
Insights into the Genetic Architecture and Genomic Prediction of Powdery Mildew Resistance in Flax (Linum usitatissimum L.)

 ,
,  ,
,
Abstract
1. Introduction
2. Results
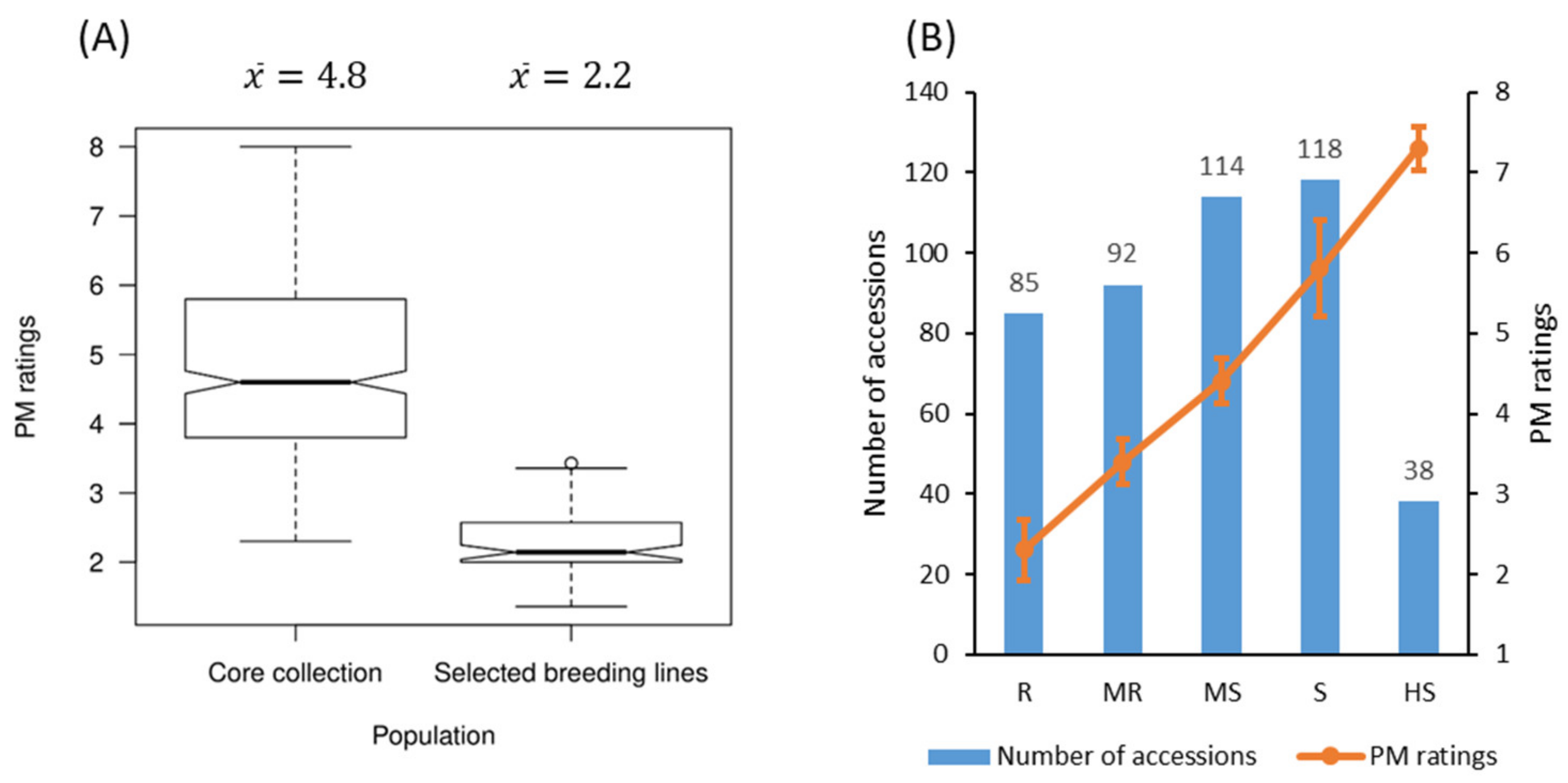
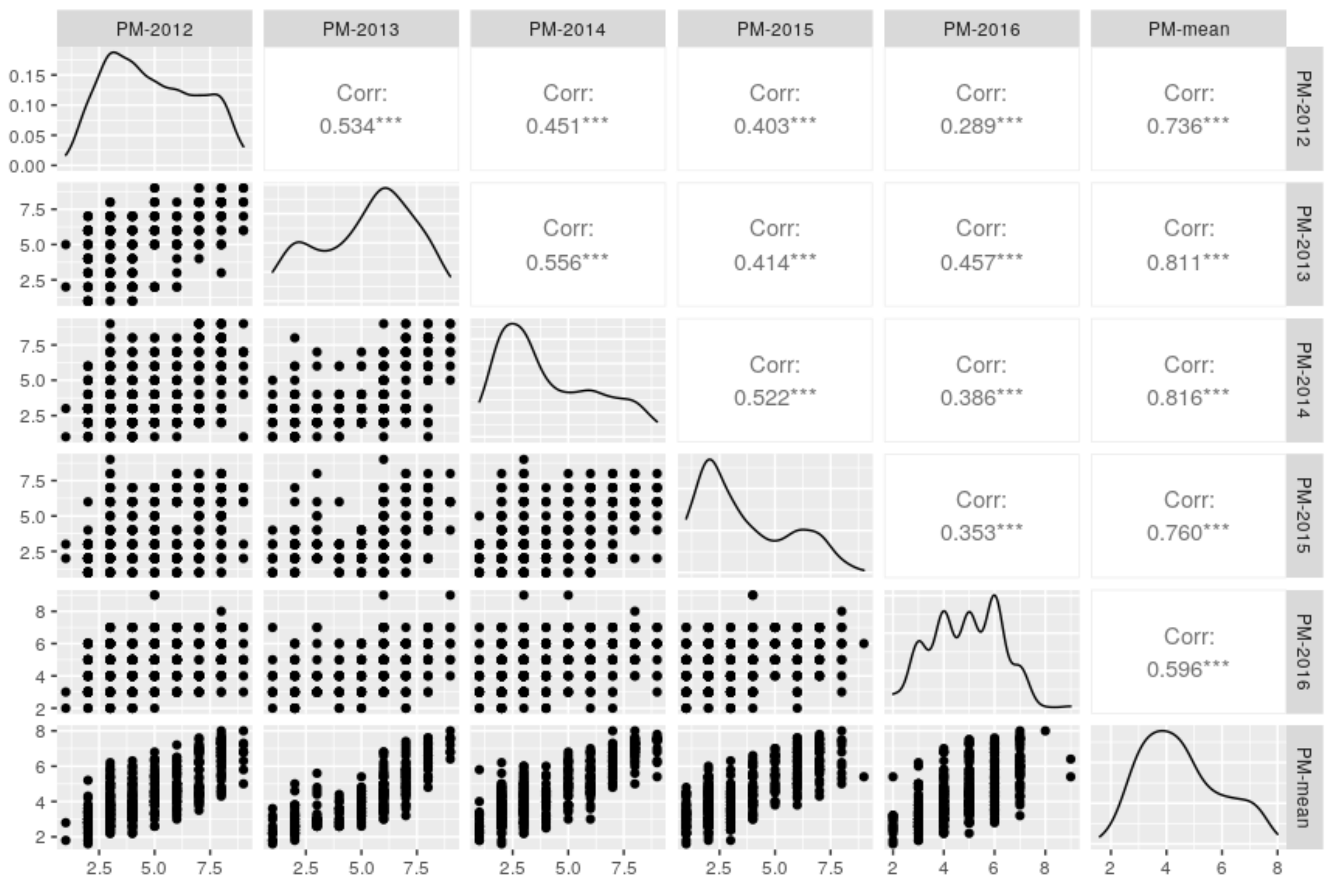
2.1. Evaluation of Powdery Mildew Resistance
2.2. Genetic Structure of the Population
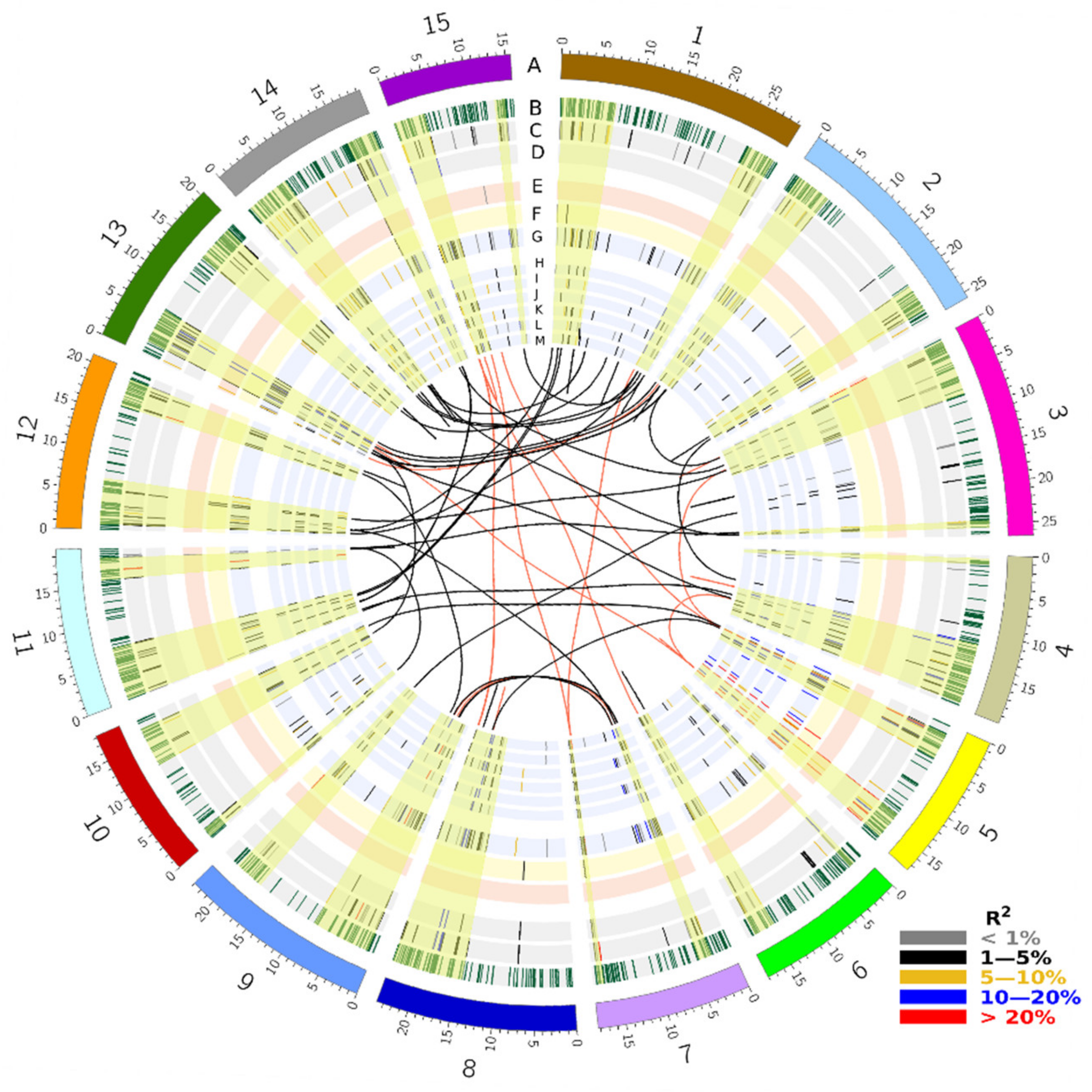
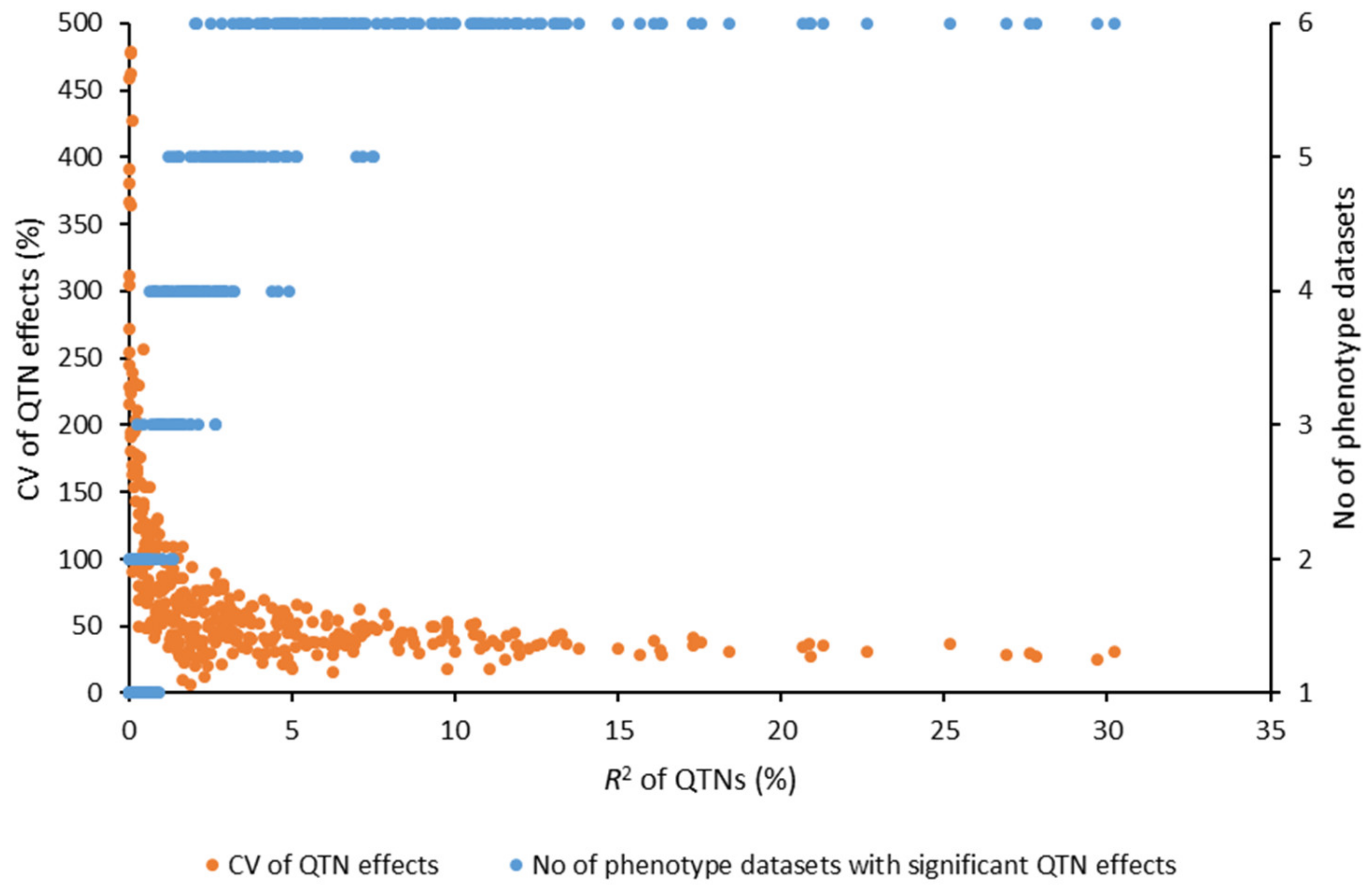
2.3. Identification of QTL
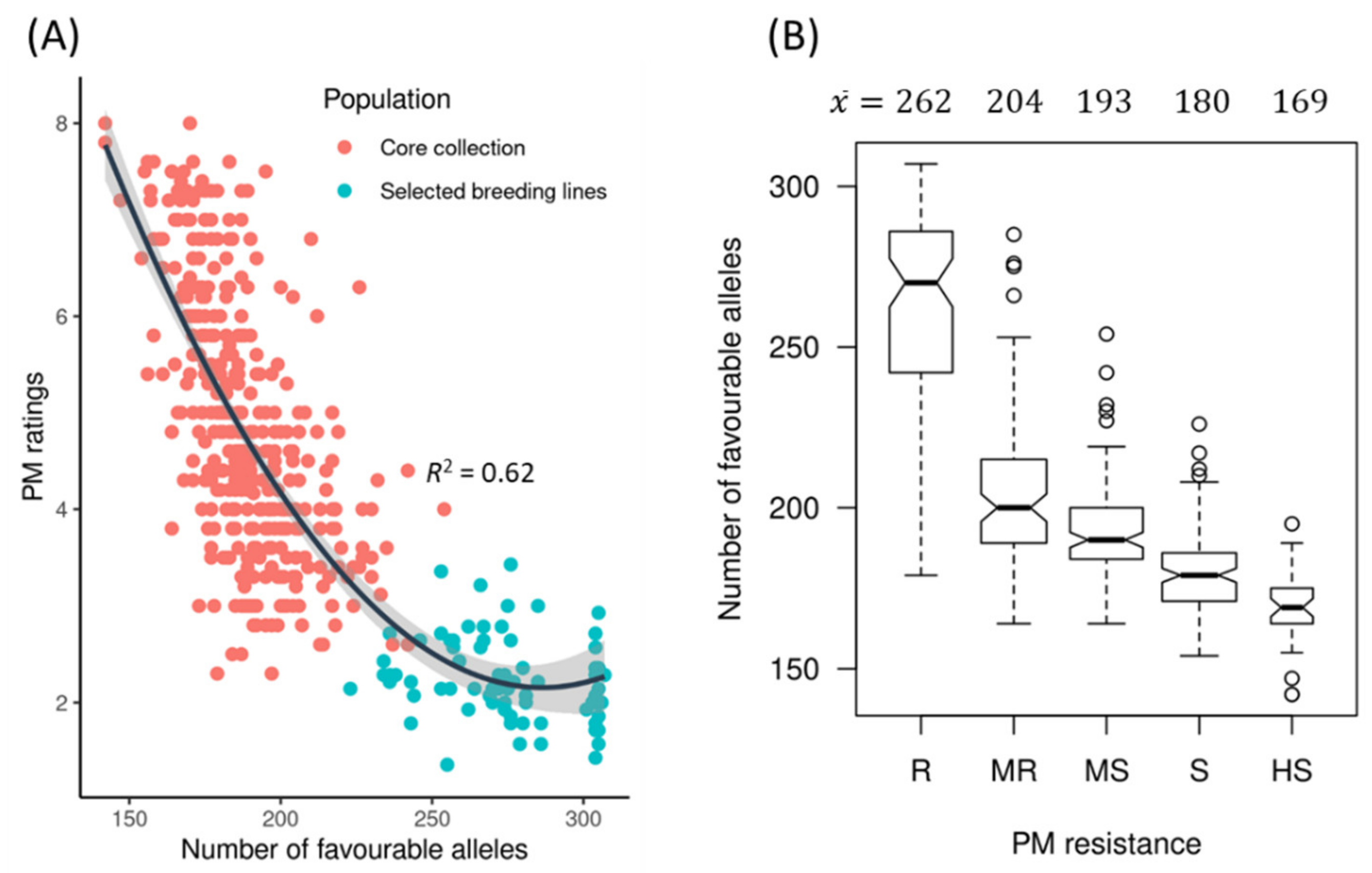
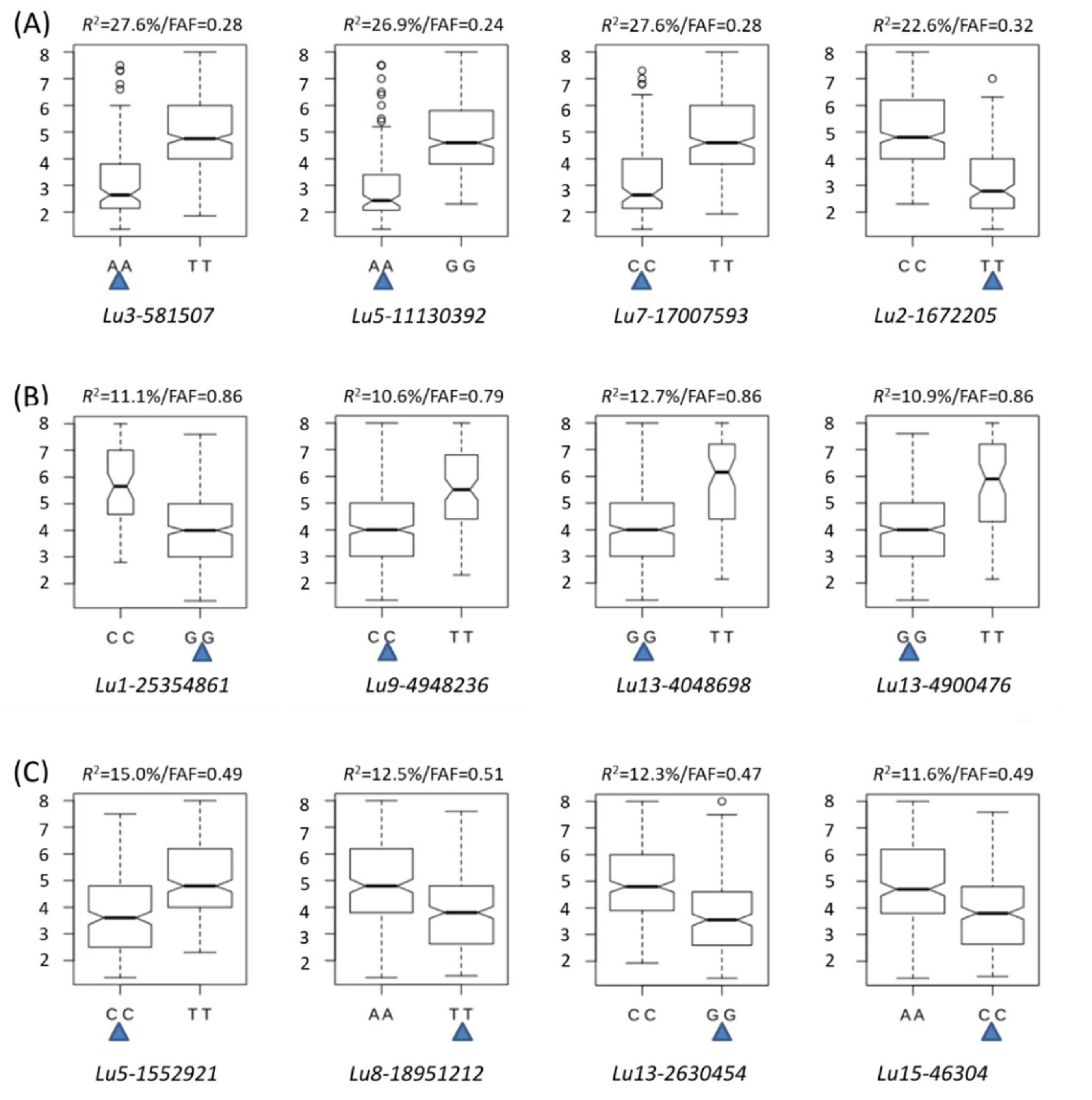
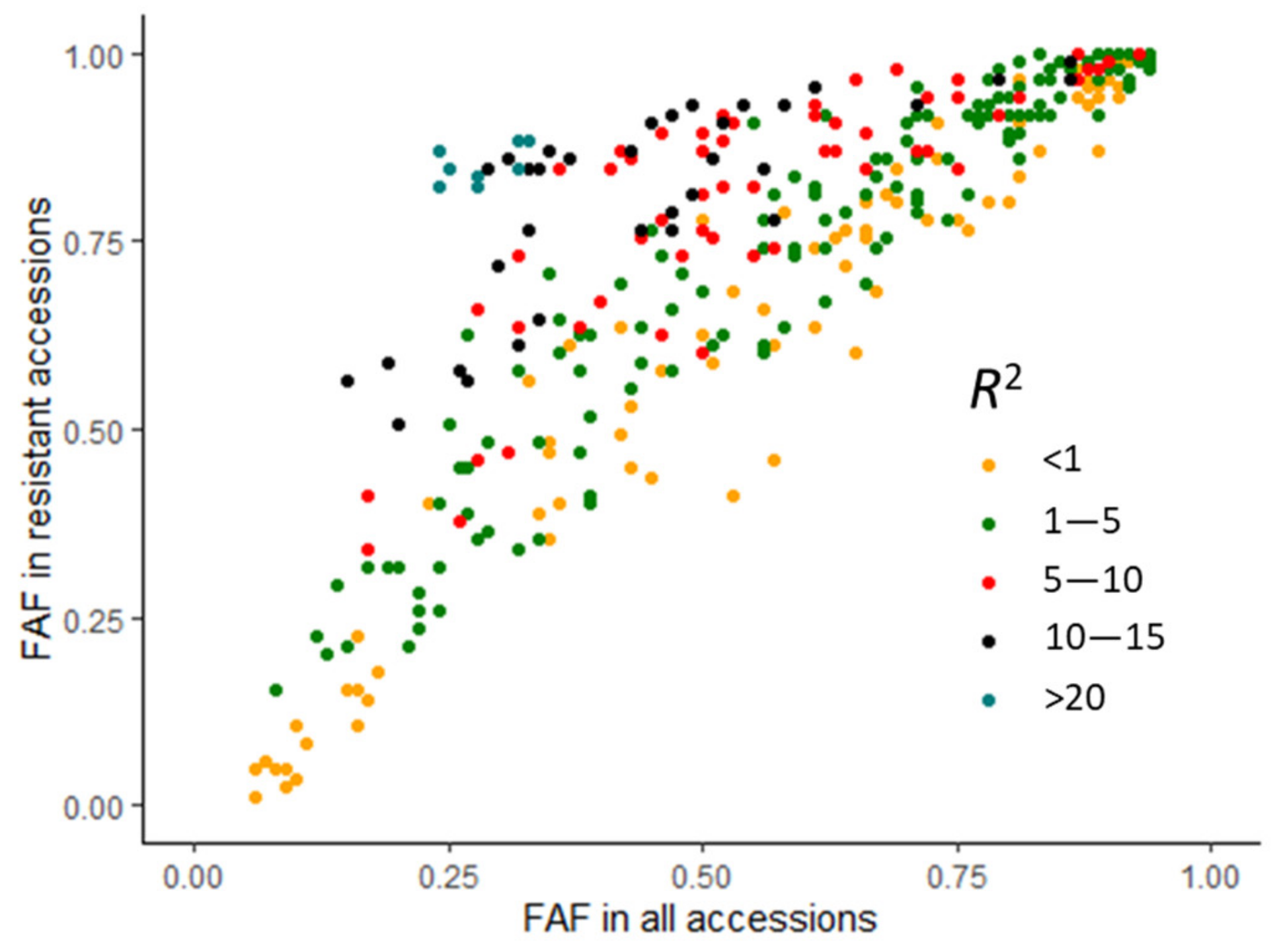
2.4. Favourable Alleles
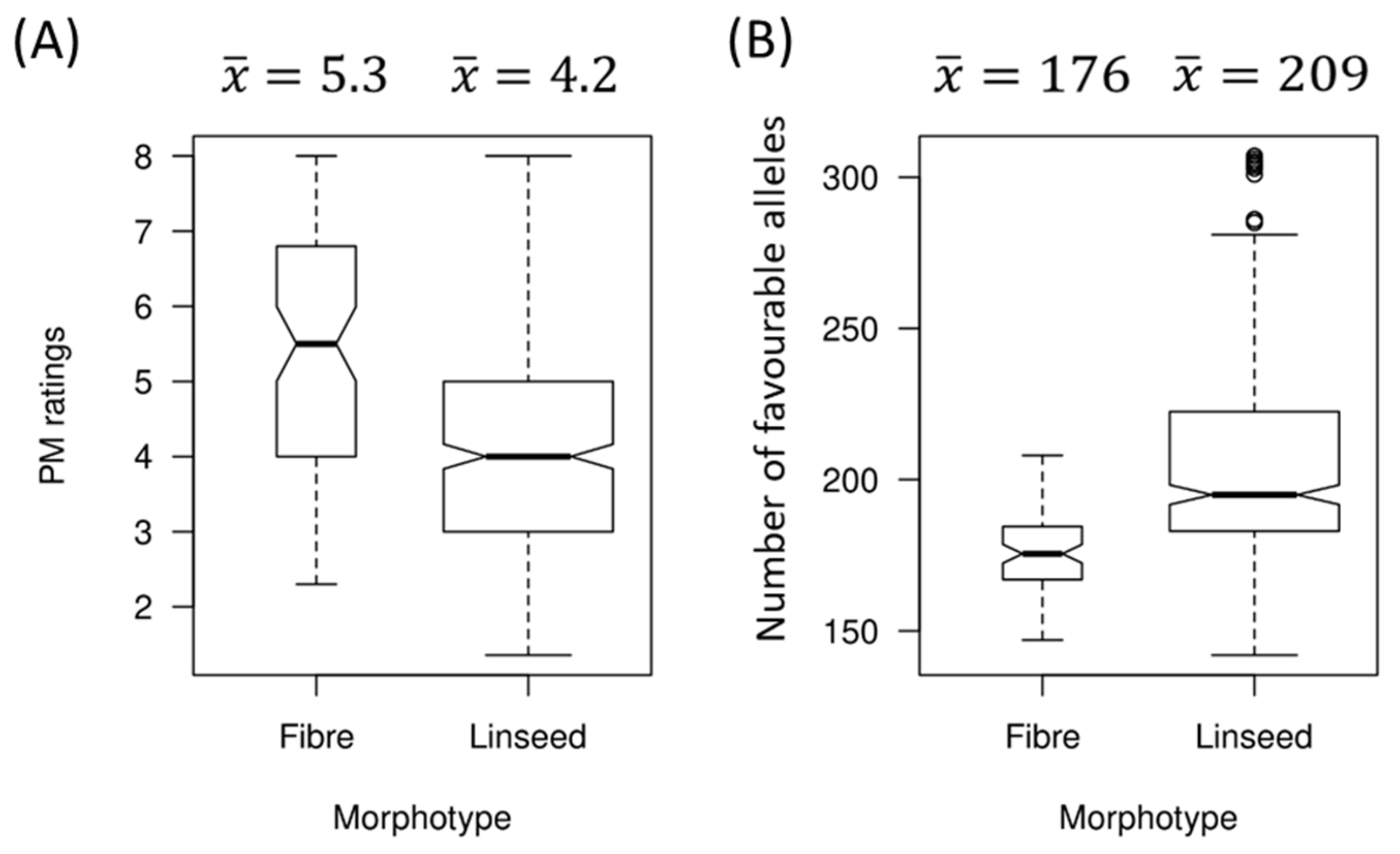
2.5. Relationship between PM Resistance and Flax Morphotypes
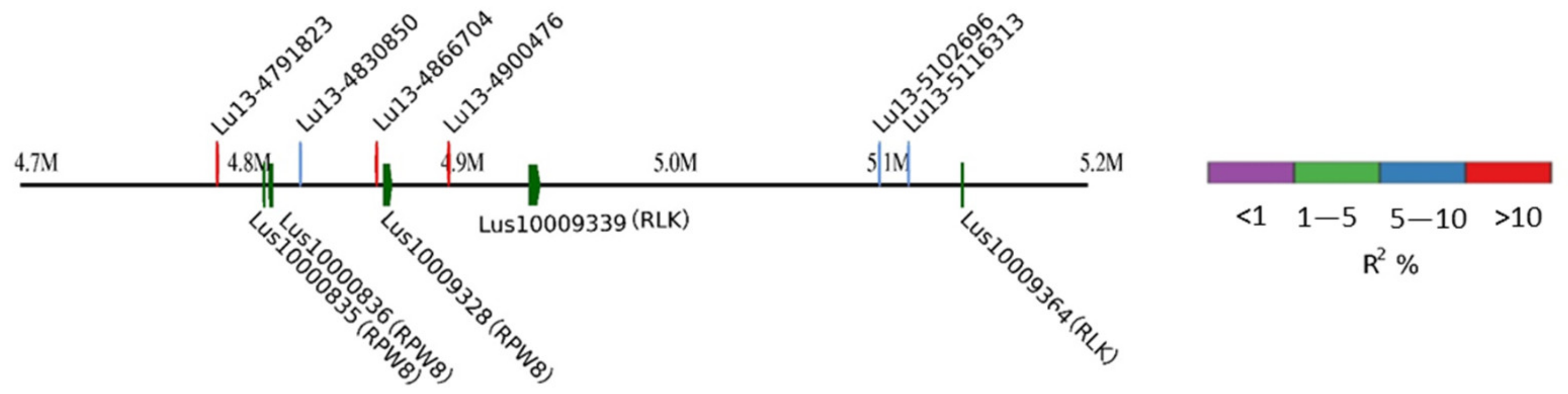
2.6. Candidate Genes
2.7. Genomic Prediction for PM Resistance
3. Discussion
4. Materials and Methods
4.1. Genetic Population
4.2. Phenotyping of Powdery Mildew Resistance and Statistical Analysis
4.3. Genotyping and SNP Identification
4.4. Genome-Wide Association Studies (GWAS)
4.5. Candidate Gene Prediction
4.6. Genomic Prediction (GP)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schulze-Lefert, P.; Panstruga, R. A molecular evolutionary concept connecting nonhost resistance, pathogen host range, and pathogen speciation. Trends Plant Sci. 2011, 16, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Panstruga, R. Establishing compatibility between plants and obligate biotrophic pathogens. Curr. Opin. Plant Biol. 2003, 6, 320–326. [Google Scholar] [CrossRef]
- Alam, M.A.; Mandal, M.S.N.; Wang, C.; Lim, W. Chromosomal location and SSR markers of a powdery mildew resistance gene in chromosome wheat line N0308. Afr. J. Micro. Res. 2013, 7, 477–482. [Google Scholar]
- Saharan, G.S.; Mehta, N.K.; Meena, P.D. Powdery Mildew Disease of Crucifers: Biology, Ecology and Disease Management; Springer Nature Singapore Pvt. Ltd.: Singapore, 2019. [Google Scholar]
- Niewoehner, A.S.; Leath, S. Virulence of Blumeria graminis f. sp. tritici on winter wheat in the Eastern United States. Plant Dis. 1998, 82, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Cowger, C.; Parks, R.; Marshall, D. Appearance of powdery mildew of wheat caused by Blumeria graminis f. sp. tritici on Pm17-bearing cultivars in North Carolina. Plant Dis. 2009, 93, 1219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.L.; Li, L.H.; He, Z.H.; Duan, X.Y.; Zhou, Y.L.; Chen, X.M.; Lillemo, M.; Singh, R.P.; Wang, H.; Xia, X.C. Seedling and adult plant resistance to powdery mildew in Chinese bread wheat cultivars and lines. Plant Dis. 2005, 89, 457–463. [Google Scholar] [CrossRef] [PubMed]
- REX Consortium. Combining selective pressures to enhance the durability of disease resistance genes. Front. Plant Sci. 2016, 7, 1916. [Google Scholar] [CrossRef]
