
Economic Separations of Organic Acidic or Basic Enantiomeric Mixtures—A Protocol Suggestion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
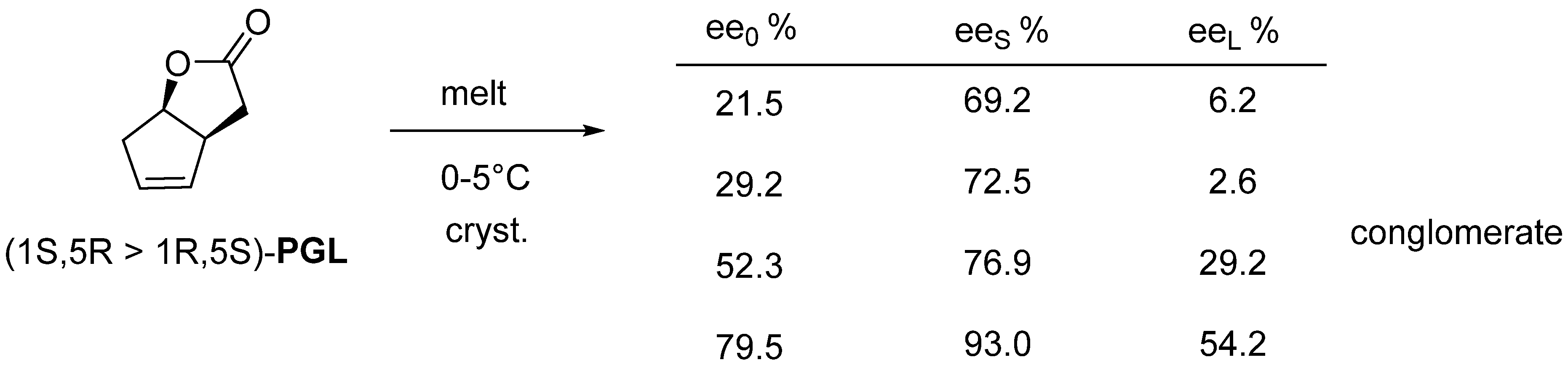{kind=link}
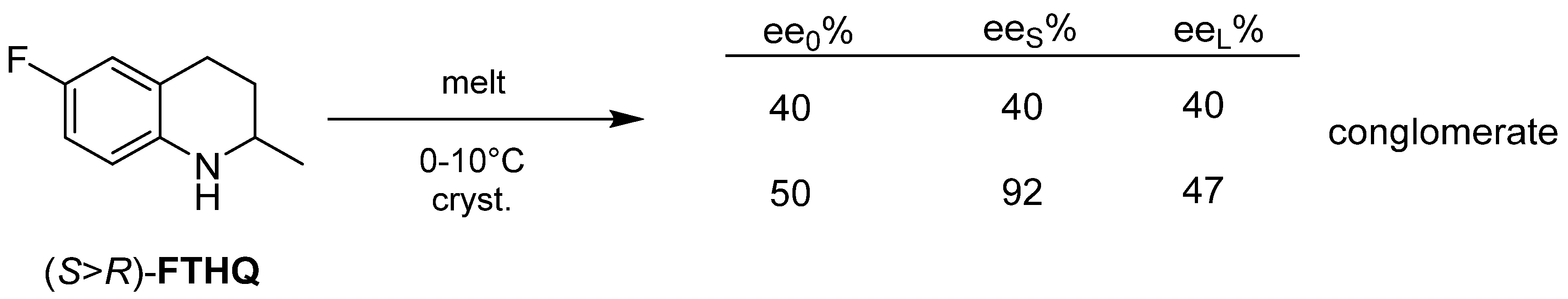{kind=link}
{kind=link}
{kind=link}
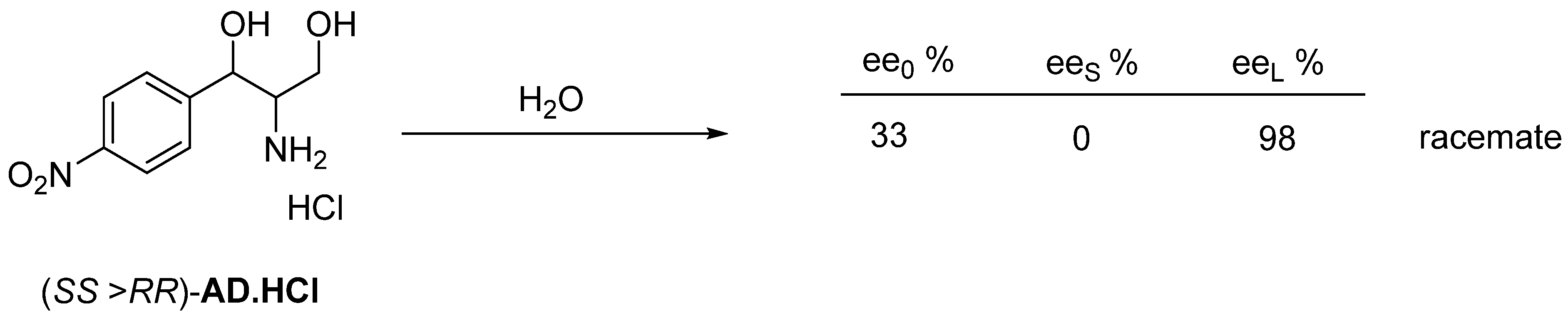{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
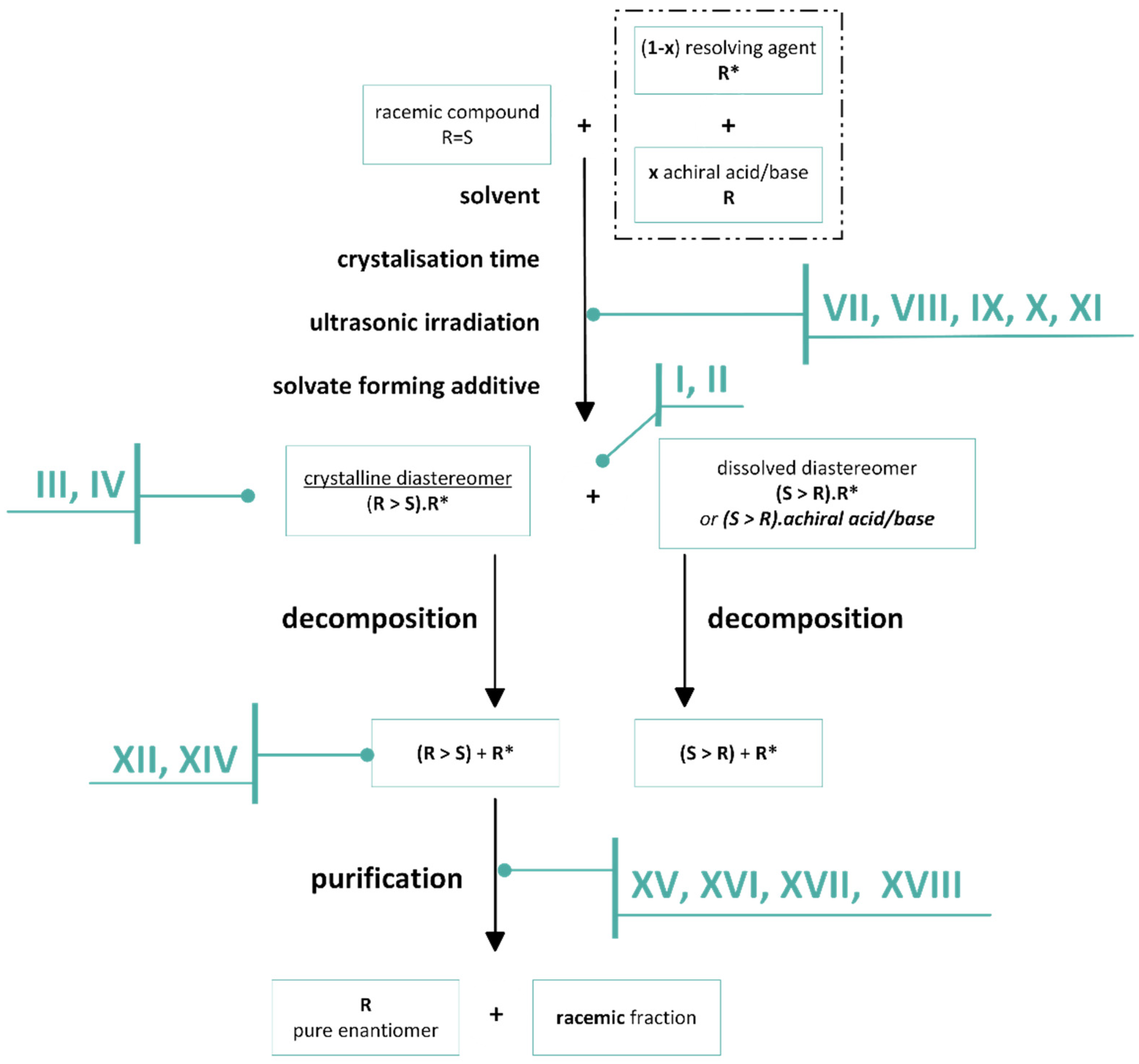
2. A protocol Suggestion for the Separation of Enantiomeric Mixtures
2.1. Design of Enantiomeric Separation from Racemic Compounds, and Non-Racemic Mixtures. Commonly Used Considerations, Protocol
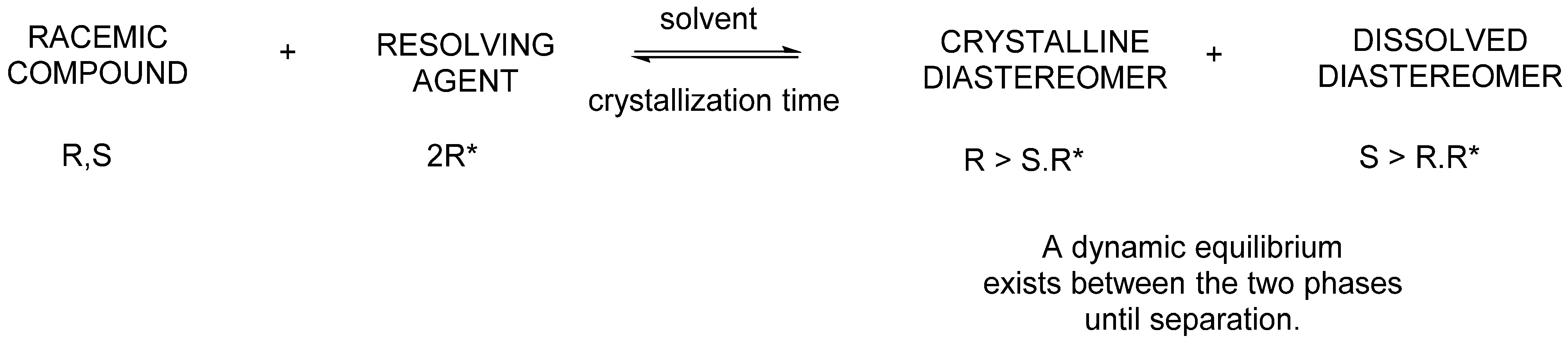
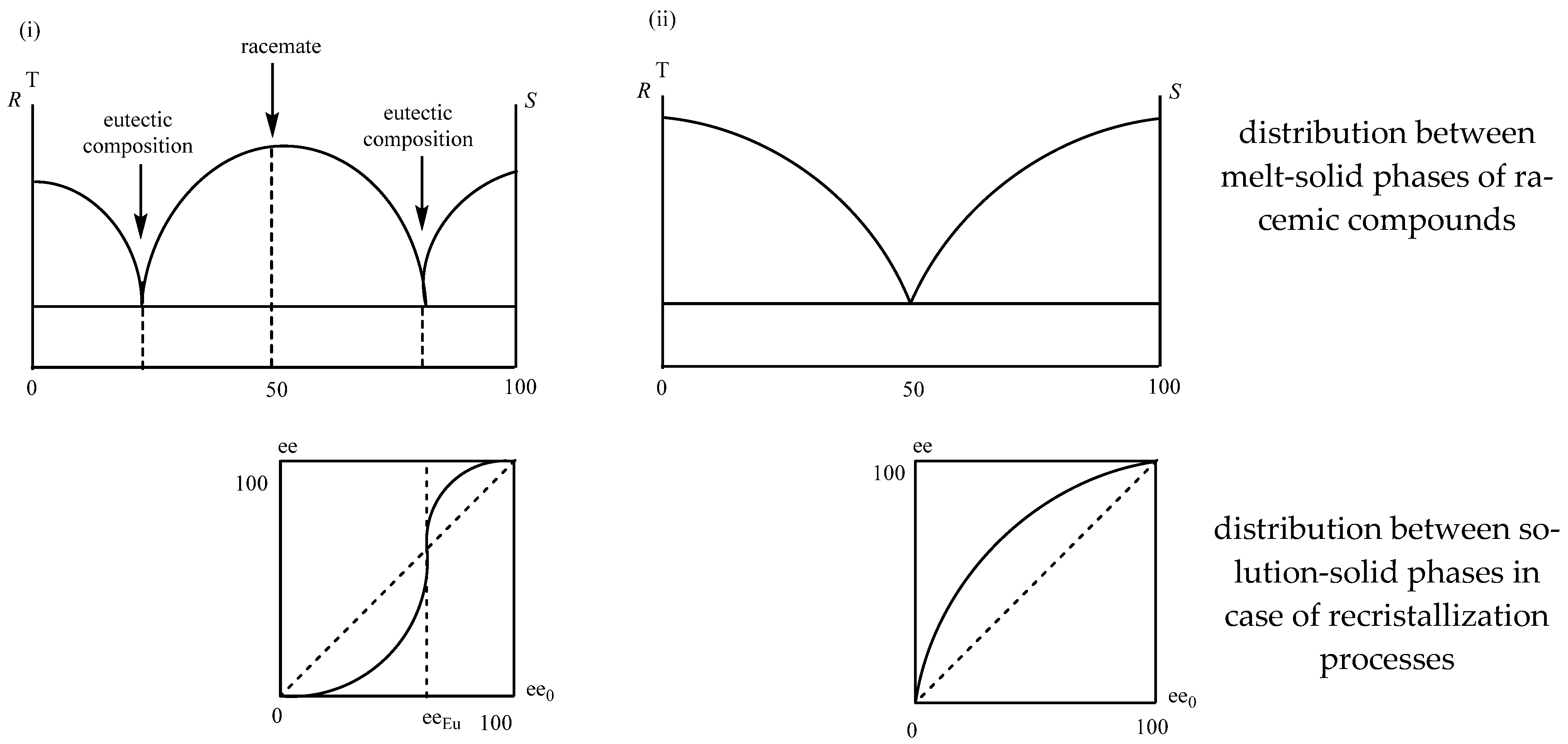
2.1.1. Separation of Racemic Compounds by Diastereomeric Salt Formation
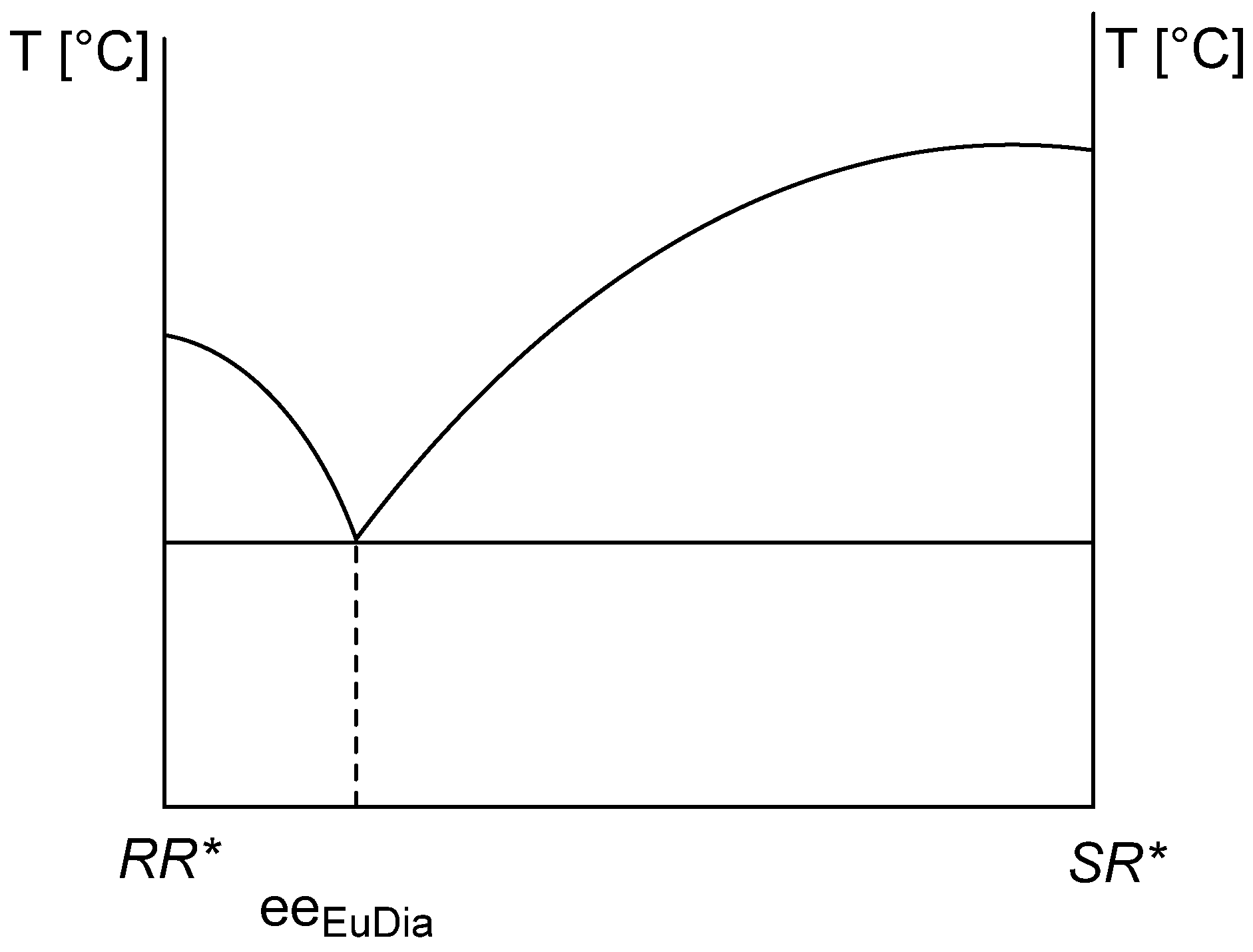
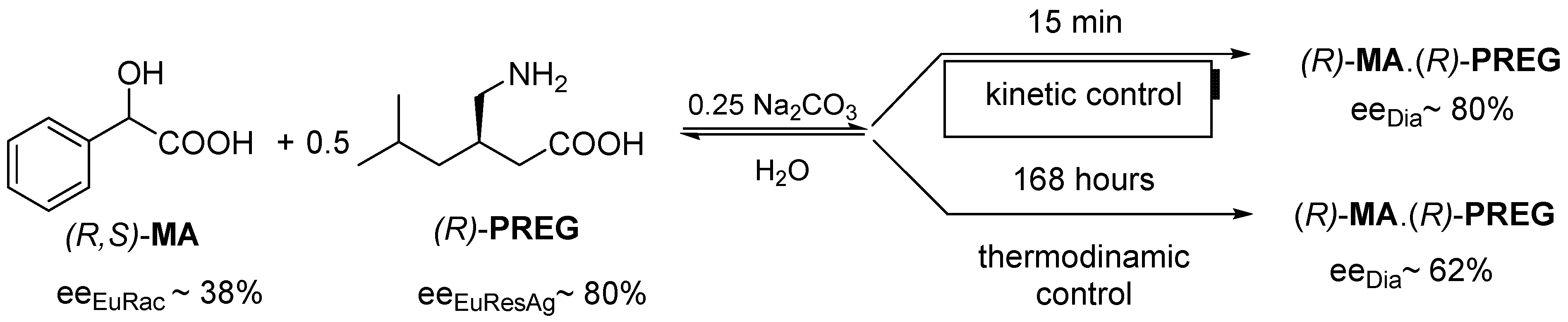
