Abstract
Glioblastoma (GBM) is the most common and malignant tumour of the central nervous system. Recent appreciation of the heterogeneity amongst these tumours not only changed the WHO classification approach, but also created the need for developing novel and personalised therapies. This systematic review aims to highlight recent advancements in understanding the molecular pathogenesis of the GBM and discuss related novel treatment targets. A systematic search of the literature in the PubMed library was performed following the PRISMA guidelines for molecular pathogenesis and therapeutic advances. Original and meta-analyses studies from the last ten years were reviewed using pre-determined search terms. The results included articles relevant to GBM development focusing on the aberrancy in cell signaling pathways and intracellular events. Theragnostic targets and vaccination to treat GBM were also explored. The molecular pathophysiology of GBM is complex. Our systematic review suggests targeting therapy at the stemness, p53 mediated pathways and immune modulation. Exciting novel immune therapy involving dendritic cell vaccines, B-cell vaccines and viral vectors may be the future of treating GBM.
1. Introduction
Glioma is an umbrella term used to describe primary brain tumours. Glioblastoma Multiforme (GBM) is the commonest glioma in adults, and it accounts for more than 60% of all adult brain tumours [1]. Traditionally, the classification of gliomas was based on its presumed cell of origin and histological features of malignancy. More recently, the updated World Health Organization (WHO) classification took a new approach by integrating molecular markers in the diagnostic process [2]. Glioblastoma remains a significant public health burden. International incidence of glioma is reported as 5.3 per 100,000 population per year and seems to be higher in the Western world [3]. Incidence peaks between ages 55 to 60 [4], and the prevalence is higher in males, with a relative sex ratio of 1.66 in England and 1.56 in the USA. Median survival for GBM has been reported as 6.1 months with 1, 2 and 5-year survivals estimated as 28.4%, 11.5% and 3.4%, respectively [5]. Treatment of GBM has been evolving. Traditionally, GBM treatment included surgery followed by radiotherapy, where the 3- and 5-year survival rates were 4.4 and 1.9%, respectively [6]. In 2005, the addition of Temozolomide (TMZ), an oral alkylating chemotherapy agent, showed significant improvement in overall survival where 3- and 5-year survival rates rose to 16% and 9.8%, respectively [7]. This led to the development of the “Stupp Protocol”, where combining TMZ with radiotherapy has been accepted as the “standard therapy” for GBM worldwide [8]. Even with these advancements, GBM remains challenging to treat. The new WHO classification of gliomas highlights the importance of molecular markers in diagnosis. Current therapy of GBM seems to have reached its limits and cannot address the genetic diversity and multiplicity of the tumor. Therefore, in order to treat GBM, we first need to understand it better. This systematic review will focus on new research regarding gliomagenesis and novel therapeutics of glioblastoma multiforme.
Background
Glioblastoma multiforme remains a notoriously hard to treat cancer associated with treatment resistance and high recurrence rates. A great deal of effort has been invested in understanding GBM pathophysiology to address the challenges in treatment. Gliomagenesis, the formation and development of gliomas, is a multistep process in which normal cells undergo genetic alterations that lead to malignant derivatives [9]. Cells progressively acquire genetic and epigenetic changes resulting in inhibiting tumour suppressor genes (TSG) and activation of proto-oncogenes. Together, these changes help cells escape the body’s regulatory mechanisms and lead to rapidly proliferating, undifferentiated cell clusters, known as tumours. These tumours become malignant when they invade and migrate into other tissues.
At a cellular level, certain tumorigenic processes have been associated with the development and progression of GBMs. These intracellular events include but are not limited to loss of cell cycle control, growth factor over-expression, angiogenesis, invasion & migration, genetic instability and disorder of apoptosis [10]. Complexity of GBMs arises at the cellular and genomic level, where they display significant inter- and intra-tumour heterogeneity [11]. In other words, GBM should not be thought of as a single cancer entity. The Cancer Genome Atlas (TCGA) network outlines four distinct molecular subtypes of GBM: Neural, Proneural, Classical and Mesenchymal [12]. Each of these subtypes harbours unique molecular and genetic aberrations that drive tumorigenesis. Some of these potential tumorigenic processes are discussed below.
2. Gliomagenesis
Loss of cell cycle control is involved in gliomagenesis. Cells go through several coordinated processes in their life cycle: cell growth, DNA replication and cell division. Regulation of the cell cycle through checkpoints is the key to normal growth and differentiation. The G1 checkpoint controls the transition from the G1 phase to the S phase, whereas the M checkpoint regulates transition through mitotic phases [13]. The G1 checkpoint has been an important topic of research in GBMs, and it mainly involves cyclins and cyclin-dependent kinases (CDK). Simply, CDKs rely on activation with cyclins to phosphorylate downstream proteins and allow initiation of DNA synthesis. The vast majority of GBMs show alteration in at least one component of such a pathway called p16INK4a/CDK-4/Retinoblastoma 1 (RB1) [14]. In normal cells, cyclin D1 activates CDK-4, which phosphorylates RB1, an oncogene, and causes the release of the E2F transcription factor, which initiates G1/S transition [15]. RB expression is found to be downregulated in 78% of GBM patients [12]. G1 checkpoint regulation is complex/multi-factorial and acts as a quality control mechanism. During the G1 phase, if DNA damage is detected at the end of the M phase, cyclin-dependent kinase inhibitors may be activated by Tumour protein p53 (p53) to halt the cell cycle and lead to apoptosis to prevent tumorigenesis [13]. Not surprisingly, 87% of GBM patients have an associated p53 mutation [12]. In summary, cell cycle checkpoints are essential in initiating DNA repair pathways in response to genotoxic stress [16].
In non-neoplastic cells, DNA repair aims to prevent genetic instability that could lead to tumorigenesis. Therefore, it is not surprising that cancer cells often display epigenetic or genetic alterations in the DNA repair pathways [17]. Most notably, p53 mutations leading to impaired apoptotic response have been linked to impaired DNA damage response (DDR) and gliomagenesis [18]. DDR describes the sophisticated cellular mechanisms that detect, signal and repair DNA lesions inflicted by endogenous and exogenous genotoxins. Paradoxically, tumour cells rely on residual DNA repair mechanisms to cope with endogenous stress induced by increased cellular metabolism, mitotic stress and hypoxia, as well as exogenous stress induced by chemotherapy and radiotherapy. Treatment strategies for glioblastoma rely on inducing DNA damage. Ionizing radiation (IR) works mainly by inflicting double-strand breaks (DSBs), base damage and single-strand breaks (SSBs), whereas Temozolomide (TMZ), an alkylating chemotherapy agent, induces N7-methylguanine (N7-meG) and N3-methyladenine [19]. DNA damage caused by these therapies is often repaired by multi-protein pathways such as homologous recombination (HR), non-homologous end joining (NHEJ), or base excision repair (BEJ), with the exception of a repair mechanism via O6- methylguanine-DNA methyltransferase (MGMT) which works as a single protein pathway [20].
Cancer cells adapt DNA repair mechanisms to play a crucial factor in development of therapy resistance and tumour relapse. DNA damage repair deficiency (DDRd) creates predictable and traceable nucleotide alterations in the tumour’s genetic code, creating mutational signatures. Understanding and taking advantage of DDRds could help develop further therapeutic strategies. An established concept of this is identifying MGMT promoter CpG methylation status in treatment of GBM. Forty-five percent of GBM cases are associated with MGMT promoter CpG methylation, which prevents the repair of DNA damage induced by TMZ and thus is associated with increased sensitivity to TMZ and better prognosis [21]. Glioblastoma cells show over-expression of certain growth factors and their receptors. This creates an autocrine growth-promoting loop which provides growth advantage to rapidly proliferating tumour cells. Tyrosine receptor kinases (RTK) such as epidermal growth factor receptor (EGFR) and platelet-derived growth factor (PDGF) have been associated with GBM [22]. Two proliferating signalling cascades have been found to be over-active in glioblastomas: PI3K/Akt/mTOR and Ras/Raf/MAPK Pathways. Interestingly, there is evidence in the literature that there is a mutually inhibitory crosstalk between these two pathways [23].
2.1. PI3K/Akt/mTOR Pathway
PIK3/Akt/mTOR Signalling Pathway is involved in various cellular processes including proliferation, growth, cytoskeletal rearrangement and apoptosis [12]. This pathway, summarised in Figure 1, involves RTKs (such as EGFR) and tumour suppressor protein PTEN (Phosphatase and Tensin Homolog Deleted from Chromosome 10). PTEN is a lipid phosphatase and leads to downregulation of the RTK/PI3K/Akt pathway via antagonizing PI3K function [24]. Protein kinase B (Akt) and mammalian target of rapamycin (mTOR) are serine/threonine-specific protein kinases (STKs) that play key roles in cellular proliferation (Figure 1) [10]. RTK/PI3K/Akt signalling is altered in 80% of human GBMs. Homozygous deletion or mutations of PTEN leading to constitutive activation of this pathway were associated with 40% of GBMs [12]. Mutations and amplifications in the PIK3CA, coding for p110 subunit, have been associated in 5% and 13% of primary GBMs, respectively [25].

Figure 1.
PI3K/Akt/mTOR Pathway.