- Marone, D.; Russo, M.A.; Laido, G.; De Vita, P.; Papa, R.; Blanco, A.; Gadaleta, A.; Rubiales, D.; Mastrangelo, A.M. Genetic basis of qualitative and quantitative resistance to powdery mildew in wheat: From consensus regions to candidate genes. BMC Genom. 2013, 14, 562. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, X.; Zhang, X.; Wang, J.; Luo, M.; Yang, M. Identification and evaluation of resistance to powdery mildew and yellow rust in a wheat mapping population. PLoS ONE 2017, 12, e0177905. [Google Scholar] [CrossRef]
- Kang, Y.; Zhou, M.; Merry, A.; Barry, K. Mechanisms of powdery mildew resistance of wheat—A review of molecular breeding. Plant Pathol. 2020, 69, 601–617. [Google Scholar] [CrossRef]
- Bonardi, V.; Cherkis, K.; Nishimura, M.T.; Dangl, J.L. A new eye on NLR proteins: Focused on clarity or diffused by complexity? Curr. Opin. Immunol. 2012, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Dangl, J.L.; Jones, J.D. Plant pathogens and integrated defence responses to infection. Nature 2001, 411, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, J.H. Discovery, characterization and exploitation of Mlo powdery mildew resistance in barley. In Breeding for Disease Resistance. Developments in Plant Pathology; Johnson, R., Jellis, G.J., Eds.; Springer: Dordrecht, The Netherland, 1992; Volume 1. [Google Scholar] [CrossRef]
- Devoto, A.; Hartmann, H.A.; Piffanelli, P.; Elliott, C.; Simmons, C.; Taramino, G.; Goh, C.S.; Cohen, F.E.; Emerson, B.C.; Schulze-Lefert, P.; et al. Molecular phylogeny and evolution of the plant-specific seven-transmembrane MLO family. J. Mol. Evol. 2003, 56, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Varallyay, E.; Giczey, G.; Burgyan, J. Virus-induced gene silencing of Mlo genes induces powdery mildew resistance in Triticum aestivum. Arch. Virol. 2012, 157, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Marone, D.; Russo, M.A.; Laido, G.; De Leonardis, A.M.; Mastrangelo, A.M. Plant nucleotide binding site-leucine-rich repeat (NBS-LRR) genes: Active guardians in host defense responses. Int. J. Mol. Sci. 2013, 14, 7302–7326. [Google Scholar] [CrossRef]
- Brun, H.; Chevre, A.M.; Fitt, B.D.; Powers, S.; Besnard, A.L.; Ermel, M.; Huteau, V.; Marquer, B.; Eber, F.; Renard, M.; et al. Quantitative resistance increases the durability of qualitative resistance to Leptosphaeria maculans in Brassica napus. New Phytol. 2010, 185, 285–299. [Google Scholar] [CrossRef]
- Delourme, R.; Bousset, L.; Ermel, M.; Duffe, P.; Besnard, A.L.; Marquer, B.; Fudal, I.; Linglin, J.; Chadoeuf, J.; Brun, H. Quantitative resistance affects the speed of frequency increase but not the diversity of the virulence alleles overcoming a major resistance gene to Leptosphaeria maculans in oilseed rape. Infect. Genet. Evol. 2014, 27, 490–499. [Google Scholar] [CrossRef]
- Tanksley, S.D. Mapping polygenes. Annu. Rev. Genet. 1993, 27, 205–233. [Google Scholar] [CrossRef]
- Liu, W.; Maccaferri, M.; Chen, X.; Laghetti, G.; Pignone, D.; Pumphrey, M.; Tuberosa, R. Genome-wide association mapping reveals a rich genetic architecture of stripe rust resistance loci in emmer wheat (Triticum turgidum ssp. dicoccum). Theor. Appl. Genet. 2017, 130, 2249–2270. [Google Scholar] [CrossRef]