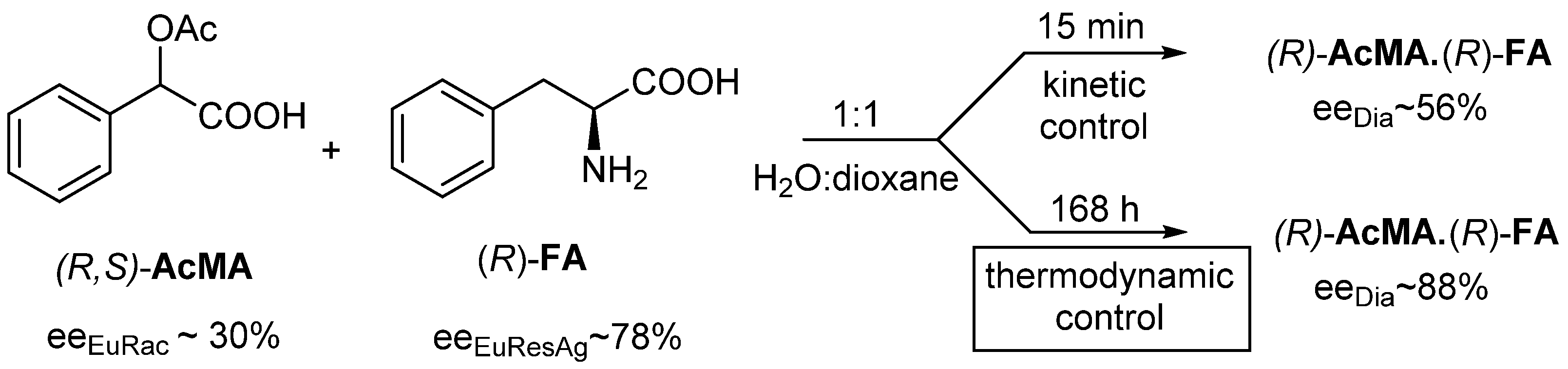
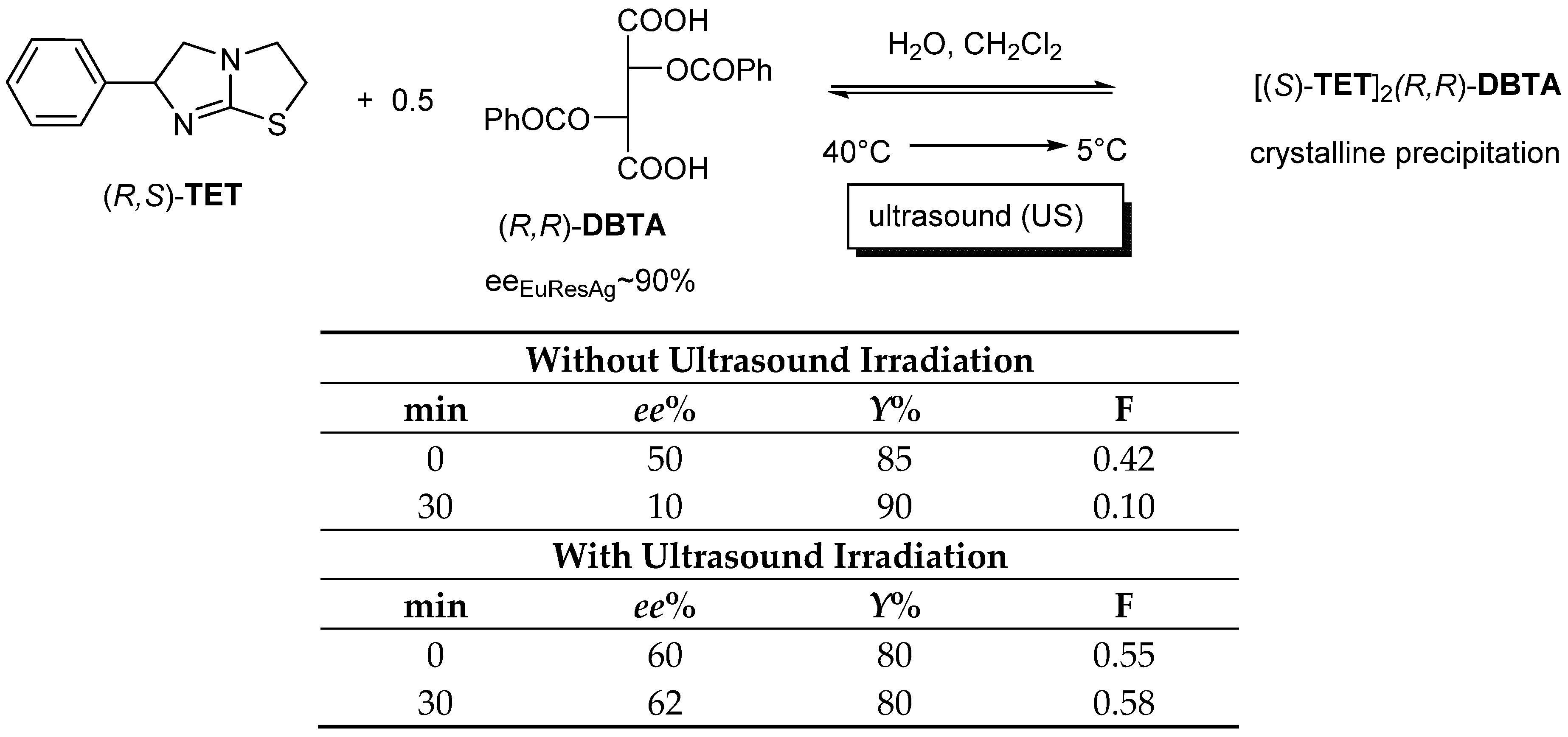
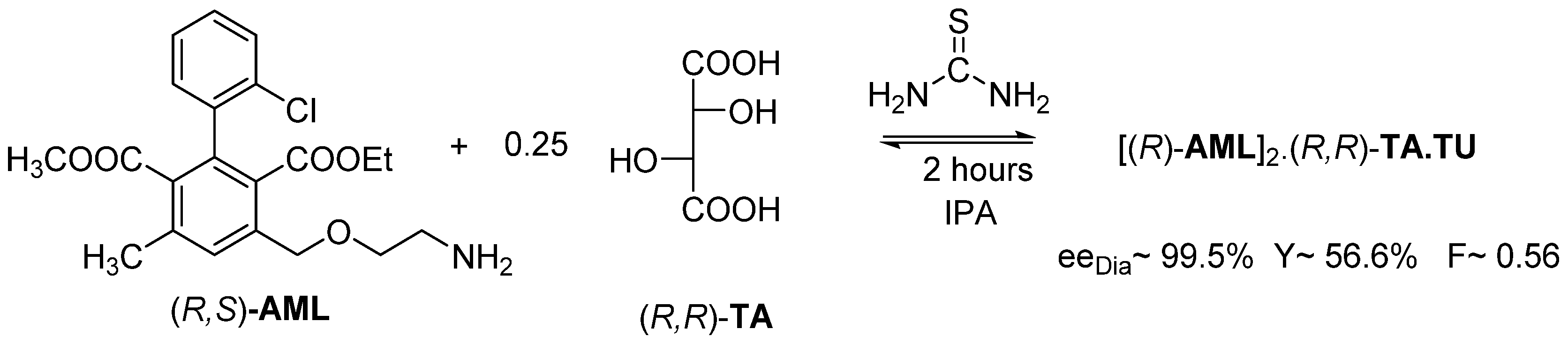
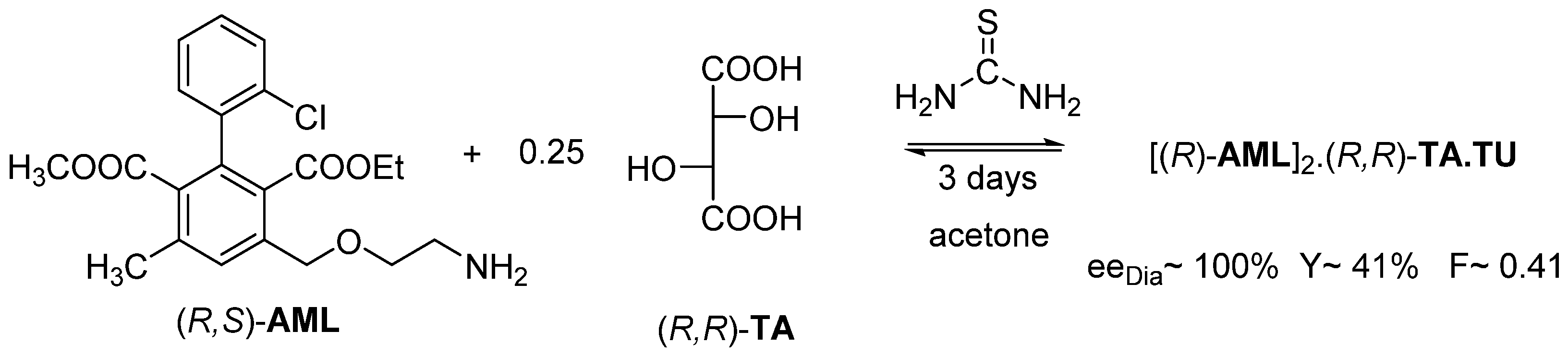
2.1.2. The Solvent and the Crystallization Time (eeDia~eeEuRac or eeEuResAg)
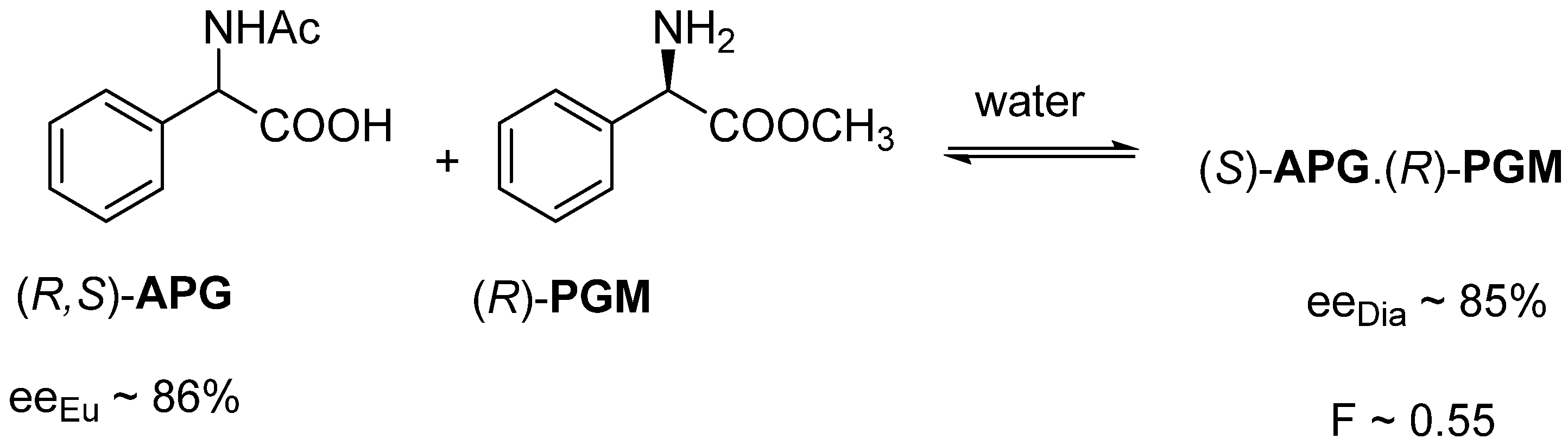
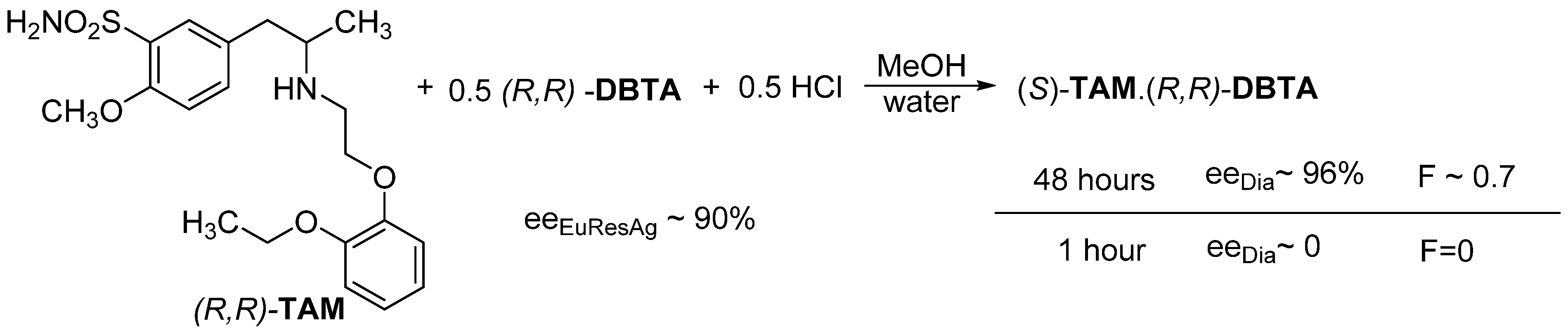
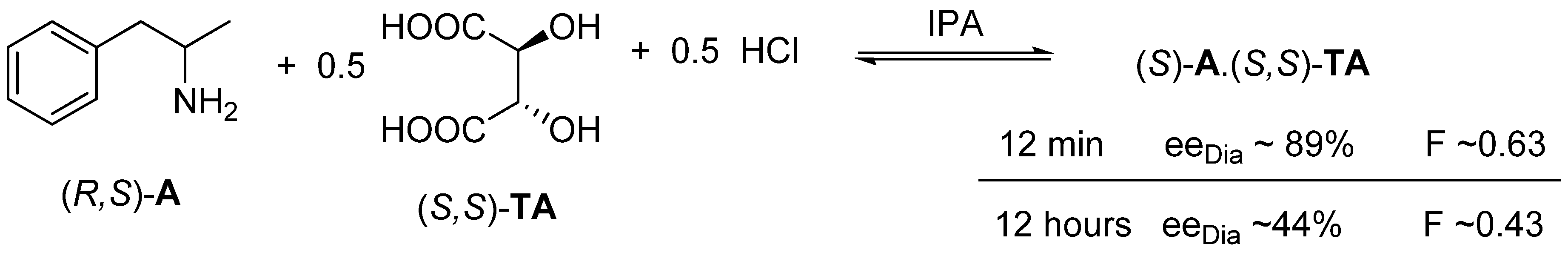
2.1.3. The Value of F Can Be Clearly Determined by the Circumstances of the Process
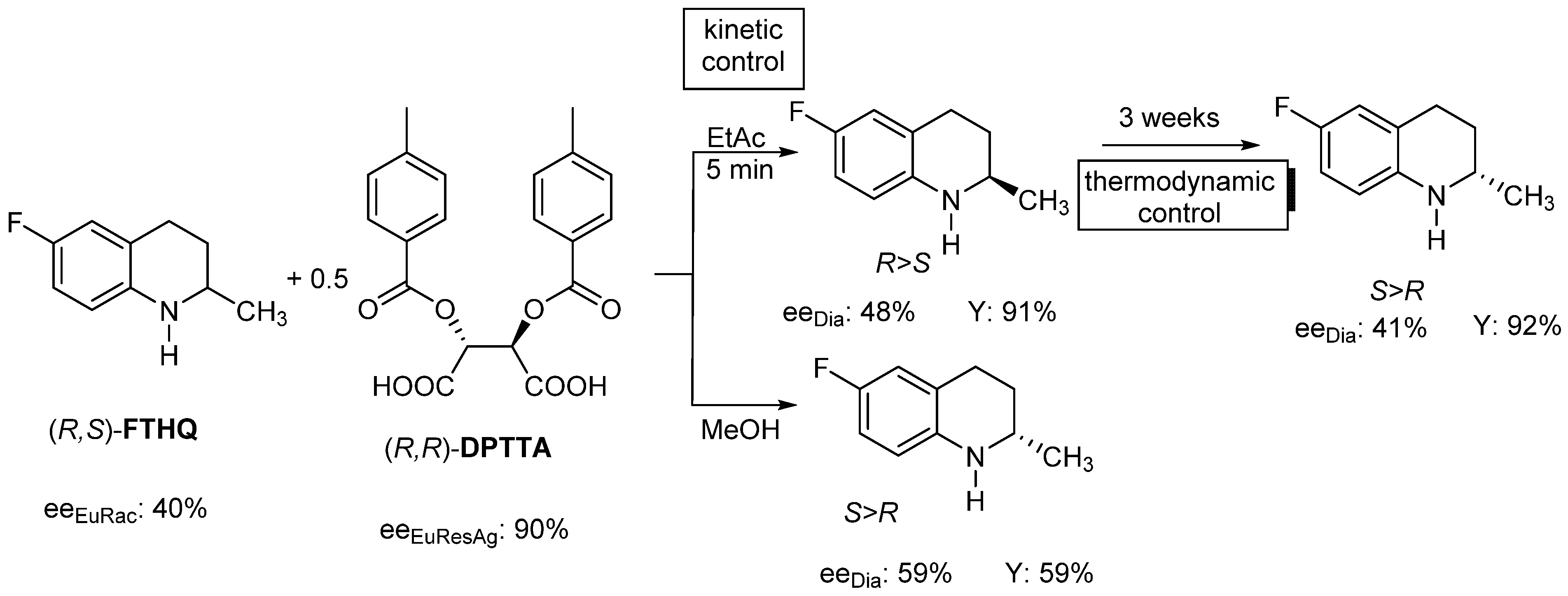
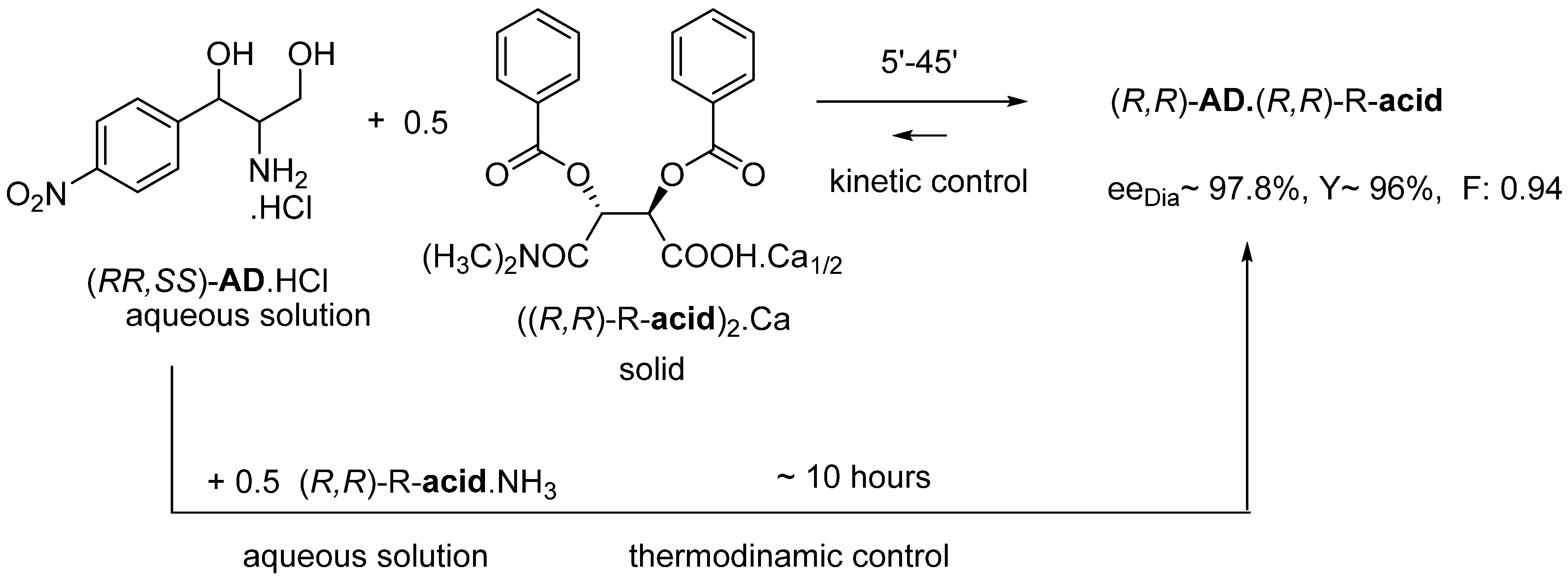
2.1.4. Thermodynamic vs. Kinetic Control
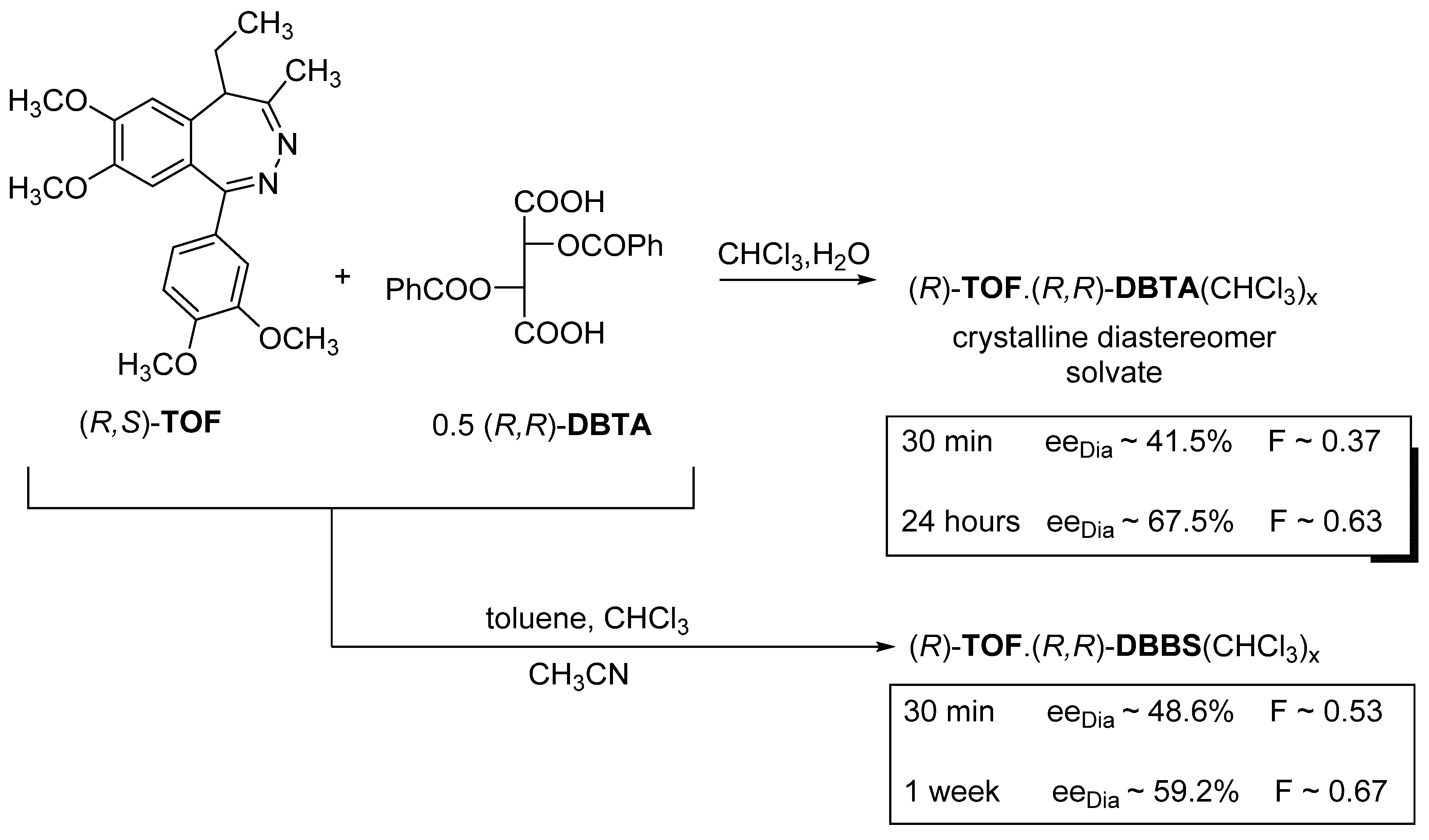
2.1.5. The Role of Solvates
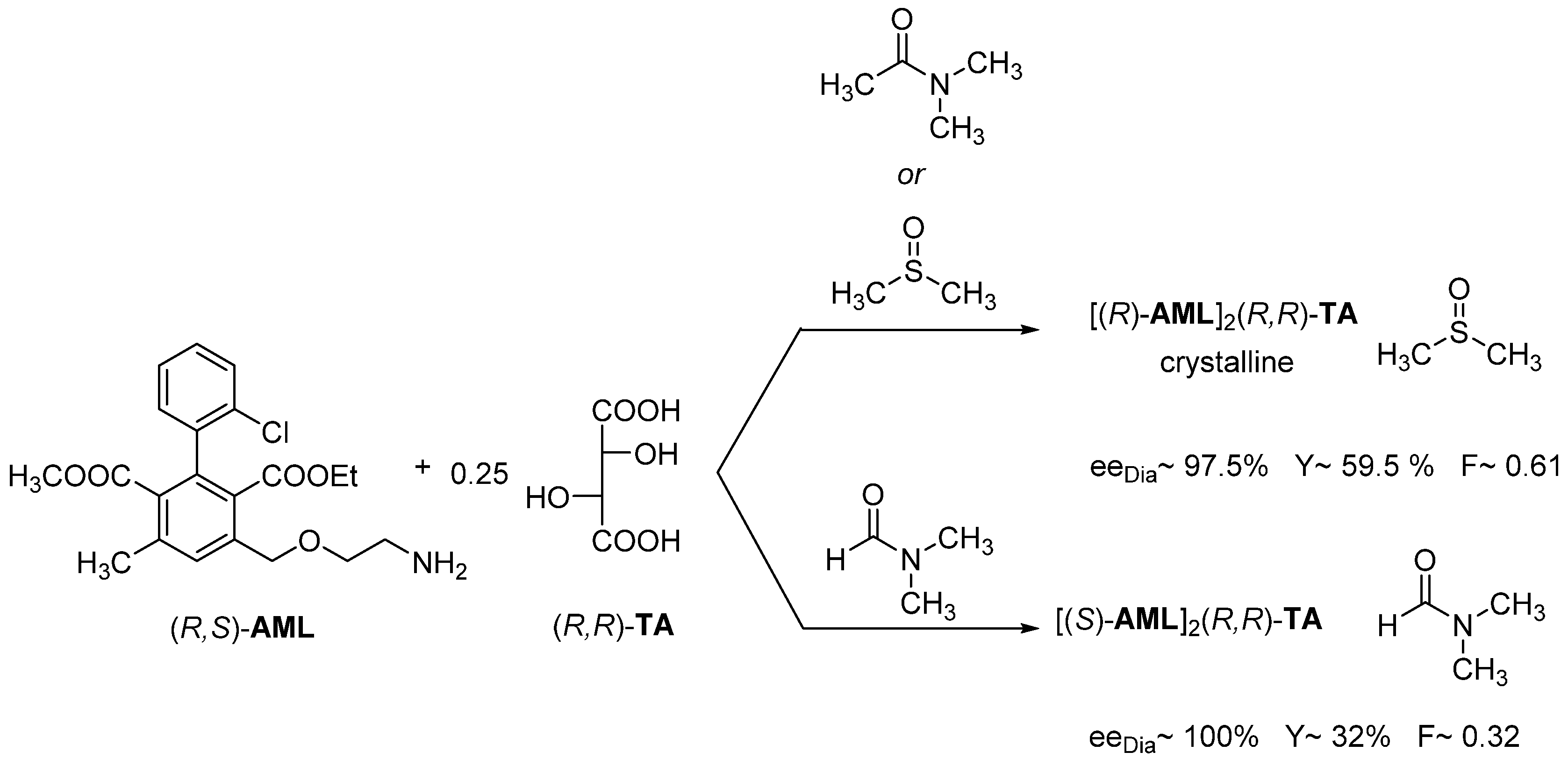
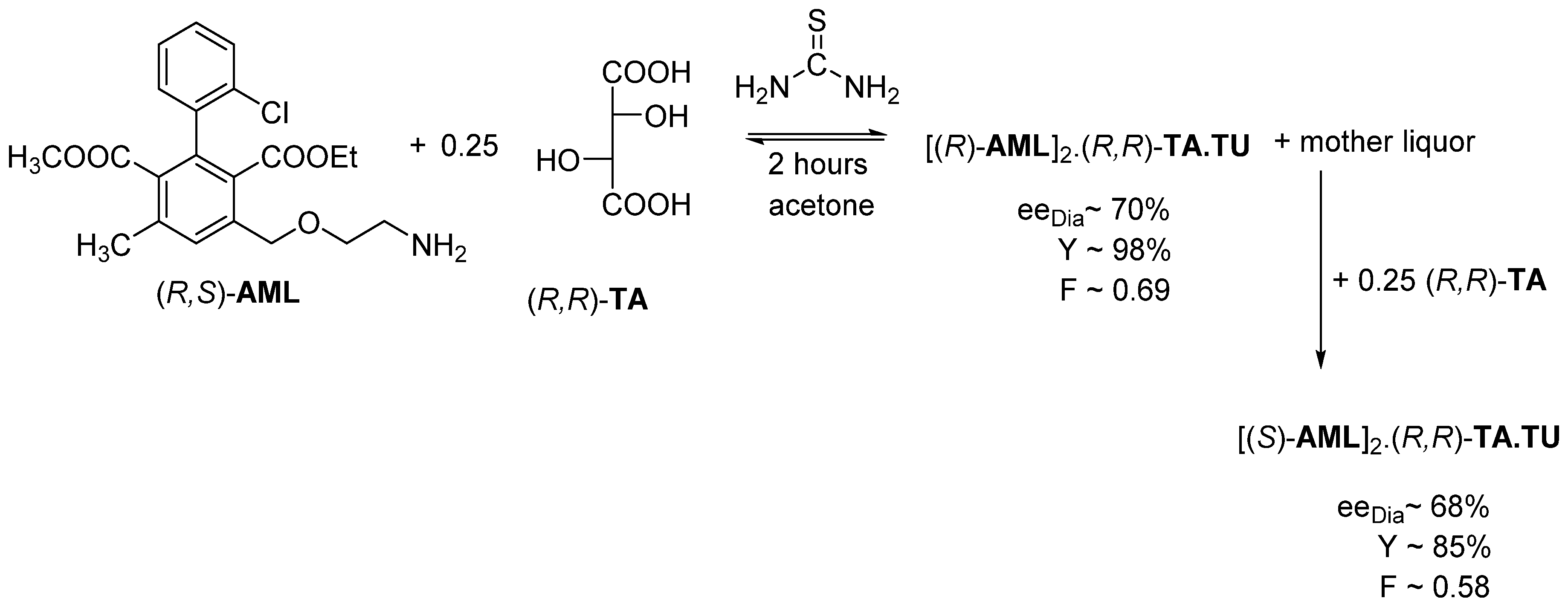
2.1.6. Tandem Resolution
2.2. Separation of Non-Racemic Mixtures
