
Application of Homology Modeling by Enhanced Profile–Profile Alignment and Flexible-Fitting Simulation to Cryo-EM Based Structure Determination


Abstract
1. Introduction
2. Materials and Methods
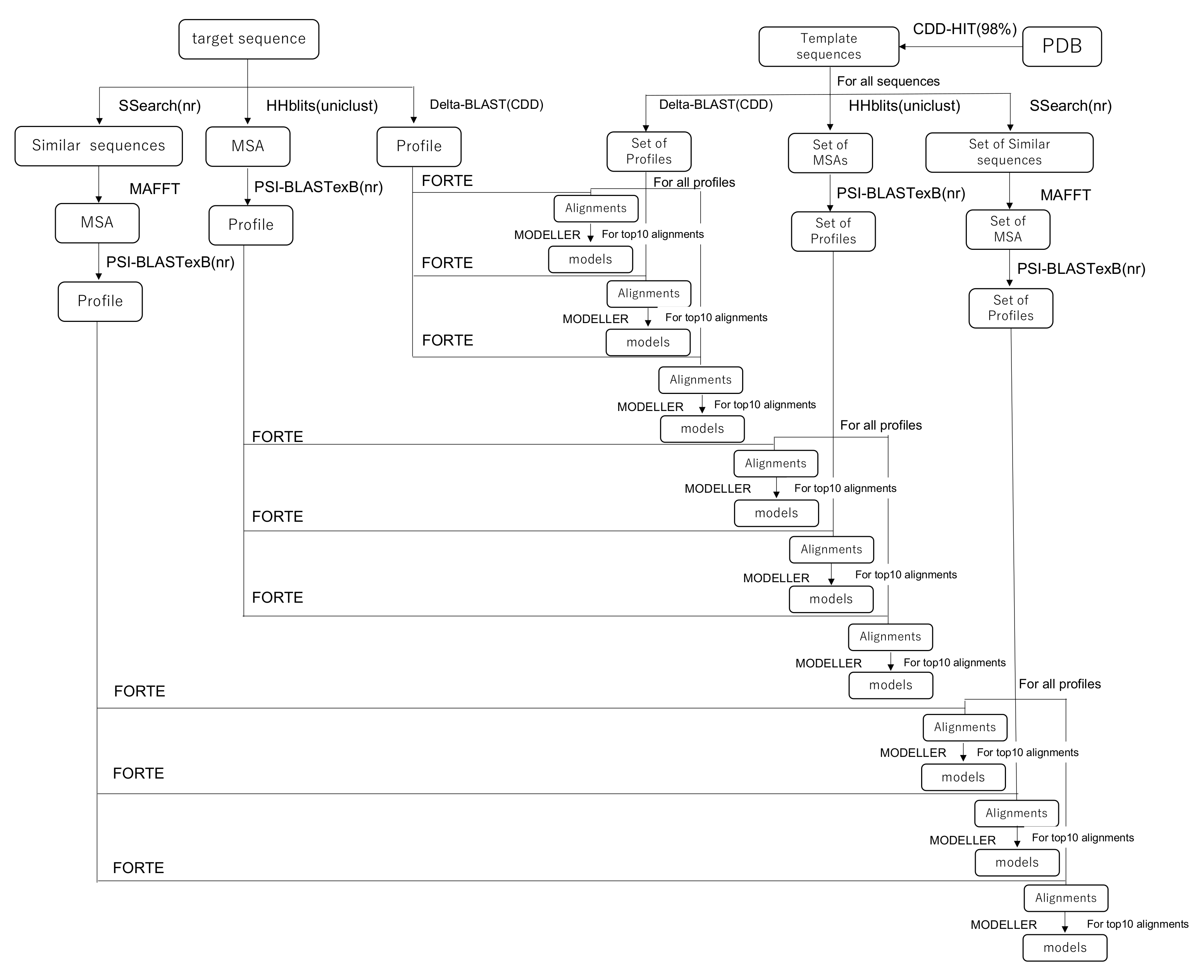
2.1. Profile–Profile-Alignment-Based Homology Modeling
