
A Short ERAP2 That Binds IRAP Is Expressed in Macrophages Independently of Gene Variation

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
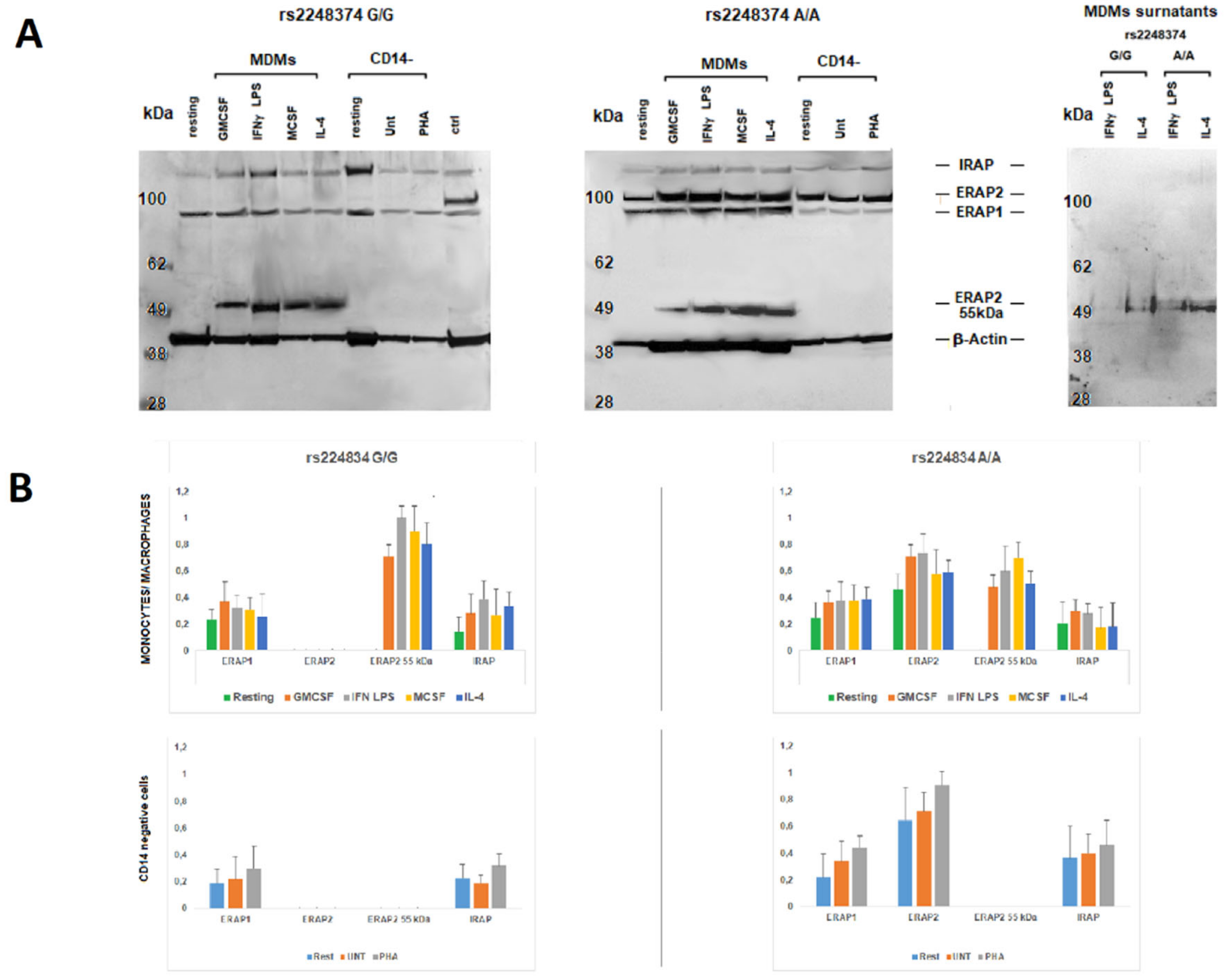
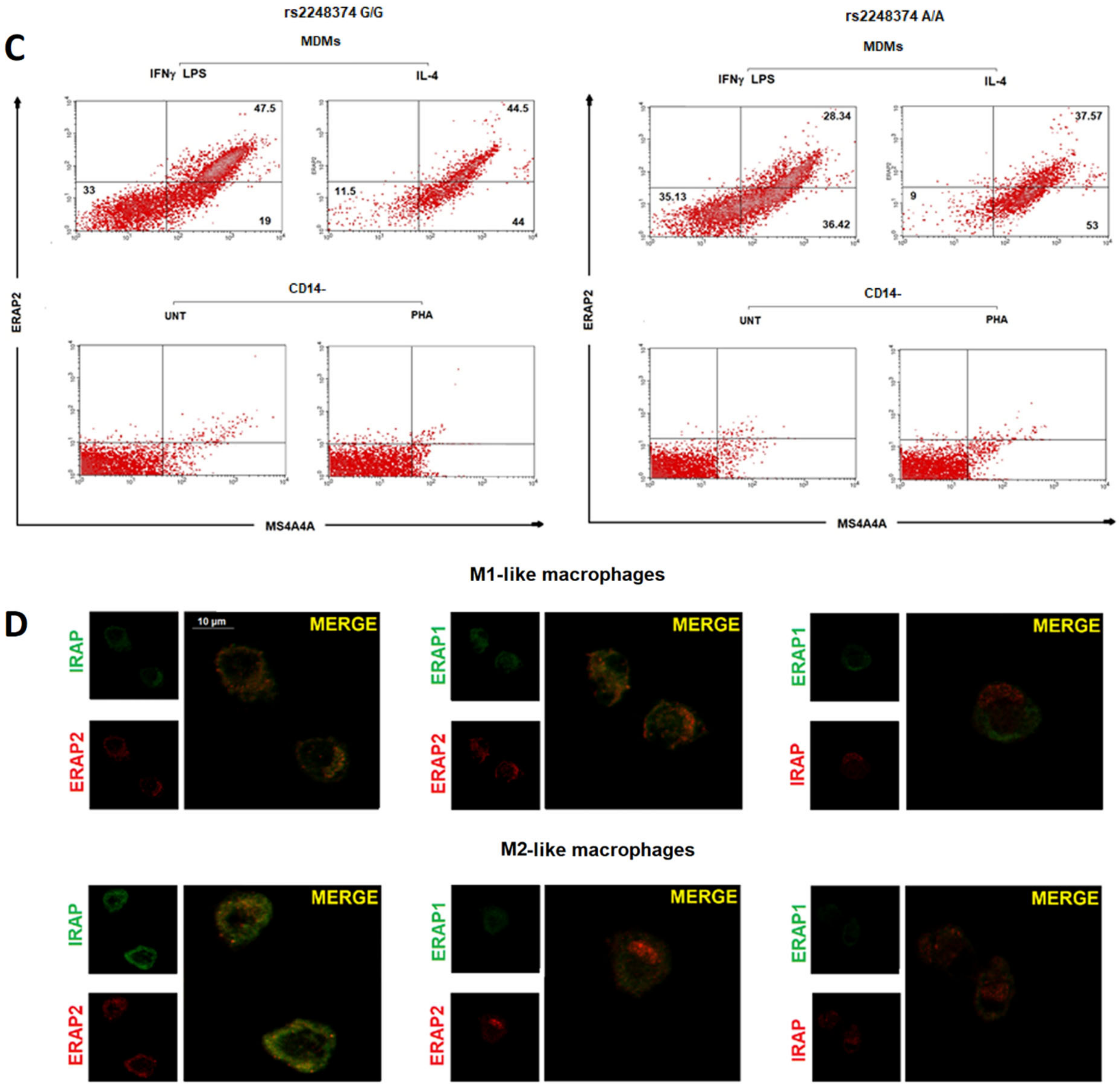
2.1. Monocyte-Derived Macrophages Express a Short ERAP2 Isoform That Binds IRAP
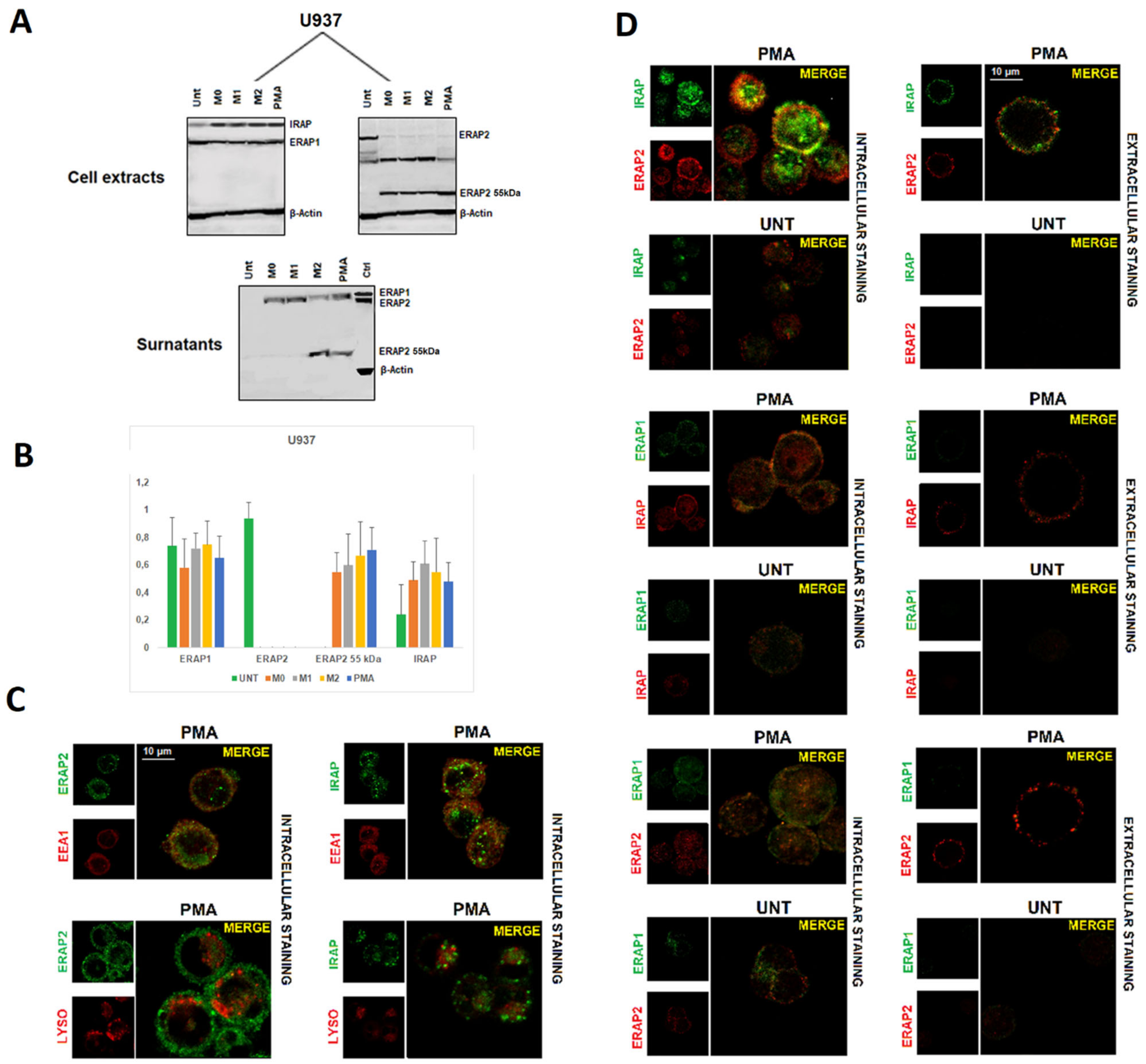
2.2. Differentiation of U937 Cells Correlates with the Loss of ERAP2 Full Length and the Occurrence of a ~55 kDa Fragment
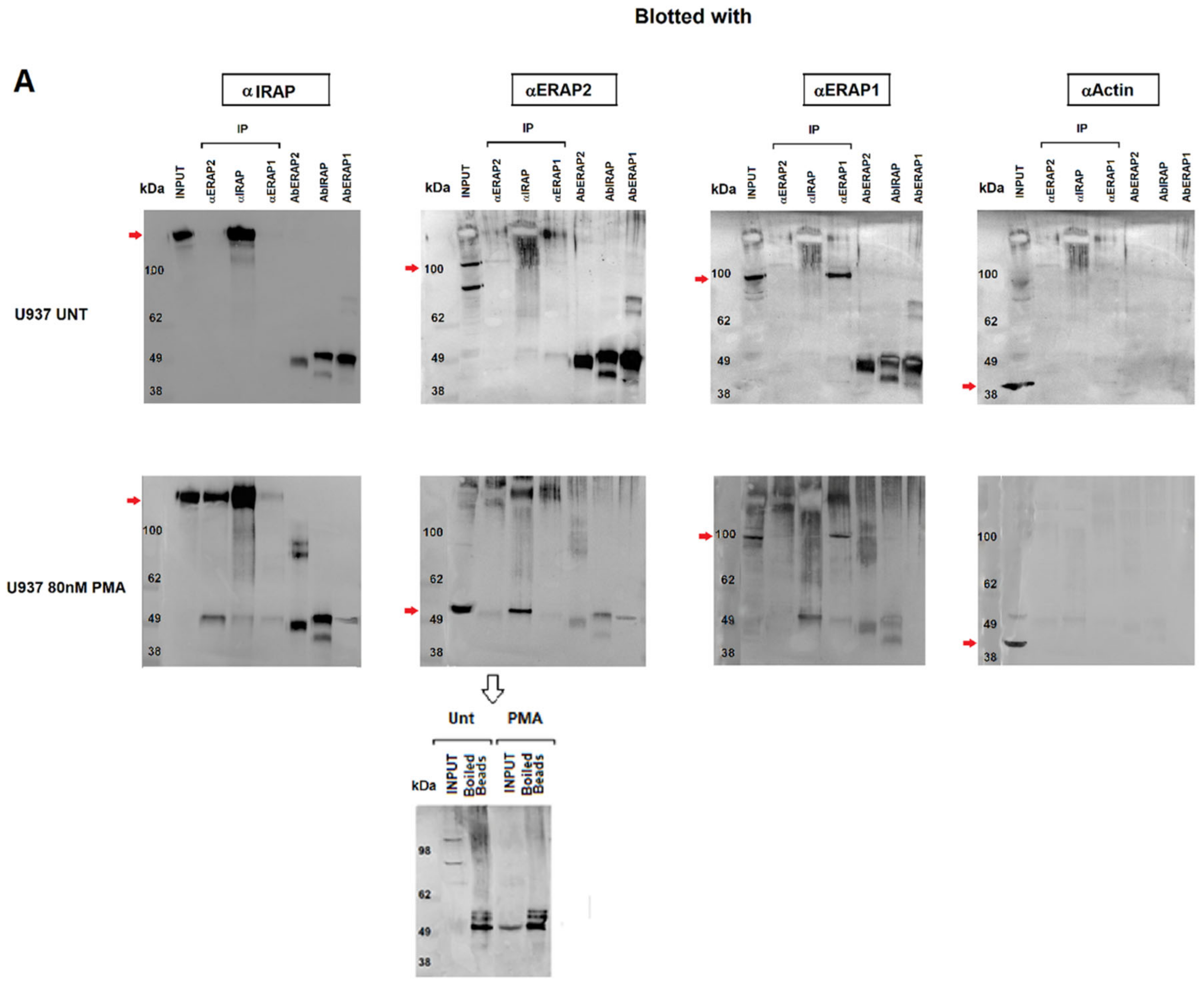
2.3. IRAP Directly Binds the ~55 kDa ERAP2
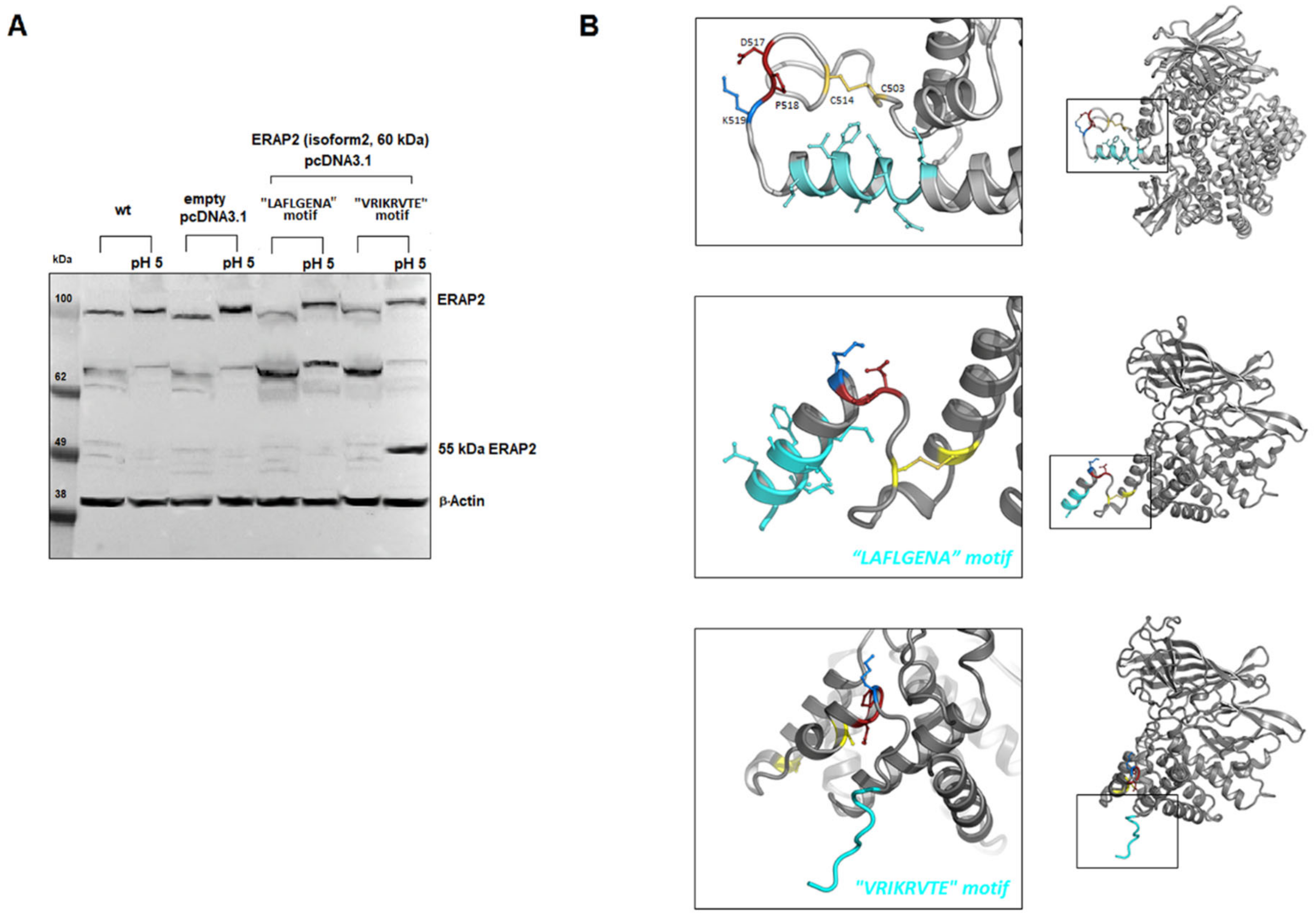
2.4. ERAP2 Contains a Potential Autocatalytic Cleavage Site
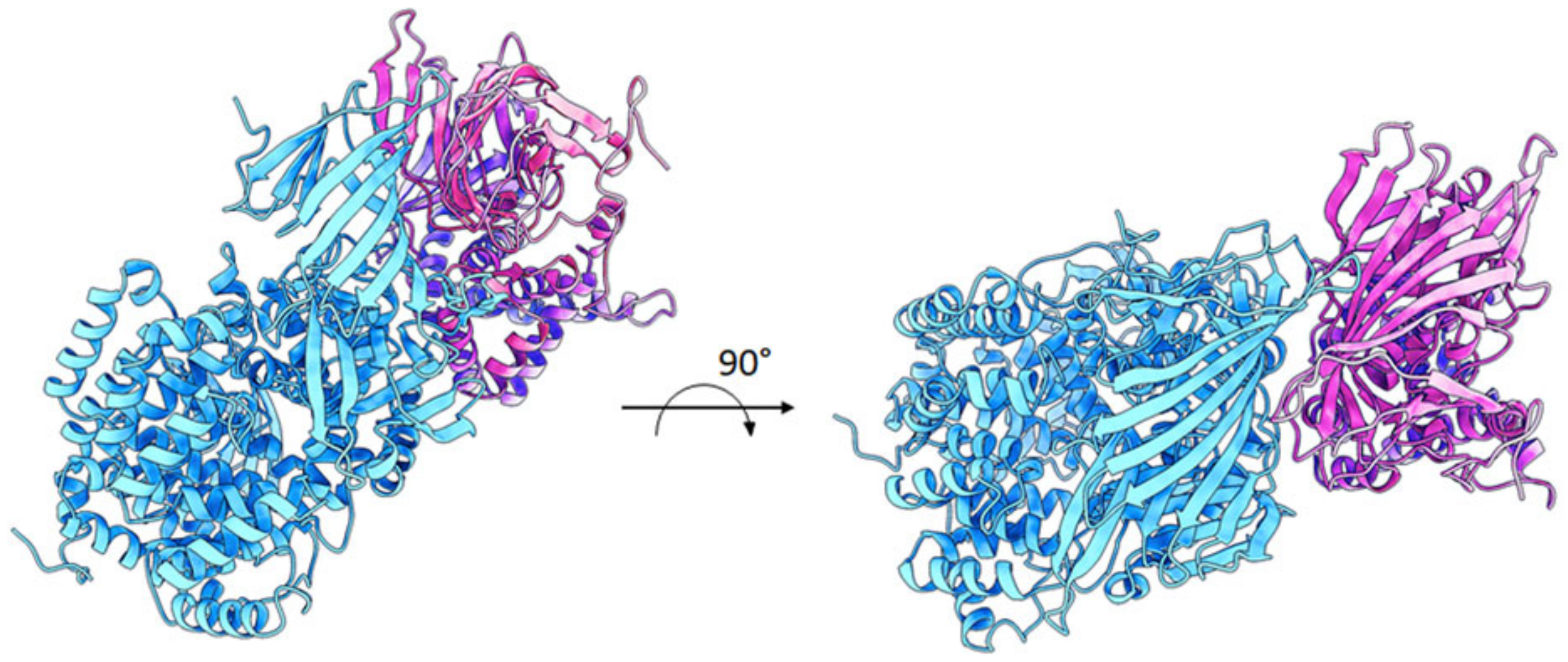
2.5. ERAP2 “Short” Derives from Longer Precursors
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Antibodies
4.3. Macrophages-Derivated-Monocytes Generation and CD14 Negative PBMCs
4.4. DNA Extraction and rs2248374 Genotyping
4.5. Flow Cytometry Analysis
4.6. Western Blot
4.7. Confocal Microscopy
4.8. Immunoprecipitation
4.9. Transfection
4.10. Modelling
4.11. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tsujimoto, M.; Hattori, A. The oxytocinase subfamily of M1 aminopeptidases. Biochim. Biophys. Acta 2005, 1751, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Paladini, F.; Fiorillo, M.T.; Tedeschi, V.; Mattorre, B.; Sorrentino, R. The Multifaceted Nature of Aminopeptidases ERAP1, ERAP2, and LNPEP: From Evolution to Disease. Front. Immunol. 2020, 11, 1576. [Google Scholar] [CrossRef] [PubMed]