2.2. RAS/RAF/MAPK Pathway
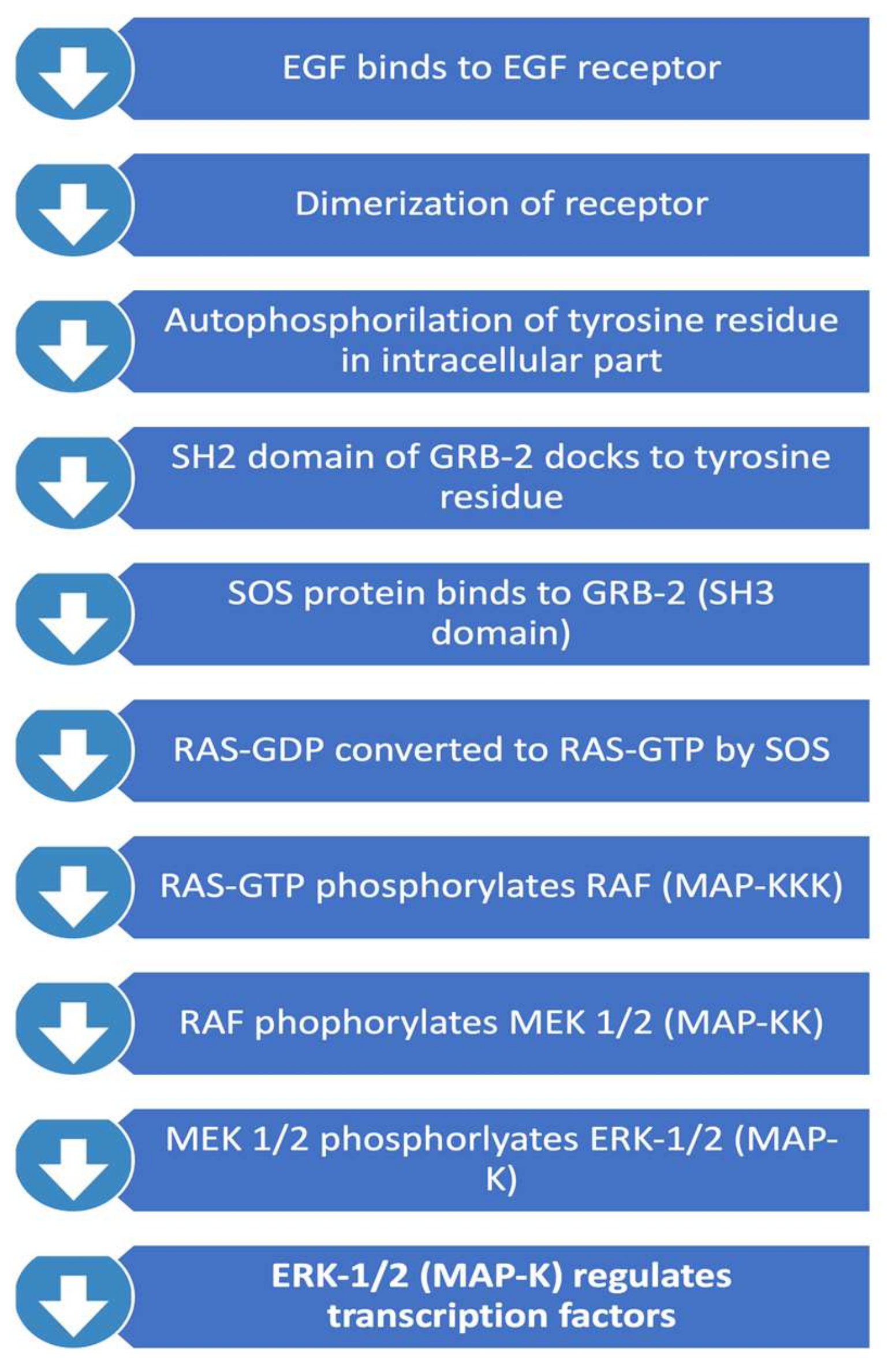
This pathway, summarised in Figure 2, is important in regulating cell proliferation, differentiation, and survival. Activated RAS protein recruits the RAF family of serine/theorinine kinase (STKs) such as ARAF, BRAF and CRAF1. Activated RAF subsequently activates mitogen-activated protein kinase kinase called MEK. Active MEK phosphorylates mitogen-activated protein kinase (MAPK). MAPK subsequently translocates into the nucleus and leads to activation of important transcription factors that promote cell proliferation. Important transcription factors activated through this pathway include c-myc, STAT (signal transducers and activators of transcriptome) and PPARγ (peroxisome proliferator-activated receptor γ) which are involved in inducing cell cycle progression and anti-apoptosis genes [26]. Interestingly, RAS mutations themselves are rare in GBMs, and therefore focus has been on the upstream regulators of the pathway. RAS activation is regulated by neurofibromin 1 (NF-1), a tumour suppressor gene. NF-1 has been identified to play a role in tumorigenesis in gliomas. 18% of GBMs contain NF-1 mutation/homozygous deletion, whereas this number is higher in mesenchymal GBMs (37%) [12].
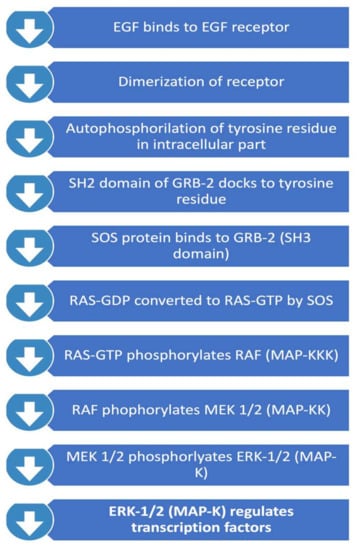
Figure 2.
RAS/RAF/MAPK Pathway.
3. Invasion and Migration
GBM remains difficult to treat because these cells employ multiple cellular and metabolic mechanisms to survive and spread. GBM can quickly adapt to adverse conditions in their environment, including those induced by radio or chemotherapy. These cells are also able to evade the immune system and continue to progress and invade. Rapidly growing GBM cells have increased metabolic and nutritional demands. This creates microenvironmental stressors such as hypoxia and metabolic scarcity. In response to these stressors, GBM cells undergo an “angiogenic switch” where new blood vessels are established through upregulation of proangiogenic factors, cytokines and relevant receptors [27]. Angiogenic factors such as vascular endothelial growth factor (VEGF) over-expression has been associated with GBM angiogenesis. Glioblastoma cells demonstrate altered expression of cytoskeletal proteins, signalling molecules and cell surface receptors [28]. Epithelial-to-mesenchymal transition (EMT) is initiated by growth factors [29] and involvement of the PI3K/AKT pathway [30]. Genetic make-up of these mesenchymal migratory cells includes high expression of proteins such as N-cadherin and fibronectin [31] which allow mobility and invasiveness of glioma cells [32].
Some GBM cells continue to adapt and evolve by preserving their stem cell abilities. These glioma stem cells (GSC) express antigens such as Nestin, CD133 (prominin-1), Musashi-1 and Bmi-1 that are specific to neural stem and progenitor cells [33]. GSCs are able to self-renew, remain multi-potent and initiate tumour growth when implanted to animal models. Expression of two main neural progenitor regulators, sonic hedgehog homolog (SHH) and Notch, have been identified to be altered or overexpressed in GSCs [34]. Similarly, aberrant IL6/JAK/STAT3 pathway has been associated in GBM. IL6 regulates Janus kinase (JAK) activation of STAT3 transcription factors (family of signal transducers and activators of transcription), which is required to maintain pluripotency of neural stem cells. STAT3 activation is observed in GBM and is associated with increased tumour grade [35].
Cancer cells use various mechanisms to circumvent host immune surveillance. GBM-associated endothelial cells create an immunological barrier around the tumour microenvironment by modulating immune checkpoints [36]. An immune checkpoint protein called CD200 has been associated with this process and found to be upregulated in GBM. CD200 causes suppression of pro-inflammatory cytokines such as IL2 and IFNy [36].
Another immunosuppressive mechanism potentially employed by GBM is through suppression of dendritic cell (DC) function. DC function plays an integral part in initiating adaptive immune response against cancer cells by antigen uptake and presentation and co-stimulation of T cells [37]. Glioblastoma microenvironments contain factors that can potentially suppress DC function; growth factor-beta (TGF-β) and indoleamine 2,3-dioxygenase (IDO) [38]. With these types of adaptations, GBMs are able to remain out of reach of the immune system and continue to proliferate.
4. Materials and Methods
A systematic search of the literature was performed in accordance with PRISMA guidelines for molecular pathogenesis and therapeutic advances. The search was done in the PubMed library and the results were filtered for the last ten years, in English, French or German language. Our study included original and meta-analysis studies that discussed (i) molecular diagnosis and prognosis, (ii) pathogenesis, (iii) gliomagenesis, (iv) infiltration/migration, (v) immune system and (vi) signalling pathways of the GBM. We have excluded duplicate articles, reviews and systematic reviews, studies about paediatric population (<18 years), low grade gliomas (WHO grade I, II) and anaplastic gliomas (WHO grade III) from our study. The search in molecular pathogenesis aimed to group and elucidate the biological mechanisms behind the pathways and the cell events that are correlated to GBMs. The search for therapeutic advances aimed to provide new knowledge that could help in the treatment of the glioblastoma. A four-stage approach was taken for our search, and it was completed with a search in the references of the articles included in our study. Therefore, a Tier-1 search was done under the terms: “(glioblastoma) AND (signaling pathways)”. The Tier-2 search was done under the search terms: (i) (gliomagenesis) AND (cell migration), (ii) (gliomagenesis) AND (cell survival), (iii) (gliomagenesis) AND (DNA repair), (iv) (gliomagenesis) AND (apoptosis), (v) (gliomagenesis) AND (G2/M arrest), (vi) (gliomagenesis) AND (G1/S progression), (vii) (gliomagenesis) AND (cell cycle progression). The Tier-3 and Tier-4 searches were done under the terms: “(glioblastoma) AND (theranostics)”, and “(glioblastoma) AND (vaccine)” respectively. Our results were screened by title and abstract for exclusion criteria. Whole articles that met our inclusion criteria were sought, or there was not enough data to apply exclusion criteria. The reference list of the articles included in our systematic review was further screened for additional studies.