2.2. Correlation-Based Flexible Fitting
2.3. Correlation-Based Metrics
2.4. Modeling of human TPC2 (T0984o in CASP13)
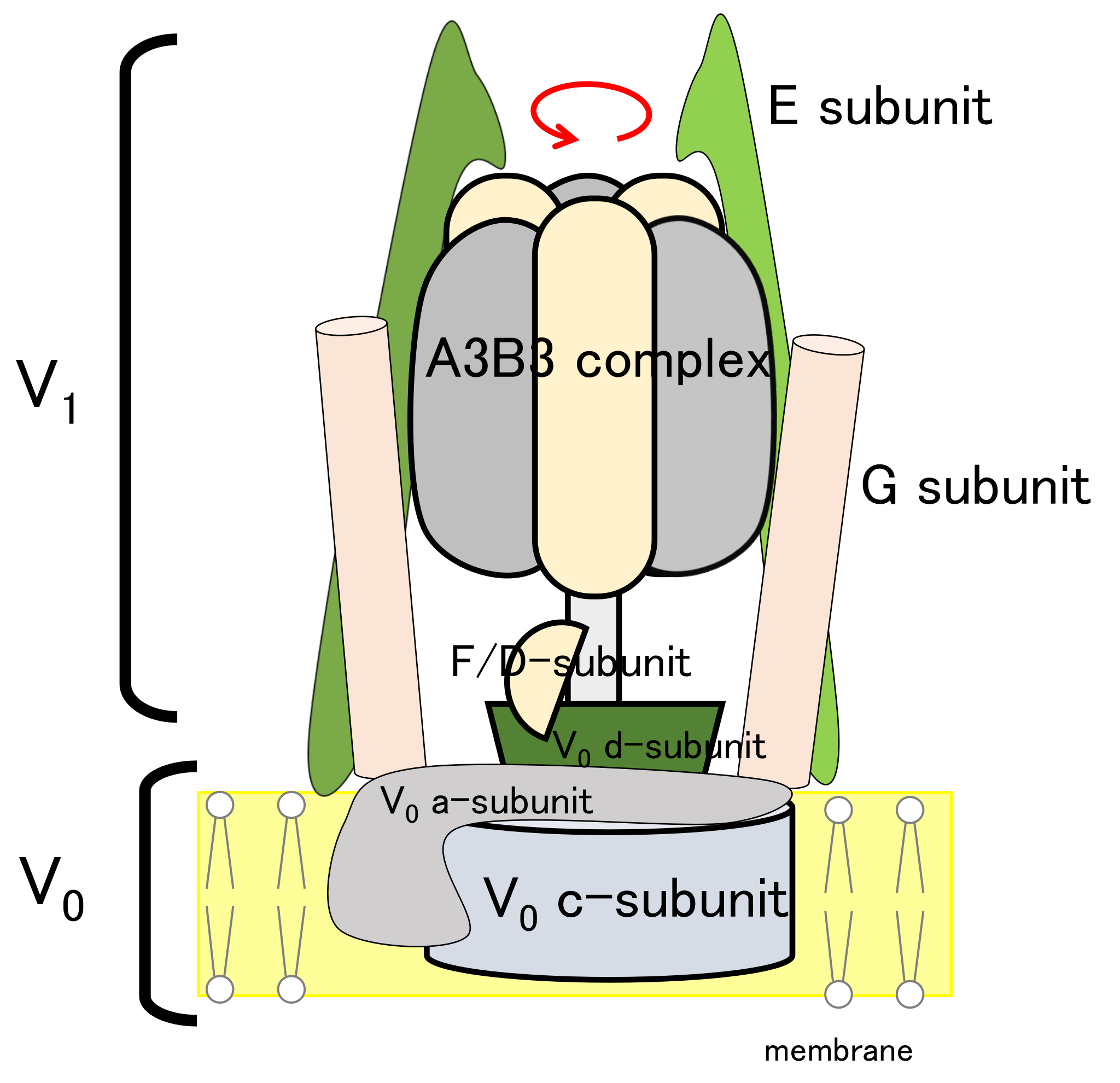
2.5. Modeling of Eh V-ATPase