- Nordestgaard, N.V.; Thach, T.; Sarup, P.; Rodriguez-Algaba, J.; Andersen, J.R.; Hovmoller, M.S.; Jahoor, A.; Jorgensen, L.N.; Orabi, J. Multi-parental populations suitable for identifying sources of resistance to powdery mildew in winter wheat. Front. Plant Sci. 2020, 11, 570863. [Google Scholar] [CrossRef]
- Pogoda, M.; Liu, F.; Douchkov, D.; Djamei, A.; Reif, J.C.; Schweizer, P.; Schulthess, A.W. Identification of novel genetic factors underlying the host-pathogen interaction between barley (Hordeum vulgare L.) and powdery mildew (Blumeria graminis f. sp. hordei). PLoS ONE 2020, 15, e0235565. [Google Scholar] [CrossRef] [PubMed]
- Montilla-Bascon, G.; Rispail, N.; Sanchez-Martin, J.; Rubiales, D.; Mur, L.A.; Langdon, T.; Howarth, C.J.; Prats, E. Genome-wide association study for crown rust (Puccinia coronata f. sp. avenae) and powdery mildew (Blumeria graminis f. sp. avenae) resistance in an oat (Avena sativa) collection of commercial varieties and landraces. Front. Plant Sci. 2015, 6, 103. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Desta, Z.A.; Ortiz, R. Genomic selection: Genome-wide prediction in plant improvement. Trends Plant Sci. 2014, 19, 592–601. [Google Scholar] [CrossRef]
- Asgarinia, P.; Cloutier, S.; Duguid, S.; Rashid, K.; Mirlohi, A.; Banik, M.; Saeidi, G. Mapping quantitative trait loci for powdery mildew resistance in flax (Linum usitatissimum L.). Crop Sci. 2013, 53, 2462–2472. [Google Scholar] [CrossRef]
- Aly, A. Examination of correlations between several biochemical components and powdery mildew resistance of flax cultivars. Korean Soc. Plant Pathol. 2012, 28, 149–155. [Google Scholar] [CrossRef]
- Rashid, K.Y. Powdery mildew on flax: A new disease in western Canada. Can. J. Plant Pathol. 1998, 20, 216. [Google Scholar]
- Beale, R.E. Studies of resistance in linseed cultivars to Oidium lini and Botrytis cinerea. In Production and Protection of Linseed; Aspects of Applied Biology Series; Association of Applied Biologists, Horticulture Research International, Wellesbourne: Warwick, UK, 1991; Volume 28, pp. 85–90. [Google Scholar]
- You, F.M.; Duguid, S.D.; Lam, I.; Cloutier, S.; Rashid, K.Y.; Booker, H. Pedigrees and genetic base of the flax varieties registered in Canada. Can. J. Plant Sci. 2016, 96, 837–852. [Google Scholar] [CrossRef]
- Rashid, K.; Duguid, S. Inheritance of resistance to powdery mildew in flax. Can. J. Plant Pathol. 2005, 27, 404–409. [Google Scholar] [CrossRef]
- Diederichsen, A.; Kusters, P.M.; Kessler, D.; Bainas, Z.; Gugel, R.K. Assembling a core collection from the flax world collection maintained by Plant Gene Resources of Canada. Genet. Resour. Crop Evol. 2013, 60, 1479–1485. [Google Scholar] [CrossRef]
- Soto-Cerda, B.J.; Diederichsen, A.; Ragupathy, R.; Cloutier, S. Genetic characterization of a core collection of flax (Linum usitatissimum L.) suitable for association mapping studies and evidence of divergent selection between fiber and linseed types. BMC Plant Biol. 2013, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xiao, J.; Rashid, K.Y.; Yao, Z.; Li, P.; Jia, G.; Wang, X.; Cloutier, S.; You, F.M. Genome-wide association studies for pasmo resistance in flax (Linum usitatissimum L.). Front. Plant Sci. 2018, 9, 1982. [Google Scholar] [CrossRef] [PubMed]