2.2.1. Separation of Non-Racemic Enantiomeric Mixtures by Recrystallization from Solvent or Melt Phase
2.2.2. Melt Crystallization of the Enantiomeric Mixture
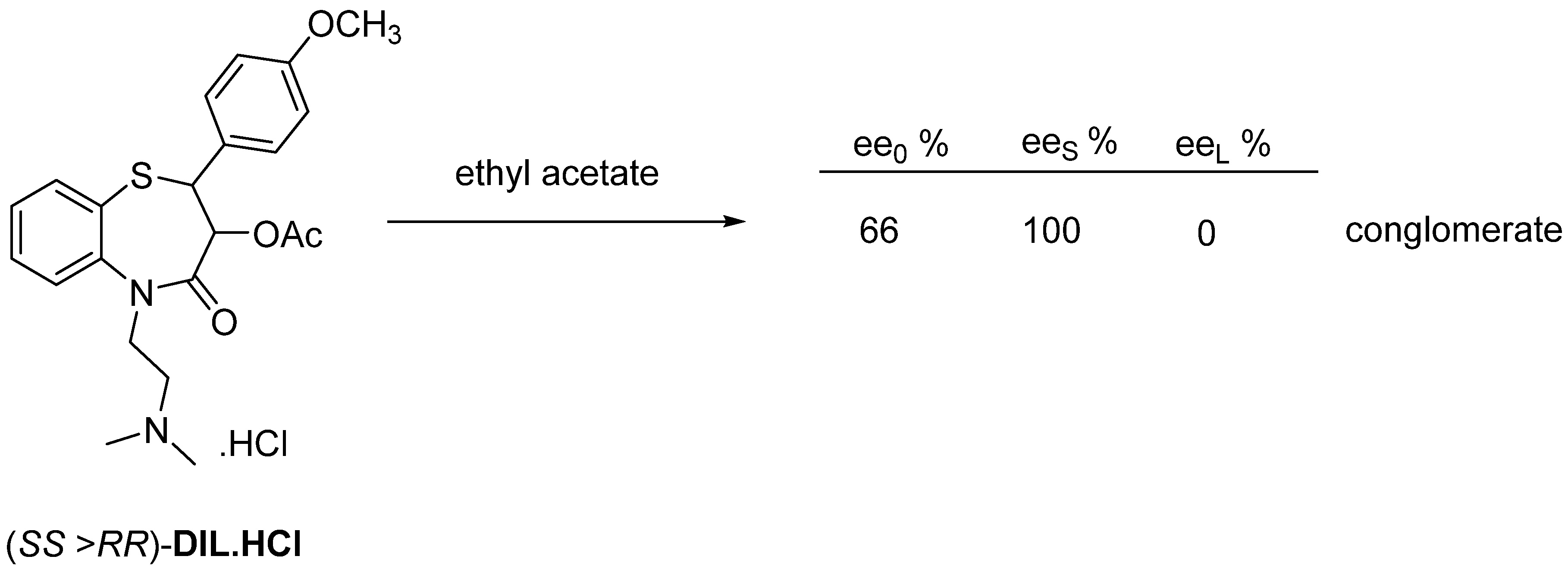
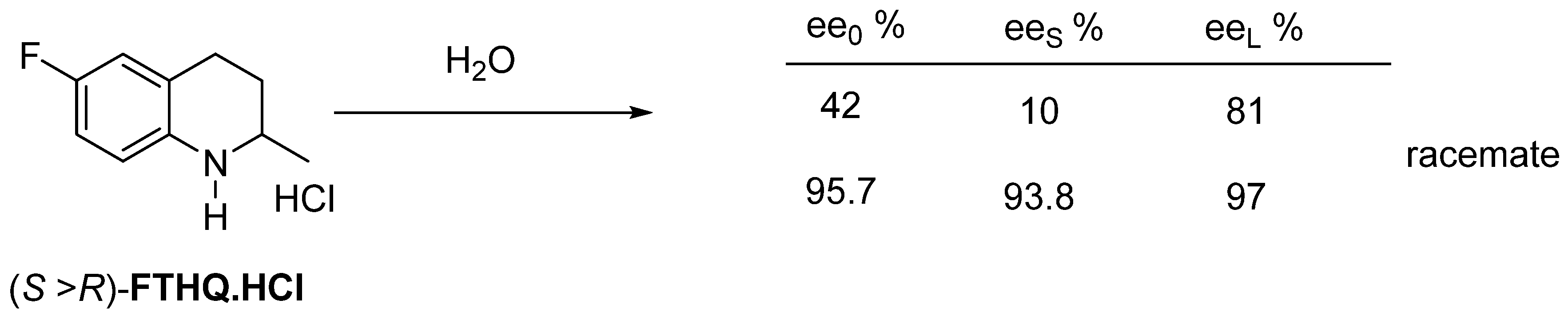
2.2.3. Recrystallization of the Enantiomeric Mixture from Solvent
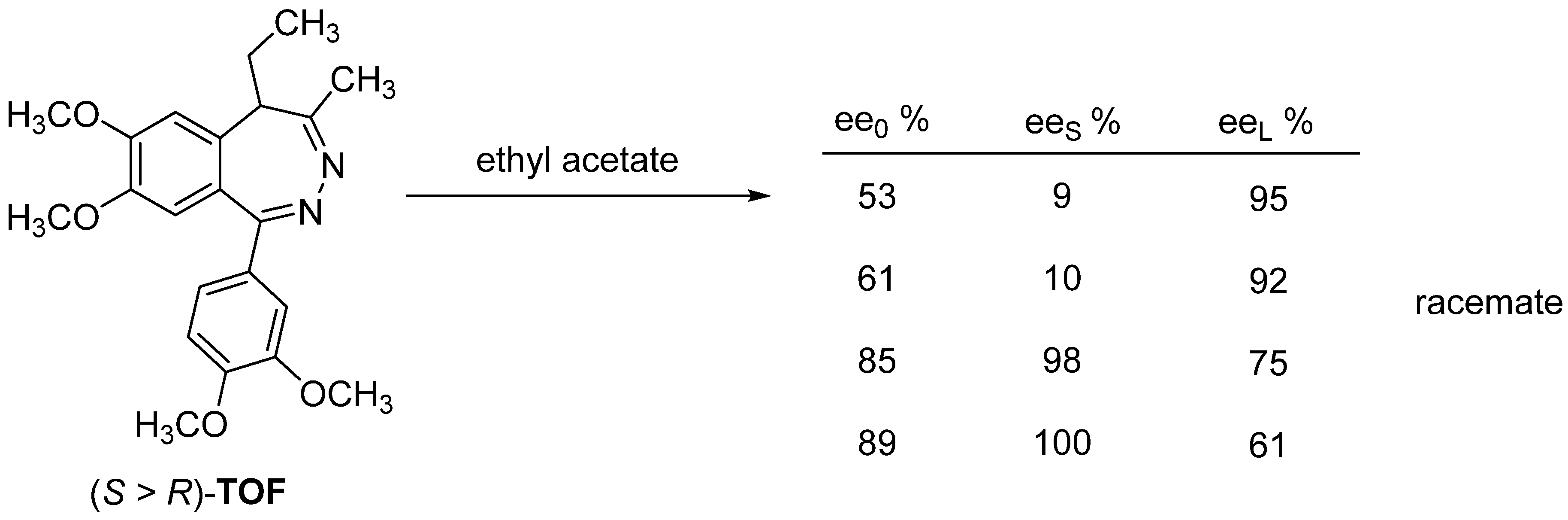
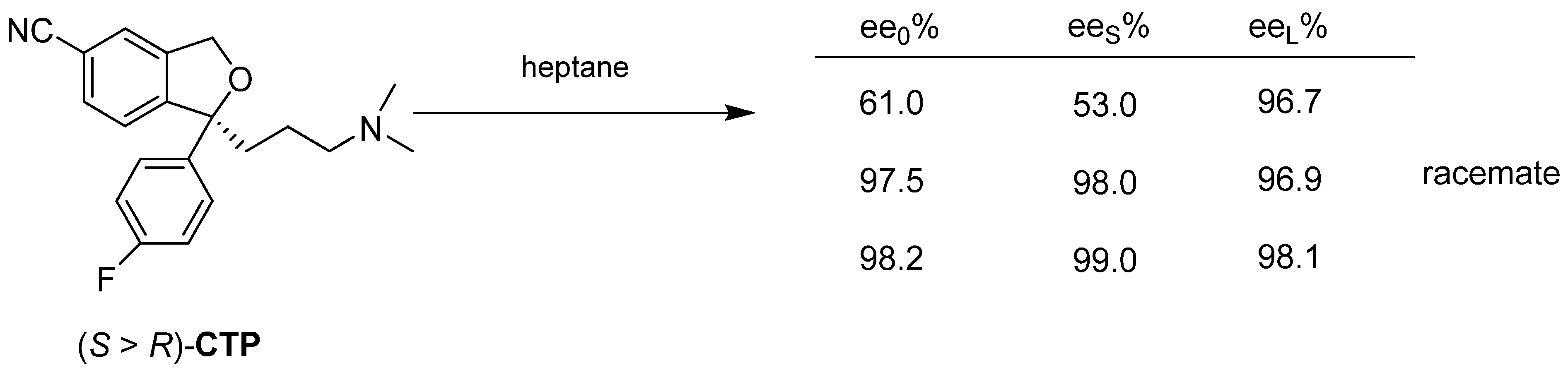
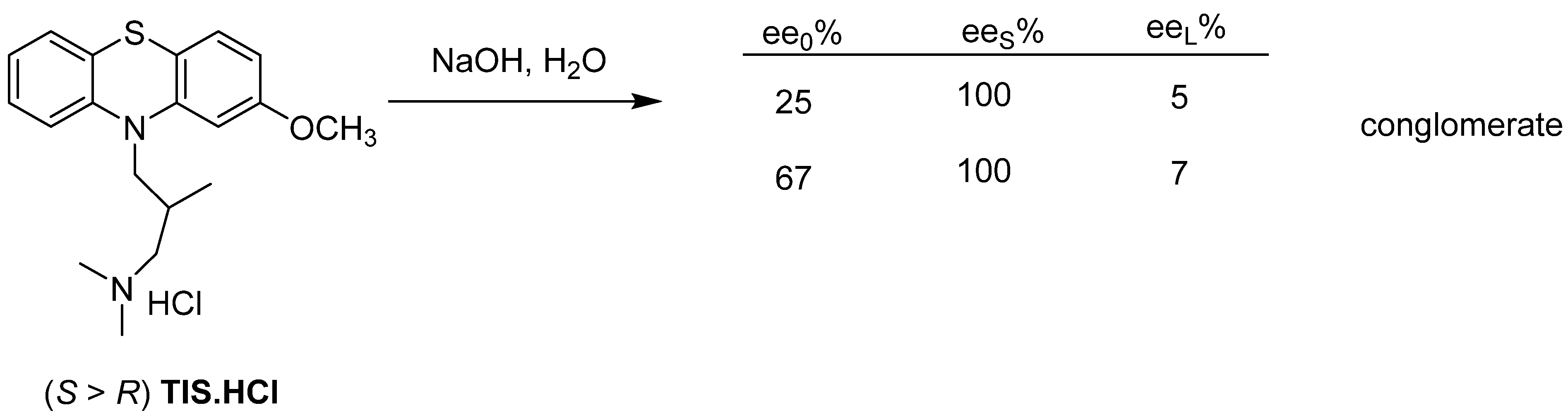
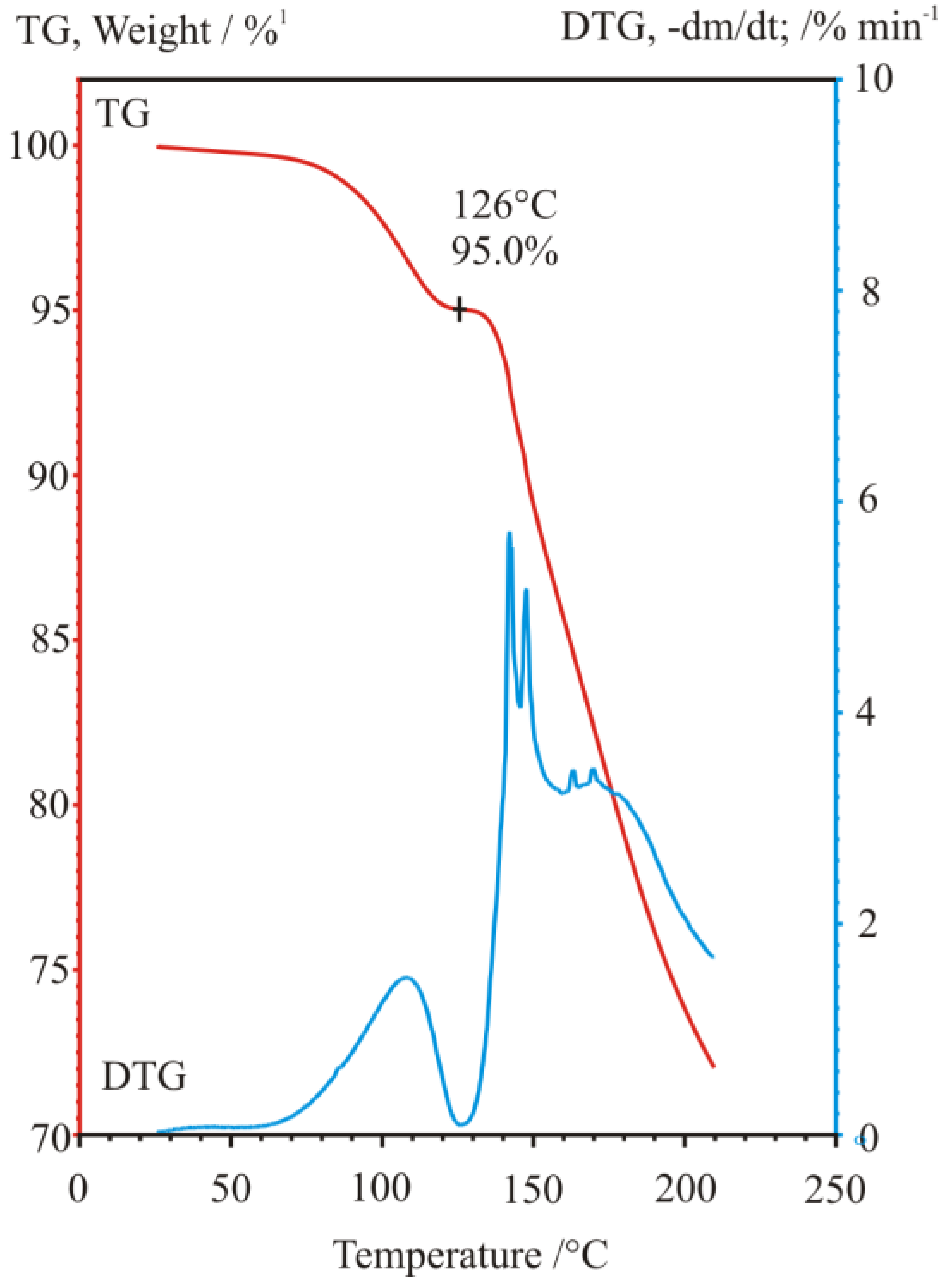
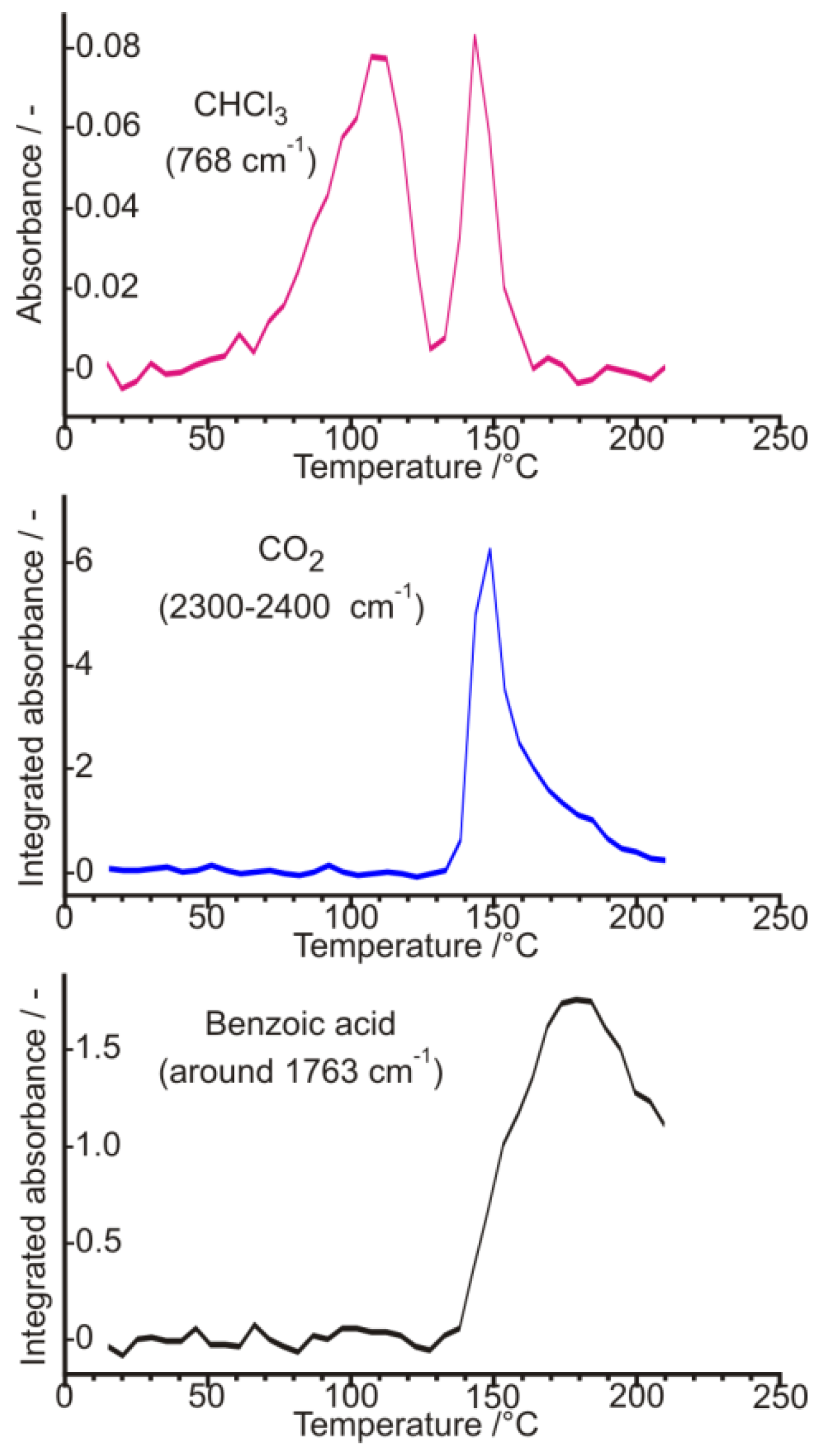
3. Three Examples of Possible Role of Solid State Analytical and Structural Investigations in Exploration and Invention Design of Resolution Processes
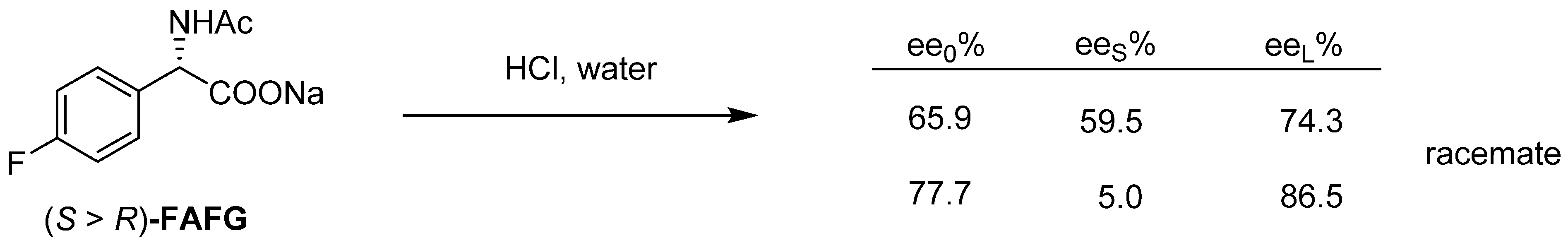
3.1. Example No. 1
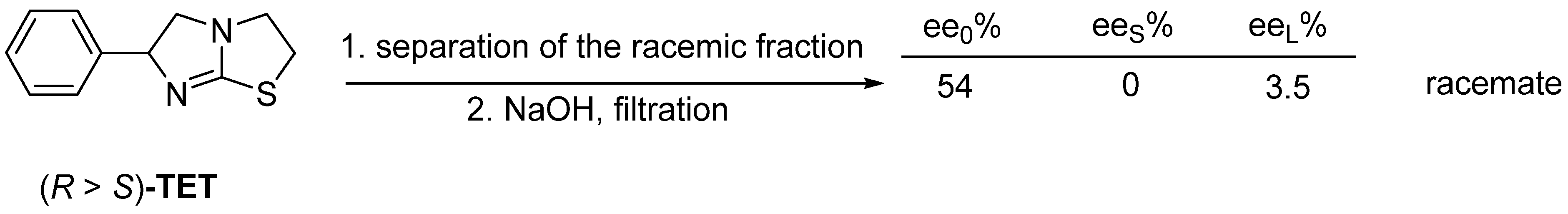
3.2. Example No. 2
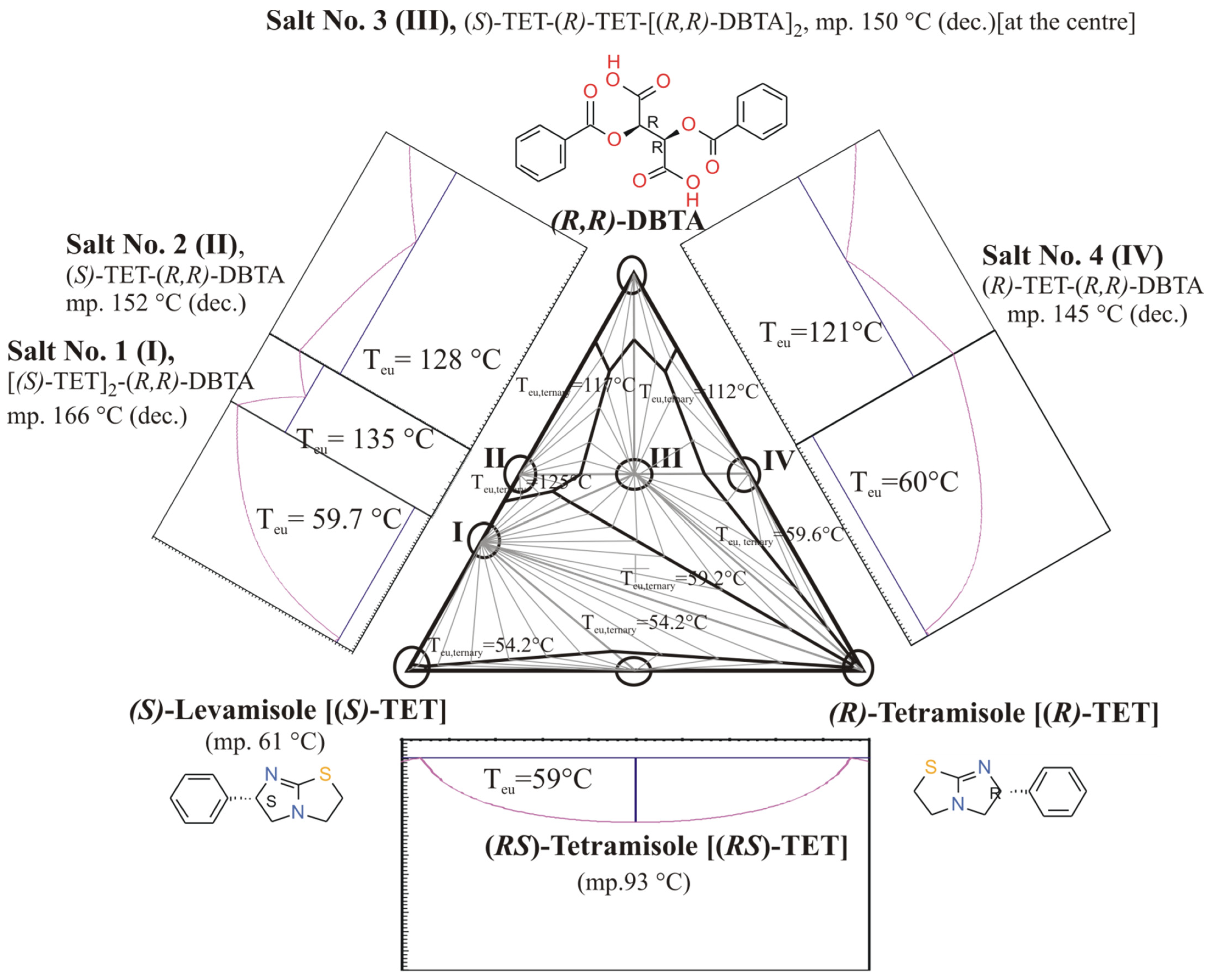
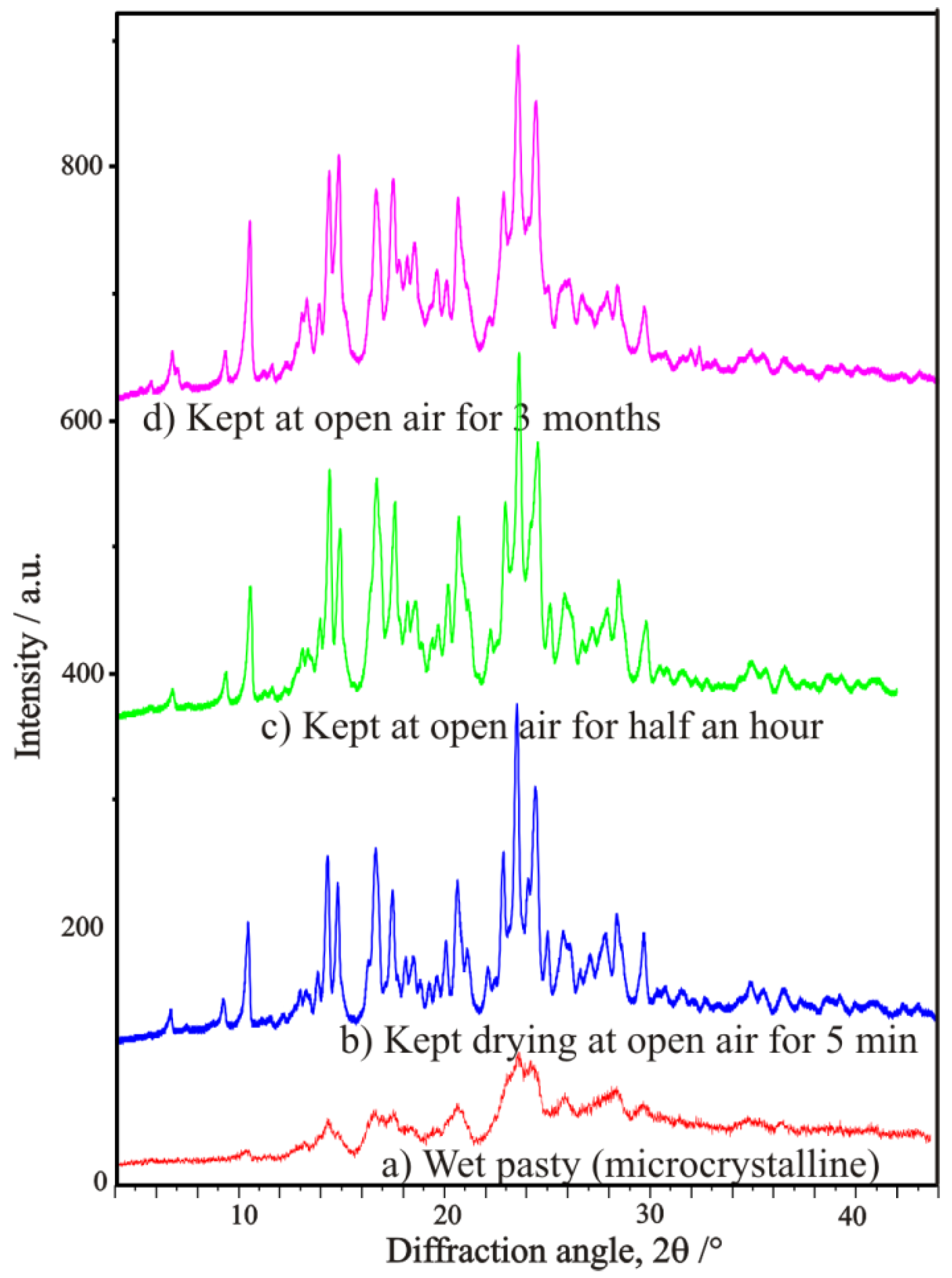