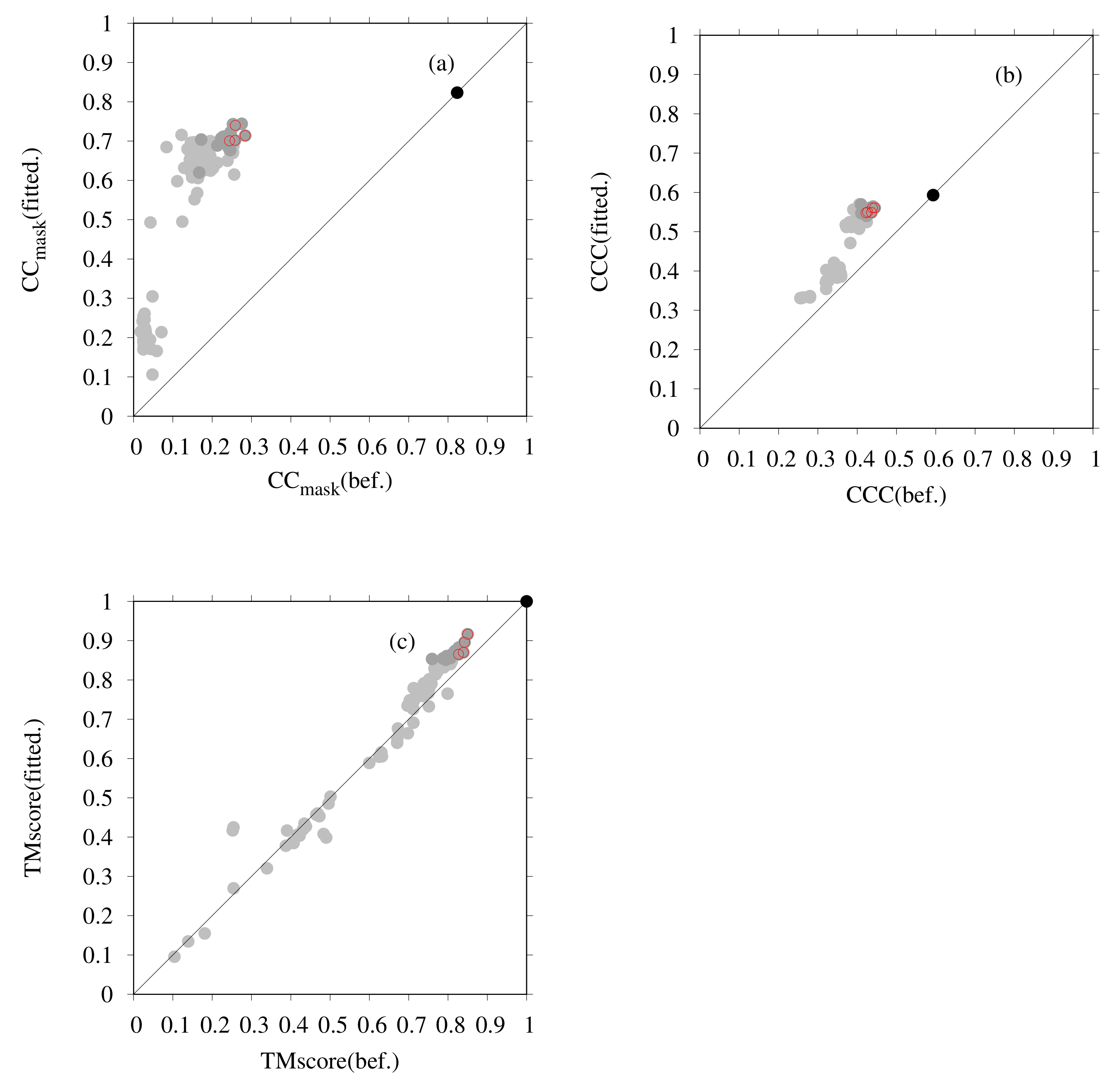
3. Results
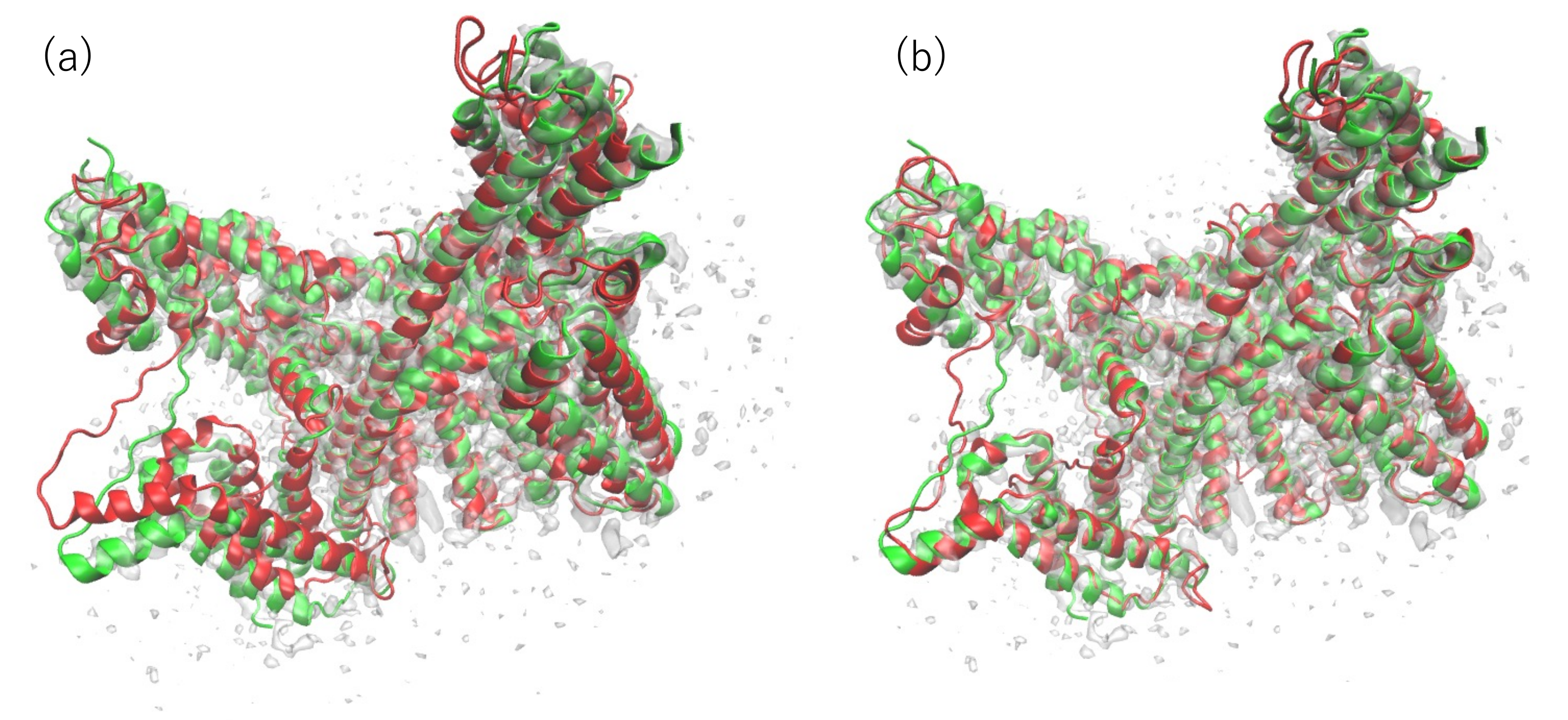