- Lan, S.; Zheng, C.; Hauck, K.; McCausland, M.; Duguid, S.D.; Booker, H.M.; Cloutier, S.; You, F.M. Genomic prediction accuracy of seven breeding selection traits improved by QTL identification in flax. Int. J. Mol. Sci. 2020, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Cloutier, S. Mapping quantitative trait loci onto chromosome-scale pseudomolecules in flax. Methods Protoc. 2020, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, B.; Wang, X.; Hu, K.; Li, K.; Li, Z.; Li, S.; Yan, L.; Guan, C.; Zhang, J.; et al. Genome-wide association study dissecting the genetic architecture underlying the branch angle trait in rapeseed (Brassica napus L.). Sci. Rep. 2016, 6, 33673. [Google Scholar] [CrossRef]
- Remington, D.L.; Thornsberry, J.M.; Matsuoka, Y.; Wilson, L.M.; Whitt, S.R.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef]
- Kumar, S.; You, F.M.; Duguid, S.; Booker, H.; Rowland, G.; Cloutier, S. QTL for fatty acid composition and yield in linseed (Linum usitatissimum L.). Theor. Appl. Genet. 2015, 128, 965–984. [Google Scholar] [CrossRef]
- Li, P.; Quan, X.; Jia, G.; Xiao, J.; Cloutier, S.; You, F.M. RGAugury: A pipeline for genome-wide prediction of resistance gene analogs (RGAs) in plants. BMC Genom. 2016, 17, 852. [Google Scholar] [CrossRef]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Gao, J.; He, L.; Zhu, T.; Luo, M.-C.; Wang, X.; Deyholos, M.K.; et al. Chromosome-scale pseudomolecules refined by optical, physical, and genetic maps in flax. Plant J. 2018, 95, 371–384. [Google Scholar] [CrossRef]
- You, F.M.; Zheng, C.; Bartaula, S.; Khan, N.; Wang, J.; Cloutier, S. Genomic cross prediction for linseed improvement. In Accelerated Plant Breeding; Gosal, S.S., Wani, S.H., Eds.; Springer Nature: Berlin/Heidelberg, Germany, 2022; Volume 4, pp. 451–480. [Google Scholar]
- He, L.; Xiao, J.; Rashid, K.Y.; Jia, G.; Li, P.; Yao, Z.; Wang, X.; Cloutier, S.; You, F.M. Evaluation of genomic prediction for pasmo resistance in flax. Int. J. Mol. Sci. 2019, 20, 359. [Google Scholar] [CrossRef]
- You, F.M.; Booker, H.M.; Duguid, S.D.; Jia, G.; Cloutier, S. Accuracy of genomic selection in biparental populations of flax (Linum usitatissimum L.). Crop J. 2016, 4, 290–303. [Google Scholar] [CrossRef]
- Pessina, S.; Lenzi, L.; Perazzolli, M.; Campa, M.; Dalla Costa, L.; Urso, S.; Vale, G.; Salamini, F.; Velasco, R.; Malnoy, M. Knockdown of MLO genes reduces susceptibility to powdery mildew in grapevine. Hortic. Res. 2016, 3, 16016. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Garcia, J.; Spencer, D.; Thieron, H.; Reinstadler, A.; Hammond-Kosack, K.; Phillips, A.L.; Panstruga, R. mlo-based powdery mildew resistance in hexaploid bread wheat generated by a non-transgenic TILLING approach. Plant Biotechnol. J. 2017, 15, 367–378. [Google Scholar] [CrossRef]