5. Results
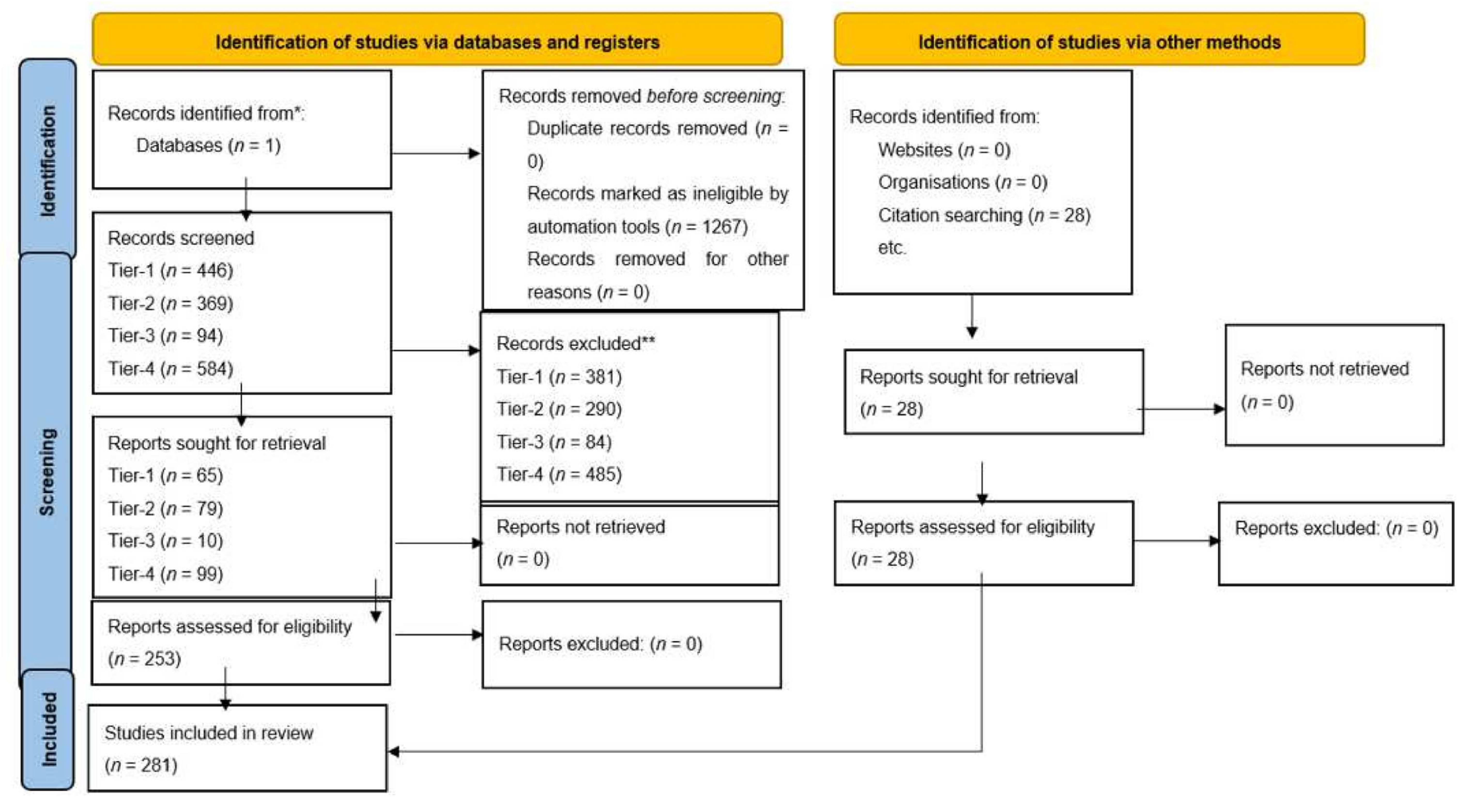
Tables demonstrate search terms used in PubMed, results from initial search with filters and articles meeting inclusion criteria (Figure 3). See Table 1 for selected articles relating to molecular pathogenesis and future treatment perspectives of GBM. All articles included in the study can be found in Tables S1–S9 in the Supplementary Materials. From our search in signalling pathways in GBM, we included eight articles which met the inclusion criteria for PI3K class 1/AKT signalling. These focused on targeting PI3K regulators such as WDR81 [39] to promote autophagy in GBM cells or inhibiting specific isoforms of PI3K to attenuate gliomagenesis. Moreover, blockade of downstream pathways in combination with PI3K had a synergistic effect on reducing tumour growth [40]. Expectedly, metabolic pathways which activate glycolysis were up-regulated secondary to PI3K dependent pathways [41]. Similarly, to PI3K Class 1, we included four articles for PI3K Class 2 which referred to effects of PI3K isoform and regulator inhibition, in addition to altered metabolism associated with this pathway. Four articles discussing the RTK/RAS pathway were analysed, and two studies highlighted the role of CIC as a tumour suppressor protein whereby preventing degradation or inhibition sensitised GBM cells to its anti-tumorigenic effects [42,43]. Four studies relating to the RB pathway were used in this review, two of which examined the use of CDK inhibitors in clinical trials, showing these agents to be ineffective monotherapy due to lack of progression free survival [44,45]. Forty-five articles examining the p53 pathway were included which reviewed the mechanisms to reactivate and enhance tumour suppressor activities of p53. For cell migration, our search yielded 13 articles which met the inclusion criteria and discussed the use of the actin cytoskeleton and MMP to promote cell migration in GBMs [46,47,48]. There is a significant role of RNA–long non-coding, micro or circular to regulate cell motility; downregulation of specific RNAs can suppress migration. EMT is augmented by alterations in the β-Catenin/Wnt pathway and expression of proteins such as E-cadherin and Vimentin [49,50,51]. Fourteen articles pertaining to cell survival were included. Glucose uptake is a key to cellular function, this is up-regulated in GBM cells by many mechanisms, one of which includes overstimulation of α-Ketoglutarate activated by NF-κB signalling, leading to increased uptake [52]. Overexpression of transcription factors such as NF-Y was associated with cell survival [53]. Only four articles related to DNA repair were analysed. DNA damage repair deficiency was most commonly found in p53 [54]. Targeting DNA repair mechanisms such as PARP attenuated gliomagenesis [55] and inhibition of DNA repair protein such as RAD51 in GBM cells resulted in greater TMZ sensitivity [56]. Two articles associated with G2/M cell cycle arrest were included, which discussed blocking zinc finger protein resulting in reduced tumour growth and enhanced apoptosis [57]. For apoptosis, 29 articles were included; the search identified specific RNAs involved in regulation and mediation of apoptosis and detection of therapeutic drugs that potentially reduce tumour growth. Proteins such as heterochromatin, including cell surface protein, have a role in apoptosis. G1/S Progression search yielded two studies, one of which identified an important role of TROAP in cell cycle regulation and enhanced tumour progression [58]. Cell Cycle Progression search yielded 19 articles relevant to cell cycle progression. In our search for cutting edge advances in therapy, the field of theragnostics contributed to our results ten articles. This new technology included specific long non-coding RNAs that could be identified and targeted by existing drugs in GBM patient population subtypes [59]. Moreover, EGFRvIII targeted chimeric antigen receptor T (CAR-T) cell therapy response was also discussed [60]. Finally, 99 articles related to vaccines in GBM were included. Vaccines induced immune response predominantly through dendritic cells and T cells to seize innate antitumorigenic characteristics and prevent tumour progression and permit longer survival.
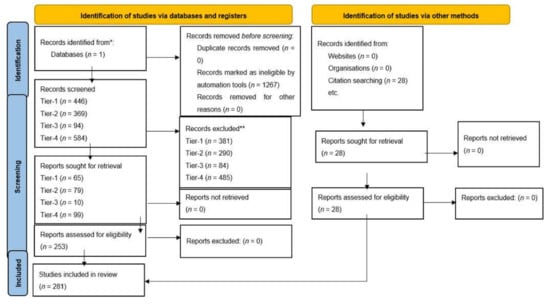
Figure 3.
Prisma 2020 Flow-chart [72].

Table 1.
Selected articles for molecular pathogenesis and future perspectives for glioblastoma.
6. Discussion
What cellular and molecular events lead to the development of glioblastoma, and can we intervene in this process at any step? The key to treating GBM and improving patient survival likely lies in answering these questions. That is why, in this systematic review, we highlight the recent advancements in our understanding of the molecular pathogenesis of GBM and discuss related novel treatment strategies.