- Weimershaus, M.; Evnouchidou, I.; Saveanu, L.; van Endert, P. Peptidases trimming MHC class I ligands. Curr. Opin. Immunol 2013, 25, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Saveanu, L.; Carroll, O.; Weimershaus, M.; Guermonprez, P.; First, E.; Lindo, V.; Fiona, G.; Jean, D.; Roland, K.; Susanna, R.K.; et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science 2009, 325, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Weimershaus, M.; Mauvais, F.X.; Evnouchidou, I.; Lawand, M.; Saveanu, L.; van Endert, P. IRAP Endosomes Control Phagosomal Maturation in Dendritic Cells. Front. Cell Dev. Biol. 2020, 8, 585713. [Google Scholar] [CrossRef] [PubMed]
- Vear, A.; Gaspari, T.; Thompson, P.; Chai, S.Y. Is There an Interplay Between the Functional Domains of IRAP? Front. Cell Dev. Biol. 2020, 8, 585237. [Google Scholar] [CrossRef]
- Descamps, D.; Evnouchidou, I.; Caillens, V.; Drajac, C.; Riffault, S.; van Endert, P.; Saveanu, L. The Role of Insulin Regulated Aminopeptidase in Endocytic Trafficking and Receptor Signaling in Immune Cells. Front. Mol. Biosci. 2020, 7, 583556. [Google Scholar] [CrossRef]
- Albiston, A.L.; Morton, C.J.; Ng, H.L.; Pham, V.; Yeatman, H.R.; Ye, S.; Fernando, R.N.; de Bundel, D.; Ascher, D.B.; Mendelsohn, F.A.O.; et al. Identification and characterization of a new cognitive enhancer based on inhibition of insulin-regulated aminopeptidase. FASEB J. 2008, 22, 4209–4217. [Google Scholar] [CrossRef] [Green Version]
- Vargas, F.; Wangesteen, R.; Rodríguez-Gómez, I.; García-Estañ, J. Aminopeptidases in Cardiovascular and Renal Function. Role as Predictive Renal Injury Biomarkers. Int. J. Mol. Sci. 2020, 21, 5615. [Google Scholar] [CrossRef]