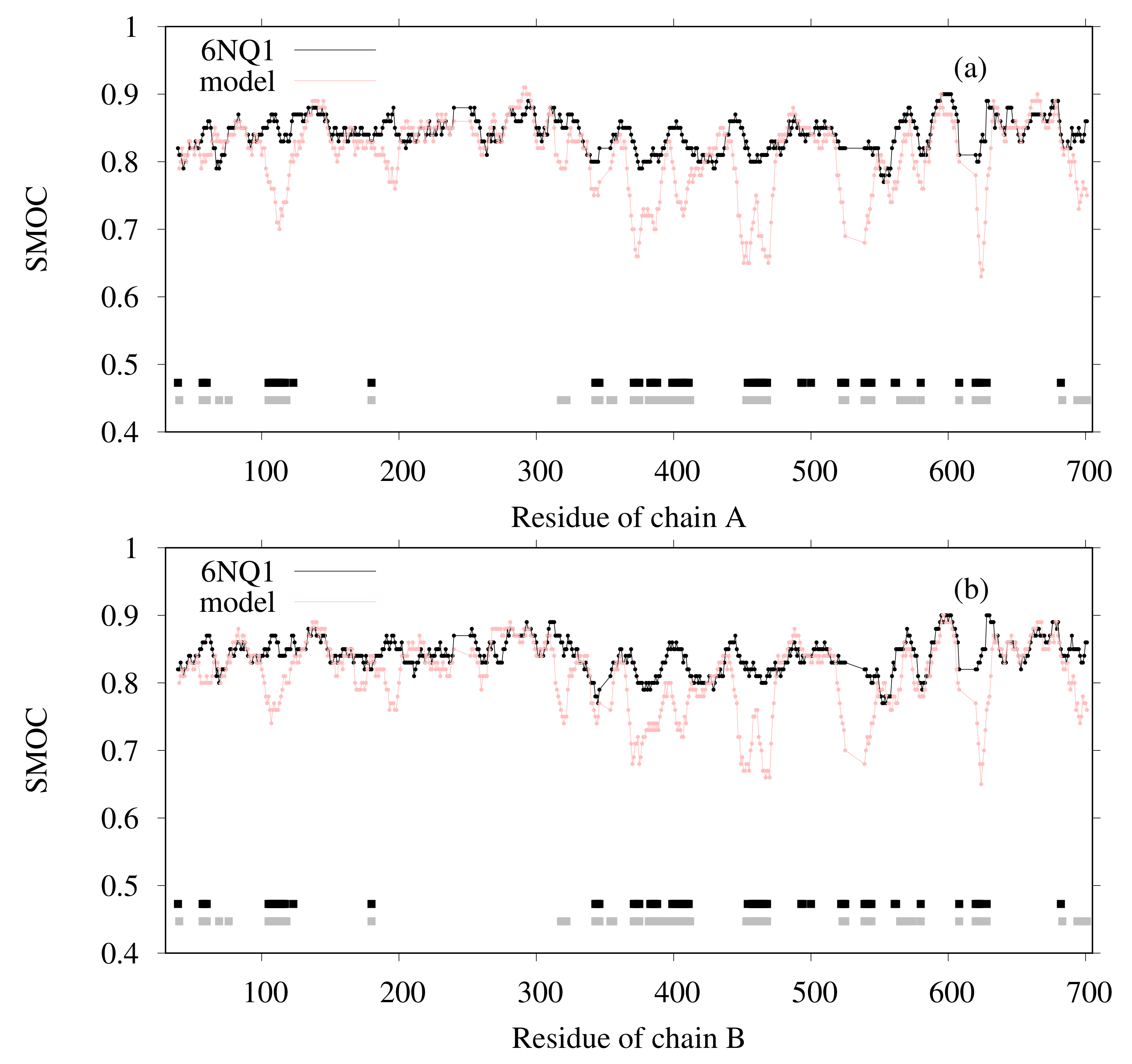
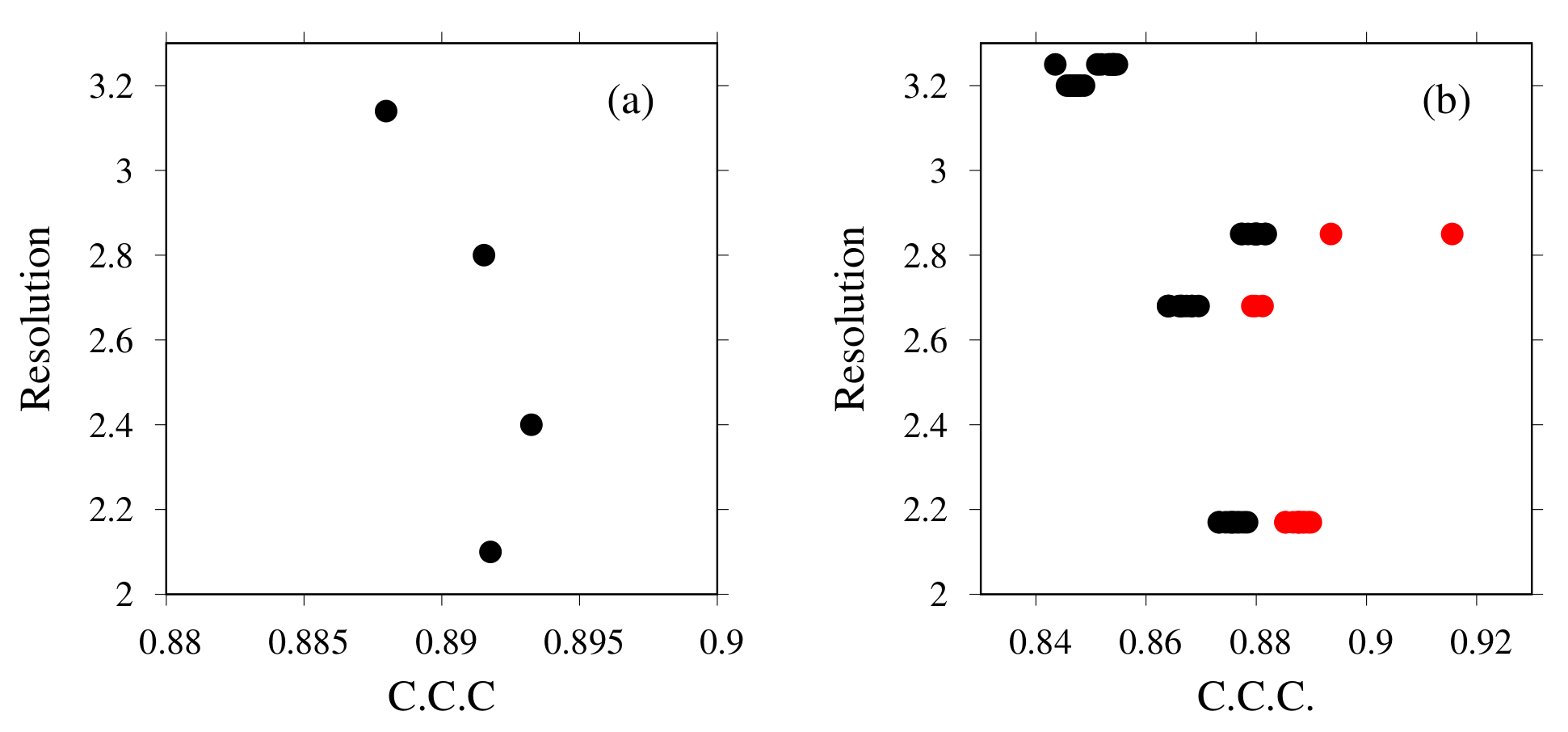
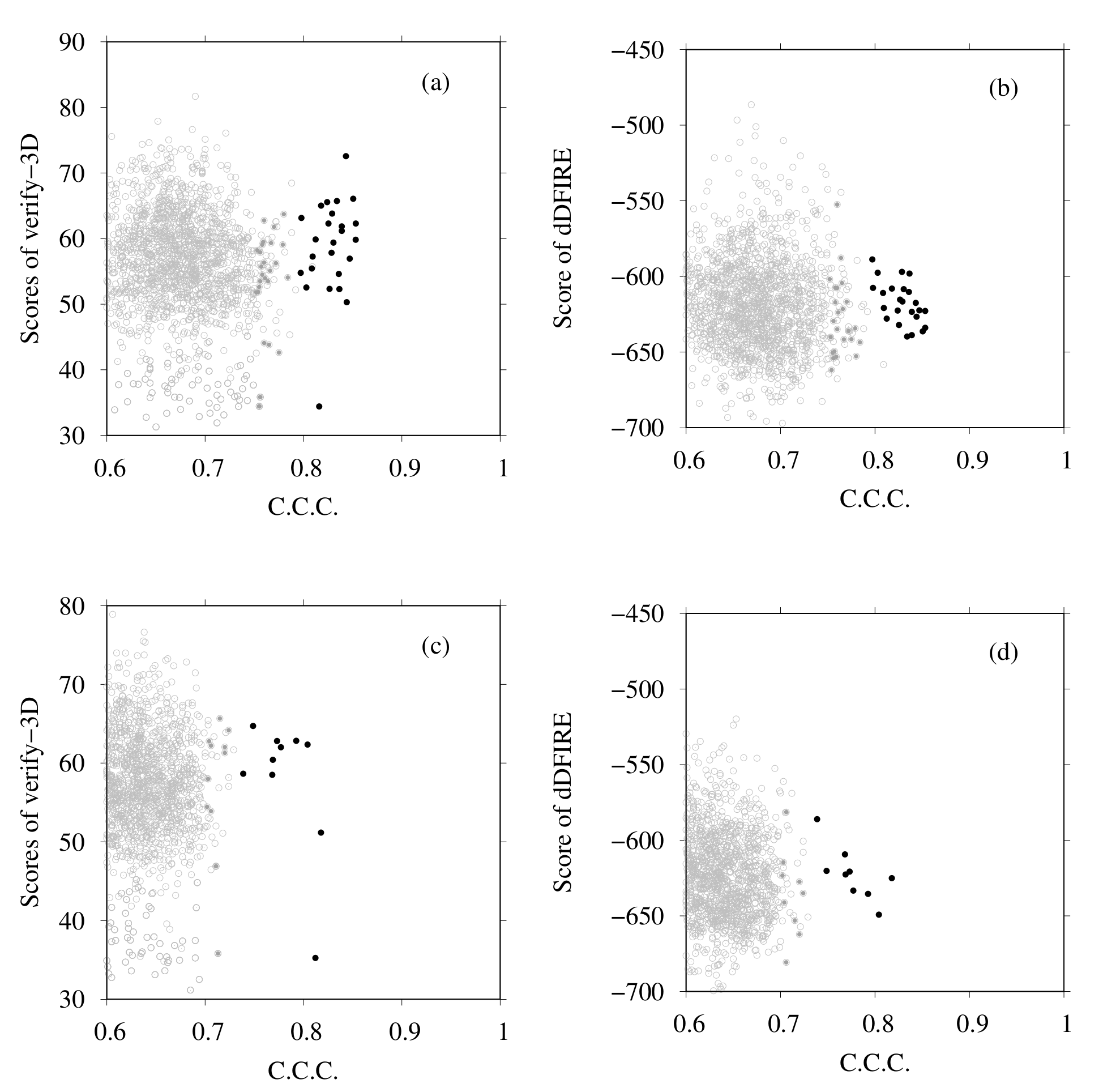
