, 474–481. [Google Scholar] [CrossRef]
- Al-Musayeib, N.M.; Mohamed, G.A.; Ibrahim, S.R.M.; Ross, S.A. New Thiophene and Flavonoid from Tagetes minuta Leaves Growing in Saudi Arabia. Molecules 2014, 19, 2819–2828. [Google Scholar] [CrossRef]
- Parejo, I.; Bastida, J.; Viladomat, F.; Codina, C. Acylated quercetagetin glycosides with antioxidant activity from Tagetes maxima. Phytochemistry 2005, 66, 2356–2362. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, J.; Jin, H.; Cao, Y.; Ming, T.; Zhang, L.; Hu, M.; Hamlati, H.; Pang, S.; Ma, X. Vasorelaxant effects of the extracts and some flavonoids from the buds of Coreopsis tinctoria. Pharm. Biol. 2013, 51, 1158–1164. [Google Scholar]
- Mohamed, G.A. Tagenols A and B: New lipoxygenase inhibitor flavonols from Tagetes minuta. Phytochem. Lett. 2016, 16, 141–145. [Google Scholar] [CrossRef]
- Dok-Go, H.; Lee, K.H.; Kim, H.J.; Lee, E.H.; Lee, J.; Song, Y.S.; Lee, Y.-H.; Jin, C.; Lee, Y.S.; Cho, J. Neuroprotective effects of antioxidative flavonoids, quercetin, (+)-dihydroquercetin and quercetin 3-methyl ether, isolated from Opuntia ficus-indica var. saboten. Brain Res. 2003, 965, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Jiang, Y.; Zhao, J.; Li, H.; Yin, X.; Wang, Y.; Xie, Y.; Chen, X.; Lu, J.; Dong, Z.; et al. Quercetin-3-methyl ether inhibits esophageal carcinogenesis by targeting the AKT/mTOR/p70S6K and MAPK pathways. Mol. Carcinog. 2018, 57, 1540–1552. [Google Scholar] [CrossRef]
- Alvarado-Sansininea, J.J.; Sánchez-Sánchez, L.; López-Muñoz, H.; Escobar, M.L.; Flores-Guzmán, F.; Tavera-Hernández, R.; Jiménez-Estrada, M. Quercetagetin and Patuletin: Antiproliferative, Necrotic and Apoptotic Activity in Tumor Cell Lines. Molecules 2018, 23, 2579. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, A.; Mesaik, M.A.; Simjee, S.U.; Lubna; Bano, S.; Faizi, S. Anti-TNF-α and anti-arthritic effect of patuletin: A rare flavonoid from Tagetes patula. Int. Immunopharmacol. 2016, 36, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Aherne, S.A.; O’Brien, N.M. Dietary flavonols: Chemistry, food content, and metabolism. Nutrition 2002, 18, 75–81. [Google Scholar] [CrossRef]
- Andersen, O.M.; Markham, K.R. Flavonoids: Chemistry, Biochemistry and Applications; CRC press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Stewart, A.J.; Chapman, W.; Jenkins, G.I.; Graham, I.; Martin, T.; Crozier, A. The effect of nitrogen and phosphorus deficiency on flavonol accumulation in plant tissues. Plant Cell Environ. 2001, 24, 1189–1197. [Google Scholar] [CrossRef]
- Brahmachari, G.; Gorai, D. Progress in the Research on Naturally Occurring Flavones and Flavonols: An Overview. Curr. Org. Chem. 2006, 10, 873–898. [Google Scholar] [CrossRef]
- Survay, N.S.; Upadhyaya, C.P.; Kumar, B.; Young, K.E.; Yoon, D.; Park, S. New Genera of Flavonols and Flavonol De-rivatives as Therapeutic Molecules. J. Korean Soc. Appl. Biol. Chem. 2011, 54, 1–18. [Google Scholar] [CrossRef]
- Tutel’Ian, V.A.; Lashneva, N.V. Biologically active substances of plant origin. Flavonols and flavones: Prevalence, dietary sourses and consumption. Vopr. Pitan. 2013, 82, 4–22. [Google Scholar]
- Ashraf, J.; Mughal, E.U.; Sadiq, A.; Naeem, N.; Muhammad, S.A.; Qousain, T.; Zafar, M.N.; Khan, B.A.; Anees, M. Design and Synthesis of New Flavonols as Dual Ɑ-Amylase and Ɑ-Glucosidase Inhibitors: Structure-Activity Relationship, Drug-Likeness, in Vitro and in Silico Studies. J. Mol. Struct. 2020, 1218, 128458. [Google Scholar] [CrossRef]