- Sharip, A.; Kunz, J. Understanding the Pathogenesis of Spondyloarthritis. Biomolecules 2020, 10, 1461. [Google Scholar] [CrossRef]
- Babaie, F.; Hosseinzadeh, R.; Ebrazeh, M.; Seyfizadeh, N.; Aslani, S.; Salimi, S.; Maryam, H.; Gholamreza, A.; Farhad, J.; Hamed, M.; et al. The roles of ERAP1 and ERAP2 in autoimmunity and cancer immunity: New insights and perspective. Mol. Immunol. 2020, 121, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, V.; Paldino, G.; Paladini, F.; Mattorre, B.; Tuosto, L.; Sorrentino, R.; Fiorillo, M.T. The Impact of the ‘Mis-Peptidome’ on HLA Class I-Mediated Diseases: Contribution of ERAP1 and ERAP2 and Effects on the Immune Response. Int. J. Mol. Sci. 2020, 21, 9608. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Q.; Yang, Z.; Wang, W.; Li, B.; Bai, M.; Wu, J.; Ge, H.; Dong, Z.; Shen, J.; Tang, H.; et al. Genetic Study on Small Insertions and Deletions in Psoriasis Reveals a Role in Complex Human Diseases. J. Invest. Dermatol. 2019, 139, 2302–2312.e14. [Google Scholar] [CrossRef] [PubMed]
- Li, D.T.; Habtemichael, E.N.; Bogan, J.S. Vasopressin inactivation: Role of insulin-regulated aminopeptidase. Vitam. Horm. 2020, 113, 101–128. [Google Scholar] [CrossRef]
- Compagnone, M.; Cifaldi, L.; Fruci, D. Regulation of ERAP1 and ERAP2 genes and their disfunction in human cancer. Hum. Immunol. 2019, 80, 318–324. [Google Scholar] [CrossRef]
- D’Amico, S.; Tempora, P.; Lucarini, V.; Melaiu, O.; Gaspari, S.; Algeri, M.; Fruci, D. ERAP1 and ERAP2 Enzymes: A Protective Shield for RAS against COVID-19? Int. J. Mol. Sci. 2021, 22, 1705. [Google Scholar] [CrossRef]