3.3. Example No. 3
4. Conclusions
- I.
- The separations exploit the different compositional distributions of mixtures of diastereoisomers and mixtures of enantiomers between two phases. A dynamic equilibrium exists between the two phases until separation.
- II.
- Diastereoisomers are salts of the racemic compound and the resolving agent (1:1 or 1:0.5).
- III.
- Enantiomeric mixtures are a mixture of the diastereomeric associates of the enantiomeric excess and the racemic moiety.
- IV.
- Mixtures of diastereoisomers and enantiomers are characterized by their melting binary phase diagrams and their eutectic compositions.
- V.
- Knowing the eutectic composition (eeDia) of the melting binary phase diagrams of the diastereomeric mixtures, the achievable result (Foptimum) of a given resolution can be calculated.
- VI.
- Either the racemic compound with its eutectic composition (eeEuRac) or the resolving agent with the eutectic composition of its enantiomeric mixtures (eeEuResAg) can be the determinant, thus encoding, depending on the circumstances, the enantiomeric enrichment of the enantiomeric mixtures, obtained from the crystallized diastereomeric salt (eeDia~eeEuRac or eeDia~eeEuResAg).
- VII.
- The composition and yield (Y) of eeDia obtained from crystalline separated diastereomers is a function of solvent and crystallization time.
- VIII.
- The optimal enantiomeric separation (F) is expected to be under the influence of kinetic or thermodynamic control. If the effect of the favorable kinetic control is rapidly reduced, it can be stabilized by ultrasonic irradiation (or other kind of energy transmission is required).
- IX.