- Buschges, R.; Hollricher, K.; Panstruga, R.; Simons, G.; Wolter, M.; Frijters, A.; van Daelen, R.; van der Lee, T.; Diergaarde, P.; Groenendijk, J.; et al. The barley Mlo gene: A novel control element of plant pathogen resistance. Cell 1997, 88, 695–705. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, X.; Shan, Q.; Zhang, Y.; Liu, J.; Gao, C.; Qiu, J.L. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 2014, 32, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, V.; Wang, C.; Win, J.; Lanz, C.; Weigel, D.; Kamoun, S. Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci. Rep. 2017, 7, 482. [Google Scholar] [CrossRef] [PubMed]
- Koseoglou, E. The Study of SlPMR4 CRISPR/Cas9-Mediated Tomato Allelic Series for Resistance against Powdery Mildew; Wageningen University: Wageningen, The Netherlands, 2017. [Google Scholar]
- Huibers, R.P.; Loonen, A.E.; Gao, D.; Van den Ackerveken, G.; Visser, R.G.; Bai, Y. Powdery mildew resistance in tomato by impairment of SlPMR4 and SlDMR1. PLoS ONE 2013, 8, e67467. [Google Scholar] [CrossRef]
- Xiao, S.; Ellwood, S.; Calis, O.; Patrick, E.; Li, T.; Coleman, M.; Turner, J.G. Broad-spectrum mildew resistance in Arabidopsis thaliana mediated by RPW8. Science 2001, 291, 118–120. [Google Scholar] [CrossRef]
- Xiao, S.; Charoenwattana, P.; Holcombe, L.; Turner, J.G. The Arabidopsis genes RPW8.1 and RPW8.2 confer induced resistance to powdery mildew diseases in tobacco. Mol. Plant Microbe Interact. 2003, 16, 289–294. [Google Scholar] [CrossRef]
- Young, L.W.; Trouve, J.P.; Speck, A.; Kutcher, R.; Rashid, K.Y.; You, F.M.; Cloutier, S.; Booker, H.M. Mapping rust and powdery mildew resistant genes in flax. In Proceedings of the XXVII Plant and Animal Genome Conference, San Diego, CA, USA, 12–16 January 2019; p. PO0871. [Google Scholar]
- You, F.M.; Jia, G.; Xiao, J.; Duguid, S.D.; Rashid, K.Y.; Booker, H.M.; Cloutier, S. Genetic variability of 27 traits in a core collection of flax (Linum usitatissimum L.). Front. Plant Sci. 2017, 8, 1636. [Google Scholar] [CrossRef]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Koh, G. Faster single-end alignment generation utilizing multi-thread for BWA. Biomed. Mater. Eng. 2015, 26 (Suppl. 1), S1791–S1796. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; You, F.M.; Cloutier, S. Genome wide SNP discovery in flax through next generation sequencing of reduced representation libraries. BMC Genom. 2012, 13, 684. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Deal, K.R.; Wang, J.; Britton, M.T.; Fass, J.N.; Lin, D.; Dandekar, A.M.; Leslie, C.A.; Aradhya, M.; Luo, M.C.; et al. Genome-wide SNP discovery in walnut with an AGSNP pipeline updated for SNP discovery in allogamous organisms. BMC Genom. 2012, 13, 354. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Huo, N.; Deal, K.R.; Gu, Y.Q.; Luo, M.C.; McGuire, P.E.; Dvorak, J.; Anderson, O.D. Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence. BMC Genom. 