- Lee, E.D. Endoplasmic Reticulum Aminopeptidase 2, a common immunological link to adverse pregnancy outcomes and cancer clearance? Placenta 2017, 56, 40–43. [Google Scholar] [CrossRef]
- Soltani, S.; Nasiri, M. Association of ERAP2 gene variants with risk of pre-eclampsia among Iranian women. Int. J. Gynaecol. Obstet. 2019, 145, 337–342. [Google Scholar] [CrossRef]
- McGonagle, D.; Aydin, S.Z.; Gül, A.; Mahr, A.; Direskeneli, H. MHC-I-opathy-unified concept for spondyloarthritis and Behçet disease. Nat. Rev. Rheumatol. 2015, 11, 731–740. [Google Scholar] [CrossRef]
- Kenna, T.J.; Robinson, P.C.; Haroon, N. Endoplasmic reticulum aminopeptidases in the pathogenesis of ankylosing spondylitis. Rheumatology 2015, 54, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Andrés, A.M.; Dennis, M.Y.; Kretzschmar, W.W.; Cannons, J.L.; Lee-Lin, S.Q.; Hurle, B.; NISC Comparative Sequencing Program; Schwartzberg, P.L.; Williamson, S.H.; Bustamante, C.D.; et al. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS Genet. 2010, 6, e1001157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Ogawa, K.; Hattori, A.; Tsujimoto, M. Secretion of endoplasmic reticulum aminopeptidase 1 is involved in the activation of macrophages induced by lipopolysaccharide and interferon-gamma. J. Biol. Chem. 2011, 286, 21906–21914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofner, L.D.; Hooper, N.M. Ectodomain shedding of cystinyl aminopeptidase from human placental membranes. Placenta 2002, 1, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Saulle, I.; Ibba, S.V.; Torretta, E.; Vittori, C.; Fenizia, C.; Piancone, F.; Minisci, D.; Lori, E.M.; Trabattoni, D.; Gelfi, C.; et al. Endoplasmic Reticulum Associated Aminopeptidase 2 (ERAP2) Is Released in the Secretome of Activated MDMs and Reduces in vitro HIV-1 Infection. Front. Immunol. 