6.1. Gliomagenesis
The most widely accepted hypothesis of GBM development, referred to as gliomagenesis, suggests that GBM develops from mature glia cells undergoing neoplastic transformation through aberrant cellular and molecular events. This theory traditionally focused on the glial cells as a cell of origin because they were thought to be the only dividing cells in the adult brain. However, we now know that the adult brain retains neural stem cells and glial progenitor cells which are also susceptible to neoplastic transformation. In fact, the vast level of heterogeneity observed amongst gliomas may suggest that they develop from a single but multipotent progenitor cell [73]. Neural stem cells exhibit characteristics similar to that of GBM cells; high motility, possession of immature antigenic phenotypes and ability to activate developmental signalling pathways [74]. This active state makes them more prone to malignant transformation requiring fewer mutations compared to differentiated glial cells [75]. Maintaining cancer stem cell (CSC) traits, often referred to as stemness, can promote and sustain tumorigenesis. Supporting this cancer stem cell theory, new evidence suggests that GBM cells often show overexpression of common neural progenitor-cell markers such as nestin, CD133 and CD163 [76,77,78]. These markers may play an essential role in tumour initiation. Singh et al., in 2004, showed that transplanting CD133-expressing GBM cells into mouse brain was able to produce a tumour with preserved parent histological appearances [79]. This, however, was not observed when CD133 negative cells were used [79].
Targeting progenitor-cell marker CD133 presents a novel therapeutic strategy to inhibit GBM stemness and potentially halt gliomagenesis early on. In 2019, Huang et al. showed that the AP-2α transcription factor, which is often downregulated in GBM cells, reduces CD133 expression through Nanog/Sox2/CD133 axis [65]. The study demonstrated that restoring AP-2α expression with the use of miR-26a inhibitors not only suppressed glioma stem cells’ self-renewal abilities in vitro, but also inhibited subcutaneous and intracranial xenograft tumour growth in vivo [65]. This study suggests AP-2α is responsible for maintaining GBM stemness via Nanog/Sox2/CD133 and could serve as a novel therapeutic target. More recently, CD133 has been a target for novel immunotherapy approaches. Do et al. developed a dendritic cell (DC)-based vaccine against CD133 expressing GBM cells by transfecting CD133 mRNA into DC [80]. Immunization in humanised mouse models led to a potent immune response against CD133 positive GBM cells and showed abrogated tumour progression in vitro and in vivo [80]. This suggests CD133 mRNA-transfected DC vaccination could be valuable as an immunotherapy against cancer stem cells.
GBM development could also be inhibited by targeting metabolic pathways involved in gliomagenesis. Relevant signalling pathways such as PI3K/Akt/mTOR and Ras/Raf/MAPK have already been discussed in detail in the background section. Our review showed extensive research on developing therapies against the PI3K signalling pathway. Xu et al. in 2019, studied the anti-proliferative and apoptotic effects of PI3Kβ isoform inhibitor AZD6482 on PTEN-mutated GBM cell lines [81]. AZD6482 administration showed a dose-dependent cytotoxicity to glioma cells leading to increased apoptosis and cell cycle arrest at the G1 phase in vitro [81]. This suggested a possible benefit of an isoform specific approach. Jones et al. studied the effects of isoform-specific PI3K inhibition in GBM cancer stem cells with a hypothesis that this inhibition would promote CSC differentiation and reduce proliferation. Two GBM models studied showed differences in their dominant catalytic PI3K isoform. Inhibiting dominant catalytic PI3K isoforms blocked AKT phosphorylation in both cell lines, but unexpectedly led to increased cancer-stem-cell-associated gene expression. They speculated that this unexpected result could have been caused by inhibitory cross-talk between PI3K and MEK/ERK pathways, where MEK/ERK pathway “corrected” the anti-tumour effects of PI3K inhibition [82]. This highlighted the need for combined targets. Supporting this idea, Iqbal et al. in 2016 demonstrated that combining p110α isoform inhibitor with a PIM kinase inhibitor can successfully reduce GBM stemness and GBM neurosphere growth in cultures [83]. Although it presents a novel therapeutic target, PI3K signalling involvement in GBM is complicated and requires further research.
Reprograming CSCs into more differentiated and less oncogenic phenotypes is a novel therapeutic approach in GBM. Undifferentiated CSCs with an uncontrolled renewal potential play a pivotal role in the development and progression of GBM [84]. The potential role of PI3K in this process has already been discussed above. In a different approach, Lee et al. demonstrated that a protein called Znf179 is involved in reprogramming GBM cells into a more differentiated phenotype by inducing cell arrest in the G0/G1 phase through p53-p21-p27 cell cycle signalling pathways. Interestingly, Znf179′s mechanism of action did not affect apoptosis or expression levels of neural progenitor markers such as CD133. It worked by reducing tumour burden via up-regulating GBM cell differentiation without depleting the neural progenitor cell population [64]. Further research is needed to better understand Znf179′s therapeutic application in human GBMs.
Inducing apoptosis is another potential approach for GBM treatment. Recently, studies have focused on the restoration of p53 function. One of the mechanisms by which p53 function is altered in GBM is through sequestration by MDM2 oncoprotein. Even in the cells with genetically functional p53, formation of the p53/MDM2 complexes reduces p53 availability, and thus, function. Therefore, multiple treatment approaches focused on disrupting these p53/MDM2 complexes. Costa et al., in 2013, demonstrated that a small MDM2 inhibitor molecule called ISA27 was able to reactivate p53 function to initiate cell cycle arrest and apoptosis in vitro. It was also effective in inhibiting cell proliferation and inducing apoptosis of tumour tissue in mouse models [85]. Another study used small-molecule antagonists of MDM2 named nutlin-3a to inhibit p53/MDM2 interaction. The use of nutlin-3a successfully induced p53-dependent apoptosis in glioma cell lines [86]. Disrupting the p53/MDM2 complex using RNA molecules has also been studied. Lou et al., in 2020, identified a circular RNA molecule called CDR1as which, by binding to p53, disrupted p53/MDM2 complex formation. Consequently, CDR1as was effective at inhibiting tumour growth both in vitro and in vivo [87]. It is important to understand that therapeutic agents focusing on p53/MDM2 interaction were only effective in treating those with functional p53. Glioma cells with absent or mutated p53 were insensitive to treatment with these agents [85,86,87].
6.2. Invasion and Migration
Cancer cells evolve to survive under stress conditions such as hypoxia and starvation. Targeting these adaptive mechanisms, which are also involved in developing therapy resistance, could be a novel treatment strategy. Wanka et al. studied the effects of a metabolism-regulator called TIGAR (Tp53-induced glycolysis and apoptosis regulator) on hypoxia-induced cell death. TIGAR was overexpressed in GBM and protected the cells against environmental glucose and oxygen restrictions. TIGAR worked by increasing respiration and improving energy yield from glucose. Reversal of TIGAR’s effects was possible with silencing RNA (siRNA)-led inhibition of TKTL1. This study suggests that targeting TKTL1 may reduce tumour survival and increase sensitivity to hypoxia-induced therapies [88]. On the other hand, Wang et al. in 2019 identified that GBM cells could up-regulate GLUT1 via α-KG/IKKβ/NF-κB signalling to cope with low glucose conditions [52]. Under low glucose conditions, phosphorylated GDH1 interacts with the IKK complex to produce α-KG, which directly activates IKKβ and NF-κB signalling and leads to GLUT1 expression. This promotes compensatory glucose uptake [52]. This novel pathway suggests that preventing phosphorylation of GDH1 or disrupting α-KG and NF-κB interaction could be targeted for future therapies. Disrupting GBM adaptations to hypoxia and starvation would decrease its survival and progression.
GBM cells show diffuse infiltration into the surrounding neural network as the disease progresses. This process is complex and include multiple signalling pathways. Here, we will focus on Wnt/β-catenin signalling pathway as a novel therapeutic target. This pathway, which is often aberrantly activated in GBM, promotes epithelial-to-mesenchymal transition (EMT) and tumour invasion through increased β-catenin phosphorylation and/or nuclear localization [89]. EMT is mediated via increased expression of downstream factors such as Twist1, ZEB1, Snail and Slug [90]. Bao et al. studied the effects of Wnt/β-catenin inhibition on GBM invasion and migration. They demonstrated that inhibition of the Wnt/β-catenin pathway with CBX7 overexpression led to reduced GBM invasive ability and EMT. This effect was facilitated through CBX7-mediated enhancement of Wnt/β-catenin inhibitor (DKK1) expression, which led to reduced downstream ZEB1 expression in vivo and in vitro [91]. This study offers a potential role of CBX7 in the inhibition of neoplastic development. On the other hand, Liu et al. tested a novel micro-RNA therapy to regulate the Wnt/β-catenin pathway in GBM. Normally, micro-RNA (miR-504) inhibits the Wnt/β-catenin pathway by directly repressing FZD7 expression. Low miR-504 expression and low miR-504/FZD7 ratio correlated with increased MET and poor survival. They demonstrated that increasing miR-504 expression could suppress GBM migration, invasion, EMT and stemness characteristics in vitro and in vivo [92]. This identifies miR-504 as a possible therapeutic candidate in malignant brain tumours.
Glioblastoma cells masterfully manipulate tumour microenvironment (TME) and the cells within to invade brain parenchyma. Astrocytic gliosis is a term used to describe reactive changes seen in previously healthy astrocytes following exposure to glioma environment. These astrocytes show increased proliferation and display properties that help GBM invasion and angiogenesis [93]. Recently, the focus has been on studying the role of GBM extracellular vesicles (EV) in pathological glioma/astrocyte interaction. Hallal et al. exposed healthy astrocytes to EVs isolated from GBM patients [94]. Internalization of GBM-EV by astrocytes led to enhanced astrocyte podosome formation, suggesting a mechanism by which astrocytes support GBM invasion (ECM remodelling and blood-brain barrier breakdown). These effects were more significant when EVs were isolated from cancer stem cells (NES+/CD133+) compared to differentiated GBM cells (NES-/CD133-). Further molecular analysis suggested GBM-EV exposure activated MYC and inhibited p53 pathways, creating a pro-inflammatory and tumour-promoting astrocyte phenotype [94]. A novel immunotherapy approach involving GBM extravesicles has been tested by Pineda et al. Vaccination with GBM microvesicles led to significant reduction in tumour volume in animal models. This was also associated with increased tumour infiltration with immune cells [69].