2011, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Altshuler, D.; Donnelly, P.A.; International HapMap Consortium. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.Y.; Ni, Y.L.; Wen, Y.J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.M. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.L.; Wen, Y.J.; Dunwell, J.M.; Zhang, Y.M. pKWmEB: Integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2017, 120, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Tamba, C.L. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018, 341784. [Google Scholar] [CrossRef]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Ni, Y.L.; Zhang, Y.M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Tamba, C.L.; Wen, Y.-J.; Li, P.; Ren, W.-L.; Ni, Y.-L.; Gao, J.; Zhang, Y.-M. mrMLM v4.0: An R platform for multi-locus genome-wide association studies. bioRxiv 2020, 18, 481–487. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de Los Campos, G.; Crossa, J.; Gianola, D. Genomic-enabled prediction based on molecular markers and pedigree using the bayesian linear regression package in R. Plant Genome 2010, 3, 106–116. [Google Scholar] [CrossRef]
- Wu, Q.; Ye, Y.; Liu, Y.; Ng, M.K. SNP selection and classification of genome-wide SNP data using stratified sampling random forests. IEEE Trans. Nanobiosci. 2012, 11, 216–227. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G. Genome-assisted prediction of quantitative traits using the R package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Dataset Code | Sample Size | Range | CV (%) | |
|---|---|---|---|---|---|
| 2012 | PM-2012 | 447 | 4.9 ± 2.0 | 1.0–9.0 | 41.1 |
| 2013 | PM-2013 | 253 | 5.2 ± 2.1 | 1.0–9.0 | 40.9 |
| 2014 | PM-2014 | 447 | 4.1 ± 2.2 | 1.0–9.0 | 53.2 |
| 2015 | PM-2015 | 391 | 3.6 ± 2.0 | 1.0–9.0 | 57.3 |
| 2016 | PM-2016 | 447 | 4.9 ± 1.4 | 2.0–9.0 | 28.1 |
| Mean | PM-Mean | 447 | 4.5 ± 1.4 | 1.6–8.0 | 31.1 |
| Gene | Chr | Gene Start Position | Gene End Position | Gene Family | Tag QTN-Position | SNP | FA | FAF | CV of QTN Effects | Effect | R2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lus10006056 | 1 | 28681727 | 28684082 | RLK | Lu1-28683876 | A/G | A | 0.39 | 27.12 | −0.41 | 4.44 |
| Lus10030587 | 2 | 23955411 | 23958647 | RLK | Lu2-23956609 | A/C | A | 0.22 | 38.27 | −0.35 | 5.68 |
| Lus10040576 | 3 | 5746000 | 5750240 | TNL | Lu3-5748445 | G/A | A | 0.78 | 208.73 | 0.18 | 1.19 |
| Lus10036891 | 4 | 12429035 | 12433792 | WRKY | Lu4-12432479 | G/T | G | 0.37 | 28.39 | −0.70 | 11.51 |
| Lus10041860 | 4 | 16212942 | 16215322 | RLK | Lu4-16213043 | A/G | A | 0.18 | 27.34 | −0.62 | 3.4 |
| Lus10004727 | 5 | 1534218 | 1535440 | TNL | Lu5-1534998 | A/G | A | 0.28 | 31.31 | −1.33 | 27.83 |
| Lus10004726 | 5 | 1535502 | 1538672 | TNL | Lu5-1535619 | C/T | C | 0.26 | 27.43 | −1.54 | 32.6 |
| Lus10004719 | 5 | 1568734 | 1573096 | TNL | Lu5-1569098 | G/T | G | 0.31 | 33.14 | −1.06 | 27.63 |
| Lus10032303 | 5 | 3005188 | 3006765 | WRKY | Lu5-3006723 | T/C | T | 0.45 | 22.08 | −0.68 | 24.08 |