2019, 10, 1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiola, I.; Tomay, F.; De Pizzol, M.; Silva-Gomes, R.; Savino, B.; Gulic, T.; Doni, A.; Lonardi, S.; Boutet, M.A.; Nerviani, A.; et al. The macrophage tetraspan MS4A4A enhances dectin-1-dependent NK cell-mediated resistance to metastasis. Nat. Immunol. 2019, 8, 1012–1022. [Google Scholar] [CrossRef]
- Fruci, D.; Ferracuti, S.; Limongi, M.Z.; Cunsolo, V.; Giorda, E.; Fraioli, R.; Sibilio, L.; Carroll, O.; Hattori, A.; van Endert, P.M.; et al. Expression of Endoplasmic Reticulum Aminopeptidases in EBV-B Cell Lines from Healthy Donors and in Leukemia/Lymphoma, Carcinoma, and Melanoma Cell Lines. J. Immunol. 2006, 176, 4869–4879. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, R.; Polyak, M.J.; Zuccolo, J.; Puri, M.; Deng, L.; Roberts, L.; Zuba, A.; Storek, J.; Luider, J.M.; Sundberg, E.M.; et al. MS4A4A: A novel cell surface marker for M2 macrophages and plasma cells. Immunol. Cell Biol. 2017, 95, 611–619. [Google Scholar] [CrossRef]
- Papakyriakou, A.; Stratikos, E. The Role of Conformational Dynamics in Antigen Trimming by Intracellular Aminopeptidases. Front. Immunol. 2017, 8, 946. [Google Scholar] [CrossRef] [Green Version]
- Lidell, M.E.; Johansson, M.E.V.; Hansson, G.C. An autocatalytic cleavage in the C terminus of the human MUC2 mucin occurs at the low pH of the late secretory pathway. J. Biol. Chem. 2003, 278, 13944–13951. [Google Scholar] [CrossRef] [Green Version]