6.3. Immune Suppression
GBM cells can evade the host immune system by modifying TME. Notch1 has been identified as a potential regulator in the development of an immune suppressive TME. Notch1 increases expression of M-CSF and SDF-1, which are important in recruitment and development of TAMs (tumour-associated macrophages). TAMs secrete pro-tumorigenic factors such as TGF-β and promote tumour growth. Gu et al. demonstrated that inhibiting an upstream regulator of Notch1 called NKAP (NF-κB activating protein) led to abrogated tumour growth and invasion both in vitro and in vivo. NKAP’s function involved regulating TME through Notch1 expression [49]. Interestingly, Hai et al. demonstrated that Notch1 and NF-κB interact in a reciprocal regulatory loop. Inhibition of Notch1 using a chemical Notch1 inhibitor (DAPT) was associated with NF-κB (p65) suppression and subsequent inhibition of GBM cell proliferation and increased apoptosis both in vitro and in vivo [95]. This study suggests that, in the future, combining drugs to inhibit both Notch1 and NF-κB pathways may be advantageous.
Multiple immunotherapy strategies have been trialed to train the host immune system against GBM cells. Our review identified that the major focus has been on development of dendritic cell vaccines. Simply, these vaccines are developed by training autologous DC to recognize tumour-specific factors. This is accomplished by pulsing them with tumour lysate or tumour-specific antigens.
One group designed an autologous DC vaccine called ICT-107 using six synthetic peptide epitopes targeting glioma-stem-cell-associated antigens. A randomized, double-blind placebo-controlled phase II trial using ICT-107 showed that the addition of ICT-107 to Stupp Protocol significantly improved progression-free survival with maintenance of quality of life [96]. They further identified that patients with HLA-A2+ tumours showed increased benefit and greater immunological response [96].
Other trials used tumour lysate in the development of autologous DC vaccines [66,97]. Phase I data for such vaccines establishes their safety and tolerability [66]. The phase II trial of such a vaccine (Audencel) showed that although well tolerated, it failed to show significant improvement in overall- or progression-free survival [97]. Blinded interim data for a phase III clinical trial using a similar DC vaccine (DCVax®-L) suggests the addition of DCVax®-L to standard therapy is safe and may extend survival [98].
Certain molecular characteristics may influence response to DC immunotherapy. Erhant et al. studied effects of Audencel immunization at molecular and cellular levels after it failed to show positive clinical outcomes. They identified that Audencel was able to stimulate the immune system, especially Th-1 related immunovariables, however its effect was influenced by the host immune system [99]. Supporting this theory, Zeng et al. demonstrated that high 90K antigen expression, a biomarker for glioma malignancy and prognosis, was associated with enhanced response to DC vaccine immunotherapy in vitro [100]. A double-blind, placebo-controlled phase II clinical trial showed that patients with wild-type IDH1, mutated TERT and low B7-H4 expression have greater response to DC-based vaccine immunotherapy [101]. Other predictors of beneficial response to autologous DC vaccine were younger age, gross resection of tumour, tumour showing infiltrating lymphocytes and peripheral blood mononuclear cells with lower PD-1+/CD8+ ratio [102].
An alternative immunotherapeutic approach is the use of B cell-based vaccines. Lee-Chang et al. developed BVax, a B cell vaccine made up of 4-1BBL+ B cells activated with CD40 agonist and FNγ stimulation [103]. BVax has been shown to migrate to secondary lymphoid organs and activate autologous CD8+ T cells, which could successfully kill autologous GBM cells. A combination of radiation, BVax and PD-L1 blockade led to 80% tumour eradication and elicited immunological memory. Excitingly, this immunological memory prevented the re-growth of new tumours upon reinjection in cured mice [103]. B cell-based vaccination in GBM treatment is a novel concept and holds great potential for the future. More experimental immunotherapy strategies include the use of viral vectors. Kim et al. developed a novel adenovirus vector carrying a GBM-specific antigen that selectively infected DC. Vaccination with this viral vector in GBM-implanted murine models was associated with prolonged survival [104]. The safety and clinical benefit of these novel therapies are still being investigated.
It is obvious that neuroscience is progressing and shows true potential in finding new drugs against glioblastomas in the future. Neuro-Researchers must deal with the molecular and cellular heterogeneity, the migration and invasion, and the immune microenvironment of the glioblastoma. Another challenge that needs to be taken into consideration is the delivery system of any therapeutic agent to the brain. The significant obstacle to overcome is the blood-brain barrier. The latter is disrupted in glioblastomas, but it is not sufficient to allow all the therapeutic agents to cross the barrier freely [105,106]. Several delivery systems have been described but the research for the optimal one is still young [107].
7. Conclusions
Recent insights into molecular pathophysiology of GBM has expanded the horizon for therapeutic strategies. In this review, we discussed novel therapeutic strategies targeting every stage of gliomagenesis and GBM progression. Inhibiting glioma cell stemness through the Nanog/Sox2/CD133 pathway can halt gliomagenesis. Potential therapeutic targets for this pathway included miR-26a inhibitors or AP-2α. Proliferation of undifferentiated GBM cells can be stopped by restoration of p53 function through disruption of p53/MDM2 complexes. Utilizing MDM2 inhibitor molecules such as ISA27 and nutlin-3a or p53-binding circular RNA molecule (CDR1as) can restore p53 function and induce apoptosis. Targeting GBM adaptation mechanisms can prevent GBM survival. GBM cells’ adaptation to hypoxia and starvation can be disrupted by targeting T53-induced glycolysis and apoptosis regulator action. Glioma invasion and migration can be targeted through multiple pathways. Glioblastoma cells masterfully manipulate tumour microenvironment to recruit astrocytes into their invasive process. Understanding the role of GBM extracellular vesicles in GBM/astrocyte interaction can help develop novel treatment strategies, including the use of vaccine immunotherapy. Exciting novel immune therapy strategies including dendritic cell vaccines, B-cell vaccines and viral vectors have been developed for the treatment of GBM. Autologous dendritic cell vaccines are already being tested in phase III clinical trials, while others, like viral vectors, are still early in their development stages. Whatever stage they may be in, these novel therapeutic approaches open a door of possibility and hope for the future of GBM treatment.
Supplementary Materials
The following are available online at www.mdpi.com/article/10.3390/ijms23052607/s1.
Author Contributions
Conceptualization, D.G.; Methodology, D.G., Y.E., M.A., A.B., R.P. and S.R.; Writing—Original Draft Preparation, Y.E., M.A. and D.G.; Writing—Review & Editing, M.A., D.G., A.B., R.P., S.R., I.M. and S.F. Literature systematic search: M.A., Y.E., R.P., S.R., A.B., I.M. and S.F. Tables/Figures: M.A., D.G., A.B., R.P., S.R., I.M. and S.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Akt | Protein kinase B |
| ALDOA | Fructose-bisphosphate aldolase A |
| ARST | ALDOA-Related Specific Transcript |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| ATIP1 | AT2 receptor interacting protein |
| BCNU | carmustine |
| BEJ | base excision repair |
| CAR-T | chimeric antigen receptor T |
| CBX 7 | Chromobox 7 |
| CCND2 | Cyclin D2 |
| CCNU | lomustine |
| CDK | Cyclin dependent kinase |
| CDR1 | Cerebellar degeneration-related protein |
| CIC | Capicua (Transcriptional suppressor) |
| CK2 | Casein kinase-2 |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CRNDE | Colorectal Neoplasia Differentially Expressed |
| CSC | cancer stem cell |
| CXCR4 | Chemokine receptor-4 |
| DAMPs | Damage-associated molecular pattern |
| DC | Dendritic cells |
| DDR | DNA damage response |
| DDRd | DNA damage repair deficiency |