- Hua, F.; Zhou, P.; Wu, H.-Y.; Chu, G.-X.; Xie, Z.-W.; Bao, G.-H. Inhibition of α-glucosidase and α-amylase by flavonoid glycosides from Lu’an GuaPian tea: Molecular docking and interaction mechanism. Food Funct. 2018, 9, 4173–4183. [Google Scholar] [CrossRef]
- Piparo, E.L.; Scheib, H.; Frei, N.; Williamson, G.; Grigorov, M.; Chou, C.J. Flavonoids for Controlling Starch Digestion: Structural Requirements for Inhibiting Human α-Amylase. J. Med. Chem. 2008, 51, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Protein-ligand interactions as the basis for drug actionn. In Multifaceted Roles of Crystallography in Modern Drug Discovery; NATO Science for Peace and Security Series A: Chemistry and Biology; Springer: Dordrecht, The Netherlands, 2015; pp. 83–92. [Google Scholar] [CrossRef]
- Martell, R.E.; Brooks, D.G.; Wang, Y.; Wilcoxen, K. Discovery of Novel Drugs for Promising Targets. Clin. Ther. 2013, 35, 1271–1281. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- Gani, M.A.; Nurhan, A.D.; Maulana, S.; Siswodihardjo, S.; Shinta, D.W.; Khotib, J. Structure-Based Virtual Screening of Bioactive Compounds from Indonesian Medical Plants Against Severe Acute Respiratory Syndrome Coronavirus-2. J. Adv. Pharm. Technol. Res. 2021, 12, 120–126. [Google Scholar]
- Philipp, D.M.; Friesner, R.A. Mixed Ab Initio QM/MM Modeling using Frozen Orbitals and Tests with Alanine Dipep-tide and Tetrapeptide. J. Comput. Chem. 1999, 20, 1468–1494. [Google Scholar] [CrossRef]
- Charanya, C.; Sampathkrishnan, S.; Balamurugan, N. Quantum Mechanical Analysis, Spectroscopic (FT-IR, FT-Raman, UV-Visible) Study, and HOMO-LUMO Analysis of (1S, 2R)-2-Amino-1-Phenylpropan-1-Ol using Density Func-tional Theory. J. Mol. Liq. 2017, 231, 116–125. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Adcock, S.A.; McCammon, J.A. Molecular Dynamics: Survey of Methods for Simulating the Activity of Proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simula-tions. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Alhakamy, N.A.; Mohamed, G.A.; Fahmy, U.A.; Eid, B.G.; Ahmed, O.A.A.; Al-Rabia, M.W.; Khedr, A.I.M.; Nasrullah, M.Z.; Ibrahim, S.R.M. New Alpha-Amylase Inhibitory Metabolites from Pericarps of Garcinia mangostana. Life 2022, 12, 384. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Ibrahim, S.R. Garcixanthone E and Garcimangophenone C: New Metabolites from Garcinia Man-gostana and their Cytotoxic and Alpha Amylase Inhibitory Potential. Life 2022, 12, 1875. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.K.; Li, C.; Withers, S.G.; Brayer, G.D. Order and Disorder: Differential Structural Impacts of Myricetin and Ethyl Caffeate on Human Amylase, an Antidiabetic Target. J. Med. Chem. 2012, 55, 10177–10186. [Google Scholar] [CrossRef]
- Tavella, D.; Ouellette, D.R.; Garofalo, R.; Zhu, K.; Xu, J.; Oloo, E.O.; Negron, C.; Ihnat, P.M. A novel method for in silico assessment of Methionine oxidation risk in monoclonal antibodies: Improvement over the 2-shell model. PLoS ONE 2022, 17, e0279689. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Tomé, S.M.; Oliveira, E.F.T.; Viegas, M.F.; Araújo, A.N.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; et al. Evaluation of a flavonoids library for inhibition of pancreatic α-amylase towards a struc-ture-activity relationship. J. Enzyme Inhib. Med. Chem. 2019, 34, 577–588. [Google Scholar] [CrossRef]
- Yao, Z.; Li, C.; Gu, Y.; Zhang, Q.; Liu, L.; Meng, G.; Wu, H.; Bao, X.; Zhang, S.; Sun, S.; et al. Dietary myricetin intake is inversely associated with the prevalence of type 2 diabetes mellitus in a Chinese population. Nutr. Res. 2019, 68, 82–91. [Google Scholar] [CrossRef]
- Schrödinger, L.L.C. Schrödinger Release 2021-4: LigPrep; New York, NY, USA, 2021.