- Saulle, I.; Vanetti, C.; Goglia, S.; Vicentini, C.; Tombetti, E.; Garziano, M.; Mario, C.; Mara, B. A New ERAP2/Iso3 Isoform Expression Is Triggered by Different Microbial Stimuli in Human Cells. Could It Play a Role in the Modulation of SARS-CoV-2 Infection? Cells 2020, 9, 1951. [Google Scholar] [CrossRef]
- Ye, C.J.; Chen, J.; Villani, A.C.; Gate, R.E.; Subramaniam, M.; Bhangale, T.; Mark, N.L.; Towfique, R.; Raktima, R.; Li, W.; et al. Genetic analysis of isoform usage in the human anti-viral response reveals influenza-specific regulation of ERAP2 transcripts under balancing selection. Genome Res. 2018, 28, 1812–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetti, C.; Picardi, E.; Ye, M.; Camilli, G.; D’Erchia, A.M.; Cucina, L.; Locatelli, F.; Fianchi, L.; Teofili, L.; Pesole, G.; et al. RNA editing signature during myeloid leukemia cell differentiation. Leukemia 2017, 12, 2824–2832. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, D.; Ziotopoulou, A.; Kaloumenou, E.; Lelis, A.; Papasava, A. The Discovery of Insulin-Regulated Aminopeptidase (IRAP) Inhibitors: A Literature Review. Front. Pharmacol. 2020, 11, 585838. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D.; et al. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Lu, C.; Gam, R.; Pandurangan, A.P.; Goug, J. Genetic risk factors for death with SARS-CoV-2 from the UK Biobank. medRxiv 2020. [Google Scholar] [CrossRef]
- Barbé-Tuana, F.; Funchal, G.; Schmitz, C.R.R.; Maurmann, R.M.; Baue, M.E. The interplay between immunosenescence and age-related diseases. Semin. Immunopathol. 2020, 42, 545–557. [Google Scholar] [CrossRef]