| Lus10032310 | 5 | 3052566 | 3053123 | DIR | Lu5-3052714 | T/G | G | 0.84 | 72.85 | 0.64 | 3.12 |
| Lus10032351 | 5 | 3223137 | 3225286 | RLK | Lu5-3224350 | A/G | A | 0.39 | 39.99 | −0.64 | 10.62 |
| Lus10034795 | 5 | 4643996 | 4646254 | RLK | Lu5-4646212 | C/T | C | 0.49 | 51.12 | −0.49 | 9.31 |
| Lus10029860 | 5 | 13270430 | 13273781 | TNL | Lu5-13271207 | A/G | A | 0.22 | 28.62 | −1.76 | 34.99 |
| Lus10017649 | 6 | 1878670 | 1885283 | RLK | Lu6-1883039 | A/G | A | 0.05 | 36.53 | −0.55 | 11.79 |
| Lus10021001 | 6 | 15376945 | 15378903 | RLP | Lu6-15378264 | G/A | A | 0.78 | 104.21 | 0.23 | 0.8 |
| Lus10025216 | 6 | 16664523 | 16666663 | WRKY | Lu6-16666521 | C/T | T | 0.58 | 52.26 | 0.12 | 1.13 |
| Lus10023199 | 7 | 17357355 | 17360952 | TM-CC | Lu7-17359522 | G/A | G | 0.44 | 61.38 | −0.15 | 0.73 |
| Lus10003971 | 7 | 17657006 | 17660870 | TM-CC | Lu7-17659649 | A/G | G | 0.91 | 26.37 | 1.12 | 4.85 |
| Lus10021849 | 8 | 7386368 | 7423024 | RLK | Lu8-7394085 | A/G | G | 0.78 | 39.93 | 0.38 | 3.65 |
| Lus10022265 | 8 | 15999079 | 16001545 | RLK | Lu8-15999956 | A/G | G | 0.87 | 27.70 | 0.47 | 1.36 |
| Lus10007812 | 8 | 18350700 | 18354799 | TNL | Lu8-18351964 | C/T | T | 0.65 | 44.47 | 0.73 | 11.49 |
| Lus10002249 | 8 | 19037487 | 19042009 | TNL | Lu8-19040276 | G/A | G | 0.19 | 28.07 | −1.3 | 15.67 |
| Lus10010221 | 9 | 911825 | 917007 | TNL | Lu9-916748 | T/G | G | 0.96 | 62.09 | 0.98 | 1.71 |
| Lus10031043 | 9 | 6265980 | 6269593 | RLK | Lu9-6266682 | T/C | C | 0.74 | 131.11 | 0.23 | 1.47 |
| Lus10006772 | 12 | 719787 | 720446 | RLK | Lu12-720013 | A/G | G | 0.80 | 61.75 | 0.5 | 2.61 |
| Lus10006732 | 12 | 890823 | 894178 | TNL | Lu12-892762 | C/G | C | 0.11 | 48.07 | −0.47 | 1.46 |
| Lus10023323 | 12 | 1893862 | 1897997 | RLK | Lu12-1896717 | A/T | A | 0.15 | 25.63 | −0.48 | 1.66 |
| Lus10018289 | 12 | 5110353 | 5112941 | TM-CC | Lu12-5111993 | G/C | G | 0.44 | 33.29 | −0.25 | 2.03 |
| Lus10027903 | 12 | 16614431 | 16619009 | RLP | Lu12-16614785 | A/G | G | 0.53 | 53.26 | 0.27 | 20.28 |
| Lus10033608 | 12 | 19122119 | 19129113 | RLK | Lu12-19127670 | G/A | A | 0.77 | 93.09 | 0.34 | 2.78 |
| Lus10001336 | 13 | 1747160 | 1750173 | MLO | Lu13-1749576 | G/A | G | 0.14 | 41.08 | −0.44 | 1.23 |
| Lus10019708 | 13 | 4520377 | 4539065 | TNL | Lu13-4531367 | T/C | T | 0.39 | 29.66 | −0.73 | 14.08 |
| Lus10009364 | 13 | 5141681 | 5142476 | RLK | Lu13-5142458 | C/T | C | 0.18 | 61.62 | −0.62 | 8.14 |
| Lus10030845 | 13 | 18211794 | 18215062 | RLK | Lu13-18212664 | T/G | G | 0.92 | 52.02 | 0.88 | 4.89 |
| Lus10028639 | 14 | 1171345 | 1174215 | CNL | Lu14-1171479 | C/T | C | 0.53 | 36.69 | −0.49 | 10.89 |
| Lus10020534 | 14 | 3457175 | 3462957 | TNL | Lu14-3458382 | C/G | C | 0.54 | 23.50 | −0.40 | 7.47 |
| Lus10021448 | 14 | 4018836 | 4024903 | TM-CC | Lu14-4021471 | A/T | T | 0.94 | 78.62 | 0.75 | 3.75 |
| Lus10014150 | 14 | 5375403 | 5382647 | RLK | Lu14-5382091 | A/G | G | 0.92 | 91.22 | 0.58 | 8.75 |
| Lus10015648 | 14 | 5955689 | 5959658 | TNL | Lu14-5959395 | G/C | C | 0.88 | 58.54 | 0.81 | 6.82 |