- If the interaction of the solvent and the resolving agent produces a mixture of resolving agents, the result of the resolution may be greater than calculated based on the melting binary phase diagram of the diastereomer with the original resolving agent.
- X.
- If the diastereomer crystallizes only as a solvate, but the result (F) is low, it is preferable to use a solvent that also contains the solvent which forms the solvate.
- XI.
- If the given diastereomer forms only solvates but the solvate-forming solvents are not favorable, it can be successfully crystallized from a conventional solvent with a solid compound with a ‘related structure’ containing the common structural part of the solvent.
- XII.
- Non-racemic enantiomeric mixtures are mixtures of diastereomerically related associates.
- XIII.
- From the enantiomeric mixture forming the conglomerate, a fraction of higher enantiomeric enrichment than the initial one is always crystallized.
- XIV.
- From a conglomerate-forming enantiomeric mixture, a fraction with higher enantiomeric enrichment than the initial one will crystallize.
- XV.
- The derivative with an achiral reagent of a conglomerate-forming enantiomeric mixture may also behave as a racemate and vice versa (racemate-forming enantiomeric mixture may also behave as a conglomerate).
- XVI.
- From conglomerate or racemate forming enantiomeric mixtures, a purer fraction of the starting mixtures with an enantiomeric enrichment above the eutectic composition is always introduced into the crystalline phase.
- XVII.
- The phase that crystallizes from a melt enantiomeric mixture and the composition of the melt can be different
- XVIII.