- Paladini, F.; Fiorillo, M.T.; Vitulano, C.; Tedeschi, V.; Piga, M.; Cauli, A.; Alessandro, M.; Rosa, S. An allelic variant in the intergenic region between ERAP1 and ERAP2 correlates with an inverse expression of the two genes. Sci. Rep. 2018, 8, 10398. [Google Scholar] [CrossRef]
- Fic, E.; Kedracka-Krok, S.; Jankowska, U.; Pirog, A.; Dziedzicka-Wasylewska, M. Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis 2010, 21, 3573–3579. [Google Scholar] [CrossRef]
- Schneider, C.; Rasband, W.; Eliceiri, K. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Zinchuk, V.; Zinchuk, O.; Okada, T. Quantitative Colocalization Analysis of Multicolor Confocal Immunofluorescence Microscopy Images: Pushing Pixels to Explore Biological Phenomena. Acta Histochem. Cytochem. 2007, 40, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Kochan, G.; Krojer, T.; Harvey, D.; Fischer, R.; Chen, L.; Vollmar, M.; von Delft, F.; Kavanagh, K.L.; Brown, M.A.; Bowness, P.; et al. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc. Natl. Acad. Sci. USA 2011, 108, 7745–7750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mpakali, A.; Giastas, P.; Mathioudakis, N.; Mavridis, I.M.; Saridakis, E.; Efstratios, S. Structural Basis for Antigenic Peptide Recognition and Processing by Endoplasmic Reticulum (ER) Aminopeptidase 2. J. Biol. Chem. 2015, 290, 26021–26032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, S.J.; Ascher, D.B.; Hancock, N.C.; Holien, J.K.; Michell, B.J.; Chai, S.Y.; Morton, C.J.; Parker, M.W. Crystal structure of human insulin-regulated aminopeptidase with specificity for cyclic peptides. Protein