- Ibrahim, S.R.M.; Omar, A.M.; Bagalagel, A.A.; Diri, R.M.; Noor, A.O.; Almasri, D.M.; Mohamed, S.G.A.; Mohamed, G.A. Thiophenes-Naturally Occurring Plant Metabolites: Biological Activities and In Silico Evaluation of Their Potential as Cathepsin D Inhibitors. Plants 2022, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.L.C. Schrödinger Suite 2009 Induced Fit Docking Protocol, Prime version; Schrödinger, Inc.: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger Release 2. Desmond Molecular Dynamics System, D.E. Shaw Research. In Maes-tro-Desmond Interoperability Tools; Schrödinger, Inc.: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger, L.L.C. Schrödinger Release 2023-1: QSite, New York, NY, USA, 2021.
- Schrödinger Release 2. Prime, Schrödinger, New York, NY, USA, 2022.
- Schrödinger, L.L.C. Schrödinger Release 2021-4: QikProp, New York, NY, USA, 2021.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docking Scores | MMGBSA | AAI | |||||
|---|---|---|---|---|---|---|---|
| Compound | Docking Score (kcal/mol) | XP Gscore (kcal/mol) | Glide Gscore (kcal/mol) | Glide Emodel (kcal/mol) | Prime Energy (kcal/mol) | dG Bind | IC50 μM |
| Acarbose | −14.668 | −15.22 | −15.22 | −79.824 | −20,790.9 | −33.97 | 7.1 |
| 2 | −13.882 | −13.889 | −13.889 | −87.396 | −21,006.9 | −51.04 | 7.8 |
| 1 | −12.171 | −13.868 | −13.868 | −97.793 | −21,071.5 | −17.09 | 10.1 |
| 5 | −13.039 | −13.039 | −13.039 | −85.651 | −21,027.7 | −48.4 | 8.1 |
| 3 | −12.32 | −12.362 | −12.362 | −89.862 | −21,074.6 | −54.74 | 8.9 |
| 4 | −12.262 | −12.305 | −12.305 | −91.452 | −21,096.1 | −64.34 | 9.2 |
| 6 | −12.22 | −12.22 | −12.22 | −88.197 | −21,043.3 | −55.43 | 9.7 |
| Myricetin (MYC) | −11.723 | −11.77 | −11.77 | −65.617 | −21,172.2 | −43.5 | − |
| 7 | −11.277 | −11.323 | −11.323 | −74.269 | −20,984 | −38.32 | 10.8 |
| 8 | −9.013 | −10.771 | −10.771 | −59.642 | −21,093.4 | −19.84 | 13.6 |
| 10 | −8.163 | −9.874 | −9.874 | −50.031 | −21,155.8 | −7.33 | 26.9 |
| 9 | −9.445 | −9.445 | −9.445 | −73.229 | −21,043.5 | −36.31 | 12.7 |
| 12 | −8.69 | −8.727 | −8.727 | −65.035 | −21,020.6 | −42.06 | 14.9 |
| 11 | −8.351 | −8.392 | −8.392 | −56.918 | −21,115.6 | −33.39 | 21.7 |
| Compound | IFD Score | Number of Canonical Orbitals | QM/MM Energy | HOMO | LUMO | Energy Gap |
|---|---|---|---|---|---|---|
| Acarbose | −1131.85 | 170 | −512.728183 | −0.108797 | 0.091728 | 0.201 |
| 2 | −1133.46 | 170 | −512.694847 | −0.10958 | 0.088141 | 0.198 |
| 1 | −1134.96 | 170 | −512.786174 | −0.068128 | 0.121556 | 0.190 |
| 5 | −1131.74 | 170 | −512.708656 | −0.103067 | 0.088501 | 0.192 |
| 3 | −1133.32 | 170 | −512.782343 | −0.118446 | 0.081654 | 0.200 |
| 4 | −1132.87 | 170 | −512.795165 | −0.094984 | 0.077919 | 0.173 |
| 6 | −1131.09 | 170 | −512.751505 | −0.108385 | 0.103096 | 0.211 |
| Myricetin (MYC) | −1132.91 | 170 | −512.7611 | −0.118972 | 0.093787 | 0.213 |
| 7 | −1131.83 | 170 | −512.775685 | −0.098428 | 0.099785 | 0.198 |
| 8 | −1130.88 | 170 | −512.753084 | −0.106348 | 0.083171 | 0.190 |
| 10 | −1129.54 | 170 | −512.687624 | −0.08808 | 0.148653 | 0.237 |
| 9 | −1131.07 | 170 | −512.787028 | −0.11625 | 0.098969 | 0.215 |
| 12 | −1127.55 | 170 | −512.724293 | −0.118931 | 0.090068 | 0.209 |