| Lus10015649 | 14 | 5960004 | 5963280 | TNL | Lu14-5960489 | G/A | A | 0.74 | 56.87 | 0.47 | 5.75 |
| Lus10008320 | 14 | 10547787 | 10552007 | TNL | Lu14-10551333 | T/C | C | 0.80 | 45.78 | 0.75 | 5.78 |
| Lus10035674 | 14 | 15357234 | 15366290 | TNL | Lu14-15360622 | T/C | C | 0.53 | 38.40 | 0.25 | 3.03 |
| Lus10039211 | 14 | 17203248 | 17203932 | TNL | Lu14-17203266 | G/A | G | 0.24 | 42.02 | −1.05 | 14.04 |
| Lus10007610 | 15 | 47907 | 50779 | RLK | Lu15-50397 | G/A | G | 0.45 | 35.62 | −0.52 | 15.63 |
| Lus10012678 | 15 | 3990588 | 4001681 | WRKY | Lu15-3991048 | T/G | G | 0.87 | 63.36 | 0.75 | 18.51 |
| Candidate Gene Family | R2 of QTNs (%) | Total | ||||
|---|---|---|---|---|---|---|
| <1 | 1–5 | 5–10 | 10–20 | >20 | ||
| DIR | 1 | 4 (1) | 1 | 6 (1) | ||
| DZC | 1 | 1 | ||||
| EDR | 1 | 1 | ||||
| MLO | 5 (1) | 1 | 6 (1) | |||
| RLK | 26 | 65 (9) | 32 (4) | 13 (3) | 2 | 138 (16) |
| RLP | 4 | 8 (1) | 6 | 2 | 2 (1) | 22 (2) |
| TM-CC | 4 | 18 (4) | 3 | 2 | 27 (4) | |
| WRKY | 2 | 8 (1) | 4 | 3 (2) | 1 (1) | 18 (4) |
| CNL | 2 | 3 | 4 (1) | 1 | 10 (1) | |
| TNL | 10 (1) | 25 (5) | 15 (2) | 7 (4) | 7 (4) | 64 (16) |
| TIR | 1 | 1 | ||||
| Total | 49 | 138 (21) | 62 (7) | 32 (10) | 13 (6) | 294 (45) |
| Marker Set | h2 | r |
|---|---|---|
| 349 QTNs (identified from 247,160 SNPs) | 0.691 ± 0.040 | 0.925 ± 0.014 a |
| 388 QTNs (identified from both 247,160 SNPs and 3230 RGA-derived SNPs) | 0.698 ± 0.041 | 0.926 ± 0.013 a |
| 132 QTNs located within genes (RGAs and non-RGAs) | 0.481 ± 0.048 | 0.850 ± 0.024 b |
| 279 QTNs/SNPs located within candidate RGAs | 0.587 ± 0.050 | 0.822 ± 0.031 c |
| 247,160 SNPs | 0.600 ± 0.070 | 0.690 ± 0.049 d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, F.M.; Rashid, K.Y.; Zheng, C.; Khan, N.; Li, P.; Xiao, J.; He, L.; Yao, Z.; Cloutier, S. Insights into the Genetic Architecture and Genomic Prediction of Powdery Mildew Resistance in Flax (Linum usitatissimum L.). Int. J. Mol. Sci. 2022, 23, 4960. https://doi.org/10.3390/ijms23094960
You FM, Rashid KY, Zheng C, Khan N, Li P, Xiao J, He L, Yao Z, Cloutier S. Insights into the Genetic Architecture and Genomic Prediction of Powdery Mildew Resistance in Flax (Linum usitatissimum L.). International Journal of Molecular Sciences. 2022; 23(9):4960. https://doi.org/10.3390/ijms23094960
Chicago/Turabian StyleYou, Frank M., Khalid Y. Rashid, Chunfang Zheng, Nadeem Khan, Pingchuan Li, Jin Xiao, Liqiang He, Zhen Yao, and Sylvie Cloutier. 2022. "Insights into the Genetic Architecture and Genomic Prediction of Powdery Mildew Resistance in Flax (Linum usitatissimum L.)" International Journal of Molecular Sciences 23, no. 9: 4960. https://doi.org/10.3390/ijms23094960
APA StyleYou, F. M., Rashid, K. Y., Zheng, C., Khan, N., Li, P., Xiao, J., He, L., Yao, Z., & Cloutier, S. (2022). Insights into the Genetic Architecture and Genomic Prediction of Powdery Mildew Resistance in Flax (Linum usitatissimum L.). International Journal of Molecular Sciences, 23(9), 4960. https://doi.org/10.3390/ijms23094960