Sci. 2015, 24, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangone, A.; Bonvin, A.M.J.J. Contact-based prediction of binding affinity in protein-protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Xue, L.; Rodrigues, J.; Kastritis, P.; Bonvin, A.M.J.J.; Vangone, A. PRODIGY: A web-server for predicting the binding affinity in protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattorre, B.; Caristi, S.; Donato, S.; Volpe, E.; Faiella, M.; Paiardini, A.; Sorrentino, R.; Paladini, F. A Short ERAP2 That Binds IRAP Is Expressed in Macrophages Independently of Gene Variation. Int. J. Mol. Sci. 2022, 23, 4961. https://doi.org/10.3390/ijms23094961
Mattorre B, Caristi S, Donato S, Volpe E, Faiella M, Paiardini A, Sorrentino R, Paladini F. A Short ERAP2 That Binds IRAP Is Expressed in Macrophages Independently of Gene Variation. International Journal of Molecular Sciences. 2022; 23(9):4961. https://doi.org/10.3390/ijms23094961
Chicago/Turabian StyleMattorre, Benedetta, Silvana Caristi, Simona Donato, Emilia Volpe, Marika Faiella, Alessandro Paiardini, Rosa Sorrentino, and Fabiana Paladini. 2022. "A Short ERAP2 That Binds IRAP Is Expressed in Macrophages Independently of Gene Variation" International Journal of Molecular Sciences 23, no. 9: 4961. https://doi.org/10.3390/ijms23094961
APA StyleMattorre, B., Caristi, S., Donato, S., Volpe, E., Faiella, M., Paiardini, A., Sorrentino, R., & Paladini, F. (2022). A Short ERAP2 That Binds IRAP Is Expressed in Macrophages Independently of Gene Variation. International Journal of Molecular Sciences, 23(9), 4961. https://doi.org/10.3390/ijms23094961