- In the case of fractional precipitation from a solution of achiral derivatives of enantiomeric mixtures, either the enantiomeric excess or the racemic fraction crystallizes into the solid phase in relation to the composition of the initial enantiomeric enrichment and the binary phase diagram of the mixture.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pasteur, L. Transformation des acides tartriques en acide racémique—Découverte de l’acide tartrique inactif. Nouvelle méthode de séparation de l’acide racémique en acides tartriques droit et gauche. Compt. Rend. 1853, 37, 162–166. [Google Scholar]
- Pope, W.J.; Peachey, S.J. CVIII—The application of powerful optically active acids to the resolution of externally compensated basic substances. Resolution of tetrahydroquinaldine. J. Chem. Soc. 1899, 75, 1066–1093. [Google Scholar] [CrossRef]
- Kozma, D.; Pokol, G.; Ács, M. Calculation of the efficiency of optical resolutions on the basis of the binary phase diagram for the diastereoisomeric salts. J. Chem. Soc. Perkin Trans. 2 1992, 3, 435–439. [Google Scholar] [CrossRef]
- Madarász, J.; Pokol, G.; Ács, M.; Fogassy, E. Merit of estimations from DSC measurements for the efficiency of optical resolutions. J. Thermal. Anal. Calorim. 1994, 42, 877–894. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates, and Resolution; John Wiley & Sons: New York, NY, USA, 1981; pp. 47–91. [Google Scholar]
- Kozma, D.; Acs, M.; Fogassy, E. Predictions of which diastereoisomeric salt precipitates during an optical resolution via diastereoisomeric salt formation. Tetrahedron 1994, 50, 6907–6912. [Google Scholar] [CrossRef]
- Fogassy, E.; Lopata, A.; Faigl, F.; Ács, M.; Darvas, F.; Tőke, L. A quantitative approach to optical resolution. Tetrah. Lett. 1980, 21, 647–650. [Google Scholar] [CrossRef]
- Pálovics, E.; Schindler, J.; Faigl, F.; Fogassy, E. Behavior of Structurally Similar Molecules in the Resolution Processes. In Comprehensive Chirality; Physical Separations; Erick, C., Hisashi, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 1, pp. 91–95. ISBN 978-0-08-095167-6. [Google Scholar]
- Pálovics, E.; Bánhegyi, D.F.; Fogassy, E. Effect of the Enantiomeric Ratio of Eutectics on the Results and Products of the Reactions Proceeding with the Participation of Enantiomers and Enantiomeric Mixtures. Chemistry 2020, 2, 51. [Google Scholar] [CrossRef]
- Bánhegyi, D.F.; Pálovics, E. The Stoichiometry, Structure and Possible Formation of Crystalline Diastereomeric Salts. Symmetry 2021, 13, 667. [Google Scholar] [CrossRef]
- Gizur, T.; Fogassy, E.; Bálint, J.; Egri, G.; Tőrley, J.; Demeter, A.; Greiner, I. New practical synthesis of tamsulosin. Chirality 2008, 2, 790–795. [Google Scholar] [CrossRef]
- Dombrády, Z.; Pálovics, E.; Fogassy, E. Separation of Diastereomers Taking Advantage for the Kinetic Control and Structure of Resolving Agent. Curr. Res. Bioorg. Org. Chem. 2019, 2, 1–6. [Google Scholar] [CrossRef][Green Version]
- Szeleczky, Z.; Kis-Mihály, E.; Semsey, S.; Pataki, H.; Bagi, P.; Pálovics, E.; Marosi, G.; Pokol, G.; Fogassy, E.; Madarász, J. Effect of ultrasound-assisted crystallization in the diastereomeric salt resolution of tetramisole enantiomers in ternary system with O,O’-dibenzoyl-(2R,3R)- tartaric acid. Ultrason. Sonochemistry 2016, 32, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Bánhegyi, D.F.; Fogassy, E.; Pálovics, E. Possibilities of Exploiting Kinetic Control in the Continuous Fractional Crystallization of Diastereomeric Mixtures. Symmetry 2021, 13, 1516. [Google Scholar] [CrossRef]
- Bosits, M.H.; Pálovics, E.; Madarász, J.; Fogassy, E. New discoveries in enantiomeric separation of racemic tofisopam. Hindawi J. Chem. 2019, 2019, 4980792. [Google Scholar] [CrossRef]
- Bunce, R.A. Recent Advances in the Use of Tandem Reactions for Organic Synthesis. Tetrahedron 1995, 51, 13103–13159. [Google Scholar] [CrossRef]
- Ho, T.-L. Tandem Organic Reactions; John Wiley & Sons: New York, NY, USA, 1992. [Google Scholar]
- Bánhegyi, D.F.; Szolcsányi, D.; Madarász, J.; Pálovics, E. Enantiomeric separation of racemic Amlodipine by sequential fractional crystallization through formation of diastereomeric salt solvates and co-crystals of solvate-like compounds with related structure—A tandem resolution. Chirality 2022, 34, 374–395. [Google Scholar] [CrossRef] [PubMed]
- Faigl, F.; Fogassy, E.; Nógrádi, M.; Pálovics, E.; Schindler, J. Separation of non-racemic mixtures of enantiomers: An essential part of optical resolution. Org. Biomol. Chem. 2010, 8, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Viedma, C.; McBride, M.; Karh, B.; Cintas, P. Enantiomer-Specific Oriented Attachment: Formation of Macroscopic Homochiral Crystal Aggregates from a Racemic System. Angewandte Chem. 2013, 125, 10739–10742. [Google Scholar] [CrossRef]
- Viedma, C. Chiral Symmetry Breaking During Crystallization: Complete Chiral Purity Induced by Nonlinear Autocatalysis and Recycling. Phys. Rev. Lett. 2005, 94, 065504. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Wang, T. Supramolecular chirality in self-assembled systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef]
- Sang, Y.; Liu, M. Symmetry breaking in self-assembled nanosystems. Symmetry 2019, 11, 950. [Google Scholar] [CrossRef]
- Avalos, M.; Babiano, M.; Cintás, P.; Imenez, J.L.; Palacios, J.C. Symmetry breaking by spontaneous crystallization. Orig. Life. Evol. Biosph. 2004, 34, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Fogassy, E.; Faigl, F.; Acs, M. A new method for designing optical resolutions and for determination of relative configurations. Tetrahedron 1985, 41, 2837–2840. [Google Scholar] [CrossRef]
- Fogassy, E.; Faigl, F.; Acs, M. Diastereoisomeric interactions and selective reactions in solutions of enantiomers. Tetrahedron 1985, 41, 2841–2845. [Google Scholar] [CrossRef]
- Pálovics, E.; Fogassy, E.; Schindler, J.; Nógrádi, M. Nonlinear chiral interactions in resolutions with benzylamine derivatives. Chirality 2006, 19, 1–4. [Google Scholar] [CrossRef]
- Nemák, K.; Kozma, D.; Fogassy, E. Study of the Mechanism of Optical Resolutions Via Diastereoisomeric Salt Formation Part 4. The Role of the Crystallization Temperature in Optical Resolution of Pipecolic Acid Xylidides. Mol. Cryst. Liq. Cryst. 1996, 276, 31–36. [Google Scholar] [CrossRef]
- Bánhegyi, D.F.; Fogassy, E.; Madarász, J.; Pálovics, E. Optical Resolution of Two Pharmaceutical Bases with Various Uses of Tartaric Acid Derivatives and Their Sodium Salts: Racemic Ephedrine and Chloramphenicol Base. Molecules 2022, 27, 3134. [Google Scholar] [CrossRef]
- Fogassy, E.; Ács, M.; Tóth, G.; Simon, K.; Láng, T.; Ladányi, L.; Párkányi, L. Clarification of anomalous chiroptical behaviour and determination of the absolute configuration of 1-(3,4-dimethoxyphenyl)-4-methyl-5-ethyl-7,8-dimethoxy-5H-2,3-benzodiazepine. J. Mol. Struct. 1986, 147, 143–154. [Google Scholar] [CrossRef]
- Brienne, M.J.; Jacques, J.; Marsó, K.; Ács, M. Sur les propriétés physiques du tétramisole, du lévamisole at de leurs mélanges (About the physical properties of tetramisole, levamisol and their mixtures). Bull. Soc. Chim. France 1985, 5, 876–880. [Google Scholar]
- Fogassy, E.; Ács, M.; Felméri, J.; Aracs, Z.; Rusznák, I. Preparation of L-(-)-6-Phenyl-2,3,5,6-tetrahydroimidazo [2,1-b]thiazole. Period. Polytech. 1976, 20, 247–253. [Google Scholar]
- Hu, M.; He, P.; Chen, Y.; Carr, G.; Guo, J.; Ye, N. Method validation and determination of enantiomers and conformers in tofisopam drug substances and drug products by chiral high-performance liquid chromatography and kinetic and thermodynamic study of the interconversion of the conformers. J. Chromatogr. A 2006, 1129, 47–53. [Google Scholar] [CrossRef]
- Foroughbakhshfasaei, M.; Szabó, Z.I.; Mirzahosseini, A.; Horváth, P.; Tóth, G. Enantiomeric quality control of R-Tofisopam by HPLC using polysaccharide-type chiral stationary phases in polar organic mode. Electrophoresis 2018, 39, 2566–2574. [Google Scholar] [CrossRef] [PubMed]
- Tőke, L.; Fogassy, E.; Láng, T.; Ács, M.; Láng, J.; Tóth, G.; Petőcz, L.; Kosóczky, I.; Grasser, K.; Reichmann, G. Eljárás az 1-(3,4-dimetoxifenil)-5-etil-7,8-dimetoxi-4-metil-5H-2,3-benzodiazepin enantiomerjei és sói előállítására. Hungary Patent HU 178516, 28 September 1981. [Google Scholar]
- Tőke, L.; Fogassy, E.; Ács, M.; Bencsik, P. Eljárás az 1-(3,4-dimetoxifenil)-5-etil-7,8-diemtoxi-4-metil-5H-2,3-benzodiazepin nagytisztaságú (−)-enantiomerjének előálítására. Hungary Patent HU 179452, 28 January 1982. [Google Scholar]
- Boultif, A.; Louer, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- David, W.I.; Shankland, K.; Van De Streek, J.; Pidcock, E.; Motherwell, W.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Gönczi, K.; Kudar, V.; Jászay, Z.; Bombicz, P.; Faigl, F.; Madarász, J. Solid state structural relation and binary melting phase diagram of (S-) and racemic 2-(2-nitro-1-phenylethyl)-1,3-diphenyl-propane-1,3-dione. Thermochim. Acta 2014, 580, 46–52. [Google Scholar] [CrossRef][Green Version]































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pálovics, E.; Madarász, J.; Pokol, G.; Fogassy, E.; Bánhegyi, D.F. Economic Separations of Organic Acidic or Basic Enantiomeric Mixtures—A Protocol Suggestion. Int. J. Mol. Sci. 2023, 24, 846. https://doi.org/10.3390/ijms24010846
Pálovics E, Madarász J, Pokol G, Fogassy E, Bánhegyi DF. Economic Separations of Organic Acidic or Basic Enantiomeric Mixtures—A Protocol Suggestion. International Journal of Molecular Sciences. 2023; 24(1):846. https://doi.org/10.3390/ijms24010846
Chicago/Turabian StylePálovics, Emese, János Madarász, György Pokol, Elemér Fogassy, and Dorottya Fruzsina Bánhegyi. 2023. "Economic Separations of Organic Acidic or Basic Enantiomeric Mixtures—A Protocol Suggestion" International Journal of Molecular Sciences 24, no. 1: 846. https://doi.org/10.3390/ijms24010846
APA StylePálovics, E., Madarász, J., Pokol, G., Fogassy, E., & Bánhegyi, D. F. (2023). Economic Separations of Organic Acidic or Basic Enantiomeric Mixtures—A Protocol Suggestion. International Journal of Molecular Sciences, 24(1), 846. https://doi.org/10.3390/ijms24010846