| 11 | −1130.52 | 170 | −512.771419 | −0.120463 | 0.098182 | 0.219 |
| Molecule | mol_MW | # Stars | # rtvFG | CNS | SASA | donorHB | accptHB | QPlogPo/w | QPlogHERG | QPPCaco | QPlogBB | # Metab | QPlogKhsa | Percent Human Oral Absorption |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recommended Range | (130–725) | (0.0–5.0) | (0–2) | (−2 Inactive) (+2 Active) | (300–1000) | (0–6) | (2.0–20.0) | (−2–6.5) | Concern Below −5 | <25 Poor, >500 Great | (−3–1.2) | (1–8) | (−1.5–1.5) | (<25% Poor; >80% High) |
| Acarbose | 807.753 | 15 | 4 | −2 | 992.409 | 17 | 40.6 | −9.2 | −5.826 | 0.012 | −6.846 | 17 | −3.359 | 0 |
| 2 | 480.381 | 5 | 1 | −2 | 683.765 | 8 | 14.5 | −2.0 | −5.313 | 1.608 | −4.141 | 9 | −1.036 | 0 |
| 5 | 494.408 | 5 | 1 | −2 | 711.037 | 7 | 14.5 | −1.4 | −5.427 | 2.135 | −4.066 | 9 | −0.934 | 0 |
| 3 | 480.381 | 5 | 1 | −2 | 706.734 | 8 | 14.5 | −2.0 | −5.561 | 1.002 | −4.503 | 9 | −1.033 | 0 |
| 4 | 578.526 | 8 | 2 | −2 | 887.136 | 7 | 14.8 | 0.3 | −6.362 | 3.315 | −4.673 | 9 | −0.656 | 0 |
| 6 | 494.408 | 5 | 1 | −2 | 719.054 | 7 | 14.5 | −1.3 | −5.397 | 2.744 | −3.978 | 9 | −0.917 | 1.3 |
| 1 | 642.526 | 8 | 2 | −2 | 813.947 | 9 | 16.3 | −1.1 | −5.482 | 0.318 | −5.127 | 10 | −0.79 | 0 |
| Myricetin | 318.239 | 0 | 0 | −2 | 517.206 | 5 | 6 | −0.3 | −4.756 | 7.701 | −2.778 | 6 | −0.496 | 28.1 |
| 7 | 510.407 | 5 | 1 | −2 | 735.455 | 8 | 15.25 | −1.7 | −5.474 | 1.863 | −4.3 | 10 | −1.043 | 0 |
| 8 | 332.266 | 0 | 0 | −2 | 543.866 | 4 | 6 | 0.7 | −4.906 | 33.094 | −2.215 | 6 | −0.317 | 58.0 |
| 9 | 508.435 | 4 | 1 | −2 | 737.814 | 6 | 14.5 | −0.6 | −5.321 | 6.203 | −3.595 | 9 | −0.805 | 0 |
| 12 | 376.319 | 0 | 0 | −2 | 643.535 | 3 | 6.75 | 1.9 | −5.595 | 105.079 | −2.005 | 7 | −0.038 | 74.3 |
| 11 | 346.293 | 0 | 0 | −2 | 570.127 | 3 | 6 | 1.5 | −4.962 | 89.029 | −1.826 | 6 | −0.133 | 70.4 |
| 10 | 316.267 | 0 | 0 | −2 | 521.456 | 3 | 5.25 | 1.1 | −4.725 | 59.31 | −1.826 | 5 | −0.182 | 65.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, G.A.; Omar, A.M.; El-Araby, M.E.; Mass, S.; Ibrahim, S.R.M. Assessments of Alpha-Amylase Inhibitory Potential of Tagetes Flavonoids through In Vitro, Molecular Docking, and Molecular Dynamics Simulation Studies. Int. J. Mol. Sci. 2023, 24, 10195. https://doi.org/10.3390/ijms241210195
Mohamed GA, Omar AM, El-Araby ME, Mass S, Ibrahim SRM. Assessments of Alpha-Amylase Inhibitory Potential of Tagetes Flavonoids through In Vitro, Molecular Docking, and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences. 2023; 24(12):10195. https://doi.org/10.3390/ijms241210195
Chicago/Turabian StyleMohamed, Gamal A., Abdelsattar M. Omar, Moustafa E. El-Araby, Shaza Mass, and Sabrin R. M. Ibrahim. 2023. "Assessments of Alpha-Amylase Inhibitory Potential of Tagetes Flavonoids through In Vitro, Molecular Docking, and Molecular Dynamics Simulation Studies" International Journal of Molecular Sciences 24, no. 12: 10195. https://doi.org/10.3390/ijms241210195
APA StyleMohamed, G. A., Omar, A. M., El-Araby, M. E., Mass, S., & Ibrahim, S. R. M. (2023). Assessments of Alpha-Amylase Inhibitory Potential of Tagetes Flavonoids through In Vitro, Molecular Docking, and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences, 24(12), 10195. https://doi.org/10.3390/ijms241210195