| DSBs | double-strand breaks base |
| Ebp1 | ErbB3-binding protein 1 |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-to-mesenchymal transition |
| EV | Extracellular vesicles |
| Fbxw7 | F-box/WD repeat-containing protein 7 |
| GAP | GTPase-activating proteins |
| GDH1 | Glutamate dehydrogenase 1 |
| GLUT 1 | Glucose transporter 1 |
| GSC | glioma stem cells |
| GSK-3β | Glycogen synthase kinase-3β |
| HMGA | High mobility group A |
| HR | homologous recombination |
| IDO | indoleamine 2:3-dioxygenase |
| IKK | IκB kinase |
| IR | Ionizing radiation |
| JNK | c-Jun N-terminal kinases |
| MAPK | mitogen-activated protein kinase |
| MCL-1 | Myeloid leukemia 1 |
| MDM2 | Mouse double minute 2 homolog |
| MDM4 | Mouse double minute 4 homolog |
| MEK | Mitogen-activated protein/extracellular signal-regulated kinase |
| MGMT | O6- methylguanine-DNA methyltransferase |
| MHY-9 | Myosin Heavy Chain 9 |
| MiR | micro RNA |
| MK 2 | MAPK-activated protein kinase 2 |
| MMP | Matrix Metalloproteinase |
| mTOR | mammalian target of rapamycin |
| MTSG1 | Mitochondrial tumour suppressor |
| MTUS1 | TUS1 (Microtubule Associated Scaffold Protein 1) |
| NDRG1 | N-myc downstream-regulated gene 1 |
| NF-1 | neurofibromin 1 |
| NHEJ | non-homologous end joining |
| NHEJ | Non-homologous end joining |
| NKAP | NF-κB activating protein |
| p53 | Tumour protein p53 |
| PARP | Poly (ADP-ribose) polymerase |
| PDGF | platelet-derived growth factor |
| PDK1 | phosphoinositide-dependent kinase-1 |
| PFKP | Platelet isoform of phosphofructokinase |
| PI3K | Phosphatidylinositol 3-kinase |
| PIM | Proto-oncogene serine/threonine-protein kinase |
| POU | Pituitary specific Pit1, Octamer transcription factor protein, Unc-86 transcription factor |
| PPARγ | peroxisome proliferator-activated receptor γ |
| PPDR | Pleiotropic Drug Resistance |
| Pt-1-DMCa | Platinum analogue Pt-MBL-III_7-D_MCa |
| PTEN | Phosphatase and Tensin Homolog Deleted from Chromosome 10 |
| RAD 51 | DNA repair protein RAD51 |
| RB1 | p16INK4a/CDK-4/Retinoblastoma 1 |
| RNAi | RNA interference |
| RTK | Receptor Tyrosine kinase |
| RTK | Tyrosine receptor kinases |
| Sh RNA | Short hairpin RNA |
| SHH | sonic hedgehog homolog |
| siRNA | Small interfering RNA |
| SIRT1 | Sirtuin 1 |
| SNRPG | Small Nuclear Ribonucleoprotein Polypeptide G |
| Sox | SRY-Box Transcription Factor |
| SSBs | Single-strand breaks |
| STAT | Signal transducers and activators of transcriptome |
| STKs | specific protein kinases |
| TAMs | tumour-associated macrophages |
| TCGA | The Cancer Genome Atlas |
| TGF-β | growth factor-beta |
| THTMP | 2-[(3:4-dihydroquinolin-1(2H)-yl)(p-tolyl)methyl]phenol |
| TIGAR | Tp53-induced glycolysis and apoptosis regulator |
| TKTL1 | Transketolase Like 1 |
| TME | tumour microenvironment |
| TMZ | Temozolomide |
| TNFα | Tumour Necrosis Factor α |
| TSG | Tumour suppressor genes |
| VEGF | vascular endothelial growth factor |
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Iacob, G.; Dinca, E.B. Current Data and Strategy in Glioblastoma Multiforme. J. Med. Life 2009, 2, 386–393. [Google Scholar]
- Ohgaki, H.; Kleihues, P. Epidemiology and Etiology of Gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef]
- Brodbelt, A.; Greenberg, D.; Winters, T.; Williams, M.; Vernon, S.; Collins, V.P. Glioblastoma in England: 2007–2011. Eur. J. Cancer 2015, 51, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Darefsky, A.S.; King, J.T.; Dubrow, R. Adult Glioblastoma Multiforme Survival in the Temozolomide Era: A Population-Based Analysis of Surveillance, Epidemiology, and End Results Registries. Cancer 2012, 118, 2163–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic Pathways to Primary and Secondary Glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J.-I. Aberrant Signaling Pathways in Glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [Green Version]
- Vartanian, A.; Singh, S.K.; Agnihotri, S.; Jalali, S.; Burrell, K.; Aldape, K.D.; Zadeh, G. GBM’s Multifaceted Landscape: Highlighting Regional and Microenvironmental Heterogeneity. Neuro-Oncol. 2014, 16, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas (TCGA) Research Network. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Taneri, B.; Asilmaz, E.; Delikurt, T.; Savas, P.; Targen, S.; Esemen, Y. Human Genetics and Genomics: A Practical Guide; Wiley: Weinheim, Germany, 2020; Volume 1. [Google Scholar]
- Ueki, K.; Ono, Y.; Henson, J.W.; Efird, J.T.; von Deimling, A.; Louis, D.N. CDKN2/P16 or RB Alterations Occur in the Majority of Glioblastomas and Are Inversely Correlated. Cancer Res. 1996, 56, 150–153. [Google Scholar] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK Inhibitors: Positive and Negative Regulators of G1-Phase Progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-Specific Defects in DNA Repair Pathways as Targets for Personalized Therapeutic Approaches. Trends Genet. 2014, 30, 326–339. [Google Scholar] [CrossRef]
- Leung, S.Y.; Yuen, S.T.; Chan, T.L.; Chan, A.S.; Ho, J.W.; Kwan, K.; Fan, Y.W.; Hung, K.N.; Chung, L.P.; Wyllie, A.H. Chromosomal Instability and P53 Inactivation Are Required for Genesis of Glioblastoma but Not for Colorectal Cancer in Patients with Germline Mismatch Repair Gene Mutation. Oncogene 2000, 19, 4079–4083. [Google Scholar] [CrossRef] [Green Version]
- Erasimus, H.; Gobin, M.; Niclou, S.; van Dyck, E. DNA Repair Mechanisms and Their Clinical Impact in Glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing Repair and Tolerance of DNA Damage Caused by Alkylating Agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased Expression of the Epidermal Growth Factor Receptor Gene in Malignant Gliomas Is Invariably Associated with Gene Amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunayama, J.; Matsuda, K.-I.; Sato, A.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Crosstalk Between the PI3K/MTOR and MEK/ERK Pathways Involved in the Maintenance of Self-Renewal and Tumorigenicity of Glioblastoma Stem-Like Cells. Stem Cells 2010, 28, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Huang, H.J.; Cavenee, W.K. The Phosphoinositol Phosphatase Activity of PTEN Mediates a Serum-Sensitive G1 Growth Arrest in Glioma Cells. Cancer Res. 1998, 58, 5002–5008. [Google Scholar] [PubMed]
- Kita, D.; Yonekawa, Y.; Weller, M.; Ohgaki, H. PIK3CA Alterations in Primary (de Novo) and Secondary Glioblastomas. Acta Neuropathol. 2007, 113, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, G.S.; O’Rourke, D.M. Mitogenic Signaling Cascades in Glial Tumors. Neurosurgery 2003, 52, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the Angiogenic Switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Onishi, M.; Ichikawa, T.; Kurozumi, K.; Date, I. Angiogenesis and Invasion in Glioma. Brain Tumor Pathol. 2011, 28, 13–24. [Google Scholar] [CrossRef]
- Lindsey, S.; Langhans, S.A. Crosstalk of Oncogenic Signaling Pathways during Epithelial-Mesenchymal Transition. Front. Oncol. 2014, 4, 358. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–Mesenchymal Transitions in Tumour Progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human Cortical Glial Tumors Contain Neural Stem-like Cells Expressing Astroglial and Neuronal Markers in Vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural Stem Cells and the Origin of Gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Weissenberger, J.; Loeffler, S.; Kappeler, A.; Kopf, M.; Lukes, A.; Afanasieva, T.A.; Aguzzi, A.; Weis, J. IL-6 Is Required for Glioma Development in a Mouse Model. Oncogene 2004, 23, 3308–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.; Ampudia-Mesias, E.; Shaver, R.; Horbinski, C.M.; Moertel, C.L.; Olin, M.R. Tumor-Derived Vaccines Containing CD200 Inhibit Immune Activation: Implications for Immunotherapy. Immunotherapy 2016, 8, 1059–1071. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Steinman, R.M. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Pinzon-Charry, A.; Maxwell, T.; López, J.A. Dendritic Cell Dysfunction in Cancer: A Mechanism for Immunosuppression. Immunol. Cell Biol. 2005, 83, 451–461. [Google Scholar] [CrossRef]
- Tadayoninia, A.; Bazi, Z.; Khosravi, A.; Oladnabi, M. WDR81 Gene Silencing Can Reduce Exosome Levels in Human U87-MG Glioblastoma Cells. J. Mol. Neurosci. 2021, 71, 1696–1702. [Google Scholar] [CrossRef]
- Zhao, H.-F.; Wang, J.; Jiang, H.-R.; Chen, Z.-P.; To, S.-S.T. PI3K P110β Isoform Synergizes with JNK in the Regulation of Glioblastoma Cell Proliferation and Migration through Akt and FAK Inhibition. J. Exp. Clin. Cancer Res. 2016, 35, 78. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Liu, R.; Li, J.; Wang, Y.; Tan, L.; Li, X.-J.; Qian, X.; Zhang, C.; Xia, Y.; Xu, D.; et al. EGFR-Phosphorylated Platelet Isoform of Phosphofructokinase 1 Promotes PI3K Activation. Mol. Cell 2018, 70, 197–210.e7. [Google Scholar] [CrossRef] [Green Version]
- Bunda, S.; Heir, P.; Li, A.S.C.; Mamatjan, Y.; Zadeh, G.; Aldape, K. C-Src Phosphorylates and Inhibits the Function of the CIC Tumor Suppressor Protein. Mol. Cancer Res. 2020, 18, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Heir, P.; Metcalf, J.; Li, A.S.C.; Agnihotri, S.; Pusch, S.; Yasin, M.; Li, M.; Burrell, K.; Mansouri, S.; et al. CIC Protein Instability Contributes to Tumorigenesis in Glioblastoma. Nat. Commun. 2019, 10, 661. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Traphagen, N.A.; Li, J.; Lewis, L.D.; Lopes, B.; Asthagiri, A.; Loomba, J.; de Jong, J.; Schiff, D.; Patel, S.H.; et al. Tumor Pharmacokinetics and Pharmacodynamics of the CDK4/6 Inhibitor Ribociclib in Patients with Recurrent Glioblastoma. J. Neuro-Oncol. 2019, 144, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Parikh, M.; Phillips, J.J.; James, C.D.; Molinaro, A.M.; Butowski, N.A.; Clarke, J.L.; Oberheim-Bush, N.A.; Chang, S.M.; Berger, M.S.; et al. Phase-2 Trial of Palbociclib in Adult Patients with Recurrent RB1-Positive Glioblastoma. J. Neuro-Oncol. 2018, 140, 477–483. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, P.; Liu, L.; Li, H.; Cheng, S.; Li, S.; Wang, Y.; Zheng, C.; Dong, J.; Zhang, L. The Circ_0001367/MiR-545-3p/LUZP1 Axis Regulates Cell Proliferation, Migration and Invasion in Glioma Cells. Front. Oncol. 2021, 11, 4856. [Google Scholar] [CrossRef]
- Sun, J.; He, D.; Fu, Y.; Zhang, R.; Guo, H.; Wang, Z.; Wang, Y.; Gao, T.; Wei, Y.; Guo, Y.; et al. A Novel LncRNA ARST Represses Glioma Progression by Inhibiting ALDOA-Mediated Actin Cytoskeleton Integrity. J. Exp. Clin. Cancer Res. 2021, 40, 187. [Google Scholar] [CrossRef]
- Wang, C.; Huang, S.; Rao, S.; Hu, J.; Zhang, Y.; Luo, J.; Wang, H. Decreased Expression of MiR-410-3p Correlates with Poor Prognosis and Tumorigenesis in Human Glioma. Cancer Manag. Res. 2019, 11, 10581–10592. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Gao, T.; Zhang, L.; Chen, X.; Pang, Q.; Wang, Y.; Wang, D.; Li, J.; Liu, Q. NKAP Alters Tumor Immune Microenvironment and Promotes Glioma Growth via Notch1 Signaling. J. Exp. Clin. Cancer Res. 2019, 38, 291. [Google Scholar] [CrossRef] [Green Version]
- Rosmaninho, P.; Mükusch, S.; Piscopo, V.; Teixeira, V.; Raposo, A.A.; Warta, R.; Bennewitz, R.; Tang, Y.; Herold-Mende, C.; Stifani, S.; et al. Zeb1 Potentiates Genome-wide Gene Transcription with Lef1 to Promote Glioblastoma Cell Invasion. EMBO J. 2018, 37, e97115. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, Y.; Chen, J.; Qiu, J.; Huang, K.; Wu, M.; Xia, C. IDH1 R132H Mutation Enhances Cell Migration by Activating AKT-MTOR Signaling Pathway, but Sensitizes Cells to 5-FU Treatment as NADPH and GSH Are Reduced. PLoS ONE 2017, 12, e0169038. [Google Scholar] [CrossRef]
- Wang, X.; Liu, R.; Qu, X.; Yu, H.; Chu, H.; Zhang, Y.; Zhu, W.; Wu, X.; Gao, H.; Tao, B.; et al. α-Ketoglutarate-Activated NF-ΚB Signaling Promotes Compensatory Glucose Uptake and Brain Tumor Development. Mol. Cell 2019, 76, 148–162.e7. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Zhang, M.; Wang, Y.; Wang, Y. NF-YC in Glioma Cell Proliferation and Tumor Growth and Its Role as an Independent Predictor of Patient Survival. Neurosci. Lett. 2016, 631, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Ülgen, E.; Can, Ö.; Bilguvar, K.; Oktay, Y.; Akyerli, C.B.; Danyeli, A.E.; Yakıcıer, M.C.; Sezerman, O.U.; Pamir, M.N.; Özduman, K. Whole Exome Sequencing–Based Analysis to Identify DNA Damage Repair Deficiency as a Major Contributor to Gliomagenesis in Adult Diffuse Gliomas. J. Neurosurg. 2020, 132, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Majuelos-Melguizo, J.; Rodríguez, M.I.; López-Jiménez, L.; Rodríguez-Vargas, J.M.; Martí Martín-Consuegra, J.M.; Serrano-Sáenz, S.; Gavard, J.; de Almodóvar, J.M.R.; Oliver, F.J. PARP Targeting Counteracts Gliomagenesis through Induction of Mitotic Catastrophe and Aggravation of Deficiency in Homologous Recombination in PTEN-Mutant Glioma. Oncotarget 2015, 6, 4790–4803. [Google Scholar] [CrossRef] [Green Version]
- Ohba, S.; Mukherjee, J.; See, W.L.; Pieper, R.O. Mutant IDH1-Driven Cellular Transformation Increases RAD51-Mediated Homologous Recombination and Temozolomide Resistance. Cancer Res. 2014, 74, 4836–4844. [Google Scholar] [CrossRef] [Green Version]
- Che Mat, M.F.; Mohamad Hanif, E.A.; Abdul Murad, N.A.; Ibrahim, K.; Harun, R.; Jamal, R. Silencing of ZFP36L2 Increases Sensitivity to Temozolomide through G2/M Cell Cycle Arrest and BAX Mediated Apoptosis in GBM Cells. Mol. Biol. Rep. 2021, 48, 1493–1503. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, X.; Cheng, Y.; Zhou, Y.; Ma, X.; Zhang, J.; Heng, X.; Feng, F. TROAP Regulates Cell Cycle and Promotes Tumor Progression through Wnt/Β-Catenin Signaling Pathway in Glioma Cells. CNS Neurosci. Ther. 2021, 27, 1064–1076. [Google Scholar] [CrossRef]
- Meng, Q.; Zhang, Y.; Li, G.; Li, Y.; Xie, H.; Chen, X. New Insights for Precision Treatment of Glioblastoma from Analysis of Single-Cell LncRNA Expression. J. Cancer Res. Clin. Oncol. 2021, 147, 1881–1895. [Google Scholar] [CrossRef]
- Wang, S.; O’Rourke, D.M.; Chawla, S.; Verma, G.; Nasrallah, M.P.; Morrissette, J.J.D.; Plesa, G.; June, C.H.; Brem, S.; Maloney, E.; et al. Multiparametric Magnetic Resonance Imaging in the Assessment of Anti-EGFRvIII Chimeric Antigen Receptor T Cell Therapy in Patients with Recurrent Glioblastoma. Br. J. Cancer 2019, 120, 54–56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ni, S.; Huang, B.; Wang, L.; Zhang, X.; Li, X.; Wang, H.; Liu, S.; Hao, A.; Li, X. Overexpression of SCLIP promotes growth and motility in glioblastoma cells. Cancer Biol. Ther. 2015, 16, 97–105. [Google Scholar] [CrossRef]
- Xu, W.; Zheng, J.; Bie, S.; Kang, L.; Mao, Q.; Liu, W.; Guo, J.; Lu, J.; Xia, R. Propofol inhibits Wnt signaling and exerts anticancer activity in glioma cells. Oncol. Lett. 2018, 16, 402–408. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.H.; Cheng, C.H.; Shih, C.M.; Ho, K.H.; Lin, C.W.; Lee, C.C.; Liu, A.J.; Chang, C.K.; Chen, K.C. The Inhibition of microRNA-128 on IGF-1-Activating mTOR Signaling Involves in Temozolomide-Induced Glioma Cell Apoptotic Death. PLoS ONE. 2016, 11, e0167096. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Chen, C.-L.; Lee, Y.-C.; Kao, T.-J.; Chen, K.-Y.; Fang, C.-Y.; Chang, W.-C.; Chiang, Y.-H.; Huang, C.-C. Znf179 Induces Differentiation and Growth Arrest of Human Primary Glioblastoma Multiforme in a P53-Dependent Cell Cycle Pathway. Sci. Rep. 2017, 7, 4787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhong, Z.; Luo, C.; Xiao, Y.; Li, L.; Zhang, X.; Yang, L.; Xiao, K.; Ning, Y.; Chen, L.; et al. The MiR-26a/AP-2α/Nanog Signaling Axis Mediates Stem Cell Self-Renewal and Temozolomide Resistance in Glioma. Theranostics 2019, 9, 5497–5516. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Omofoye, O.A.; Rudnick, J.D.; Kim, S.; Tighiouart, M.; Phuphanich, S.; Wang, H.; Mazer, M.; Ganaway, T.; Chu, R.M.; et al. A Phase I Study of Autologous Dendritic Cell Vaccine Pulsed with Allogeneic Stem-like Cell Line Lysate in Patients with Newly Diagnosed or Recurrent Glioblastoma. Clin. Cancer Res. 2021, 28, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, L.; Ji’an Yang, P.H.; Zhang, C.; Shi’ao Tong, Z.L.; Tian, D. Identification of Tumor Antigens and Immune Landscape in Glioblastoma for mRNA Vaccine Development. Front Genet. 2021, 12, 701065. [Google Scholar] [CrossRef] [PubMed]
- Pineda, B.; Sánchez García, F.J.; Olascoaga, N.K.; Pérez de la Cruz, V.; Salazar, A.; Moreno-Jiménez, S.; Hernández Pedro, N.; Márquez-Navarro, A.; Ortiz Plata, A.; Sotelo, J. Malignant Glioma Therapy by Vaccination with Irradiated C6 Cell-Derived Microvesicles Promotes an Antitumoral Immune Response. Mol. Ther. 2019, 27, 1612–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245, Erratum in Nature 2019, 566, E13. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Malo, C.S.; Khadka, R.H.; Ayasoufi, K.; Jin, F.; AbouChehade, J.E.; Hansen, M.J.; Iezzi, R.; Pavelko, K.D.; Johnson, A.J. Immunomodulation Mediated by Anti-angiogenic Therapy Improves CD8 T Cell Immunity Against Experimental Glioma. Front. Oncol. 2018, 8, 320. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Chekenya, M.; Pilkington, G.J. NG2 Precursor Cells in Neoplasia: Functional, Histogenesis and Therapeutic Implications for Malignant Brain Tumours. J. Neurocytol. 2002, 31, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, Y.; Nishiyama, A.; Chang, A.; Mork, S.; Barnett, G.H.; Cowell, J.K.; Trapp, B.D.; Staugaitis, S.M. Expression of Oligodendrocyte Progenitor Cell Antigens by Gliomas: Implications for the Histogenesis of Brain Tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 10361–10366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, T.; Lee, V.M.; Rorke, L.B.; Marvin, M.; McKay, R.D.; Trojanowski, J.Q. Nestin Expression in Embryonic Human Neuroepithelium and in Human Neuroepithelial Tumor Cells. Laboratory investigation. J. Tech. Methods Pathol. 1992, 66, 303–313. [Google Scholar]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; de Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Chen, J.; Zhu, Y.; Li, Y.; Wang, Y.; Chen, H.; Wang, J.; Li, X.; Liu, Y.; Li, B.; et al. CD163, a Novel Therapeutic Target, Regulates the Proliferation and Stemness of Glioma Cells via Casein Kinase 2. Oncogene 2019, 38, 1183–1199. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Do, A.S.-M.S.; Amano, T.; Edwards, L.A.; Zhang, L.; de Peralta-Venturina, M.; Yu, J.S. CD133 MRNA-Loaded Dendritic Cell Vaccination Abrogates Glioma Stem Cell Propagation in Humanized Glioblastoma Mouse Model. Mol. Ther. Oncolytics 2020, 18, 295–303. [Google Scholar] [CrossRef]
- Xu, P.-F.; Yang, J.-A.; Liu, J.-H.; Yang, X.; Liao, J.-M.; Yuan, F.-E.; Liu, B.-H.; Chen, Q.-X. PI3Kβ Inhibitor AZD6482 Exerts Antiproliferative Activity and Induces Apoptosis in Human Glioblastoma Cells. Oncology Rep. 2018, 41, 125–132. [Google Scholar] [CrossRef] [Green Version]
- JONES, N.M.; ROWE, M.R.; SHEPHERD, P.R.; McCONNELL, M.J. Targeted Inhibition of Dominant PI3-Kinase Catalytic Isoforms Increase Expression of Stem Cell Genes in Glioblastoma Cancer Stem Cell Models. Int. J. Oncol. 2016, 49, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, A.; Eckerdt, F.; Bell, J.; Nakano, I.; Giles, F.J.; Cheng, S.-Y.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Targeting of Glioblastoma Cell Lines and Glioma Stem Cells by Combined PIM Kinase and PI3K-P110α Inhibition. Oncotarget 2016, 7, 33192–33201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct Isolation of Human Central Nervous System Stem Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [Green Version]
- Costa, B.; Bendinelli, S.; Gabelloni, P.; da Pozzo, E.; Daniele, S.; Scatena, F.; Vanacore, R.; Campiglia, P.; Bertamino, A.; Gomez-Monterrey, I.; et al. Human Glioblastoma Multiforme: P53 Reactivation by a Novel MDM2 Inhibitor. PLoS ONE 2013, 8, e72281. [Google Scholar] [CrossRef] [PubMed]
- Villalonga-Planells, R.; Coll-Mulet, L.; Martínez-Soler, F.; Castaño, E.; Acebes, J.-J.; Giménez-Bonafé, P.; Gil, J.; Tortosa, A. Activation of P53 by Nutlin-3a Induces Apoptosis and Cellular Senescence in Human Glioblastoma Multiforme. PLoS ONE 2011, 6, e18588. [Google Scholar] [CrossRef] [Green Version]
- Lou, J.; Hao, Y.; Lin, K.; Lyu, Y.; Chen, M.; Wang, H.; Zou, D.; Jiang, X.; Wang, R.; Jin, D.; et al. Circular RNA CDR1as Disrupts the P53/MDM2 Complex to Inhibit Gliomagenesis. Mol. Cancer 2020, 19, 138. [Google Scholar] [CrossRef]
- Wanka, C.; Steinbach, J.P.; Rieger, J. Tp53-Induced Glycolysis and Apoptosis Regulator (TIGAR) Protects Glioma Cells from Starvation-Induced Cell Death by Up-Regulating Respiration and Improving Cellular Redox Homeostasis. J. Biol. Chem. 2012, 287, 33436–33446. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Zhu, B.; Liu, X.; Yu, H.; Yong, L.; Liu, X.; Shao, J.; Liu, Z. Inhibition of Oleandrin on the Proliferation and Invasion of Osteosarcoma Cells in Vitro by Suppressing Wnt/β-Catenin Signaling Pathway. J. Exp. Clin. Cancer Res. 2015, 34, 115. [Google Scholar] [CrossRef] [Green Version]
- Kim, K. direct evidence for a role of β-catenin/lef-1 signaling pathway in induction of emt. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef]
- Bao, Z.; Xu, X.; Liu, Y.; Chao, H.; Lin, C.; Li, Z.; You, Y.; Liu, N.; Ji, J. CBX7 Negatively Regulates Migration and Invasion in Glioma via Wnt/β-Catenin Pathway Inactivation. Oncotarget 2017, 8, 39048–39063. [Google Scholar] [CrossRef]
- Liu, Q.; Guan, Y.; Li, Z.; Wang, Y.; Liu, Y.; Cui, R.; Wang, Y. MiR-504 Suppresses Mesenchymal Phenotype of Glioblastoma by Directly Targeting the FZD7-Mediated Wnt–β-Catenin Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 358. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of Astrocyte–Vascular Coupling and the Blood–Brain Barrier by Invading Glioma Cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via P53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hai, L.; Zhang, C.; Li, T.; Zhou, X.; Liu, B.; Li, S.; Zhu, M.; Lin, Y.; Yu, S.; Zhang, K.; et al. Notch1 Is a Prognostic Factor That Is Distinctly Activated in the Classical and Proneural Subtype of Glioblastoma and That Promotes Glioma Cell Survival via the NF-ΚB(P65) Pathway. Cell Death Dis. 2018, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.-J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Preusser, M.; Stockhammer, G.; Nowosielski, M.; Iglseder, S.; Freyschlag, C.; Oberndorfer, S.; et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers 2018, 10, 372. [Google Scholar] [CrossRef] [Green Version]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First Results on Survival from a Large Phase 3 Clinical Trial of an Autologous Dendritic Cell Vaccine in Newly Diagnosed Glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [Green Version]
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological Analysis of Phase II Glioblastoma Dendritic Cell Vaccine (Audencel) Trial: Immune System Characteristics Influence Outcome and Audencel up-Regulates Th1-Related Immunovariables. Acta Neuropathol. Commun. 2018, 6, 135. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, X. 90K Predicts the Prognosis of Glioma Patients and Enhances Tumor Lysate-Pulsed DC Vaccine for Immunotherapy of GBM in Vitro. Aging 2021, 13, 8355–8368. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, F.; Tang, C.; Chen, D.; Qin, Z.; Hua, W.; Xu, M.; Zhong, P.; Yu, S.; Chen, D.; et al. Molecular Subgroups and B7-H4 Expression Levels Predict Responses to Dendritic Cell Vaccines in Glioblastoma: An Exploratory Randomized Phase II Clinical Trial. Cancer Immunol. Immunother. 2018, 67, 1777–1788. [Google Scholar] [CrossRef]
- Jan, C.-I.; Tsai, W.-C.; Harn, H.-J.; Shyu, W.-C.; Liu, M.-C.; Lu, H.-M.; Chiu, S.-C.; Cho, D.-Y. Predictors of Response to Autologous Dendritic Cell Therapy in Glioblastoma Multiforme. Front. Immunol. 2018, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Lee-Chang, C.; Miska, J.; Hou, D.; Rashidi, A.; Zhang, P.; Burga, R.A.; Jusué-Torres, I.; Xiao, T.; Arrieta, V.A.; Zhang, D.Y.; et al. Activation of 4-1BBL+ B Cells with CD40 Agonism and IFNγ Elicits Potent Immunity against Glioblastoma. J. Exp. Med. 2021, 218, 913. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kane, J.R.; Panek, W.K.; Young, J.S.; Rashidi, A.; Yu, D.; Kanojia, D.; Hasan, T.; Miska, J.; Gómez-Lim, M.A.; et al. A Dendritic Cell-Targeted Adenoviral Vector Facilitates Adaptive Immune Response Against Human Glioma Antigen (CMV-IE) and Prolongs Survival in a Human Glioma Tumor Model. Neurotherapeutics 2018, 15, 1127–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Shusta, E.V. Blood–Brain Barrier Modulation to Improve Glioma Drug Delivery. Pharmaceutics 2020, 12, 1085. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Sundgren, P.C.; Tsien, C.I.; Chenevert, T.T.; Junck, L. Physiologic and Metabolic Magnetic Resonance Imaging in Gliomas. J. Clin. Oncol. 2006, 24, 1228–1235. [Google Scholar] [CrossRef]
- Mathew, E.N.; Berry, B.C.; Yang, H.W.; Carroll, R.S.; Johnson, M.D. Delivering Therapeutics to Glioblastoma: Overcoming Biological Constraints. Int. J. Mol. Sci. 2022, 23, 1711. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).