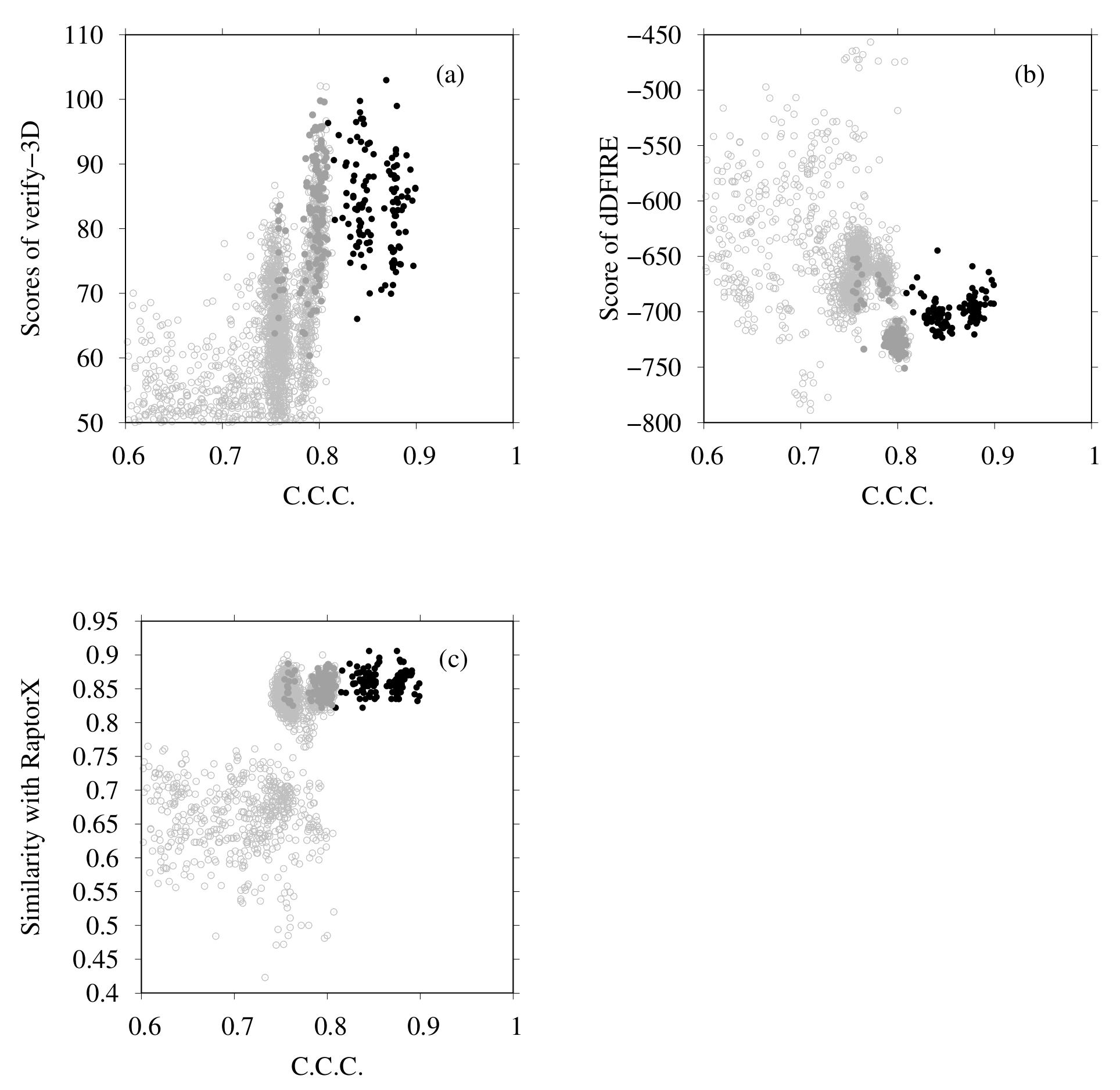
3.1. Modeling of human TPC2 (T0984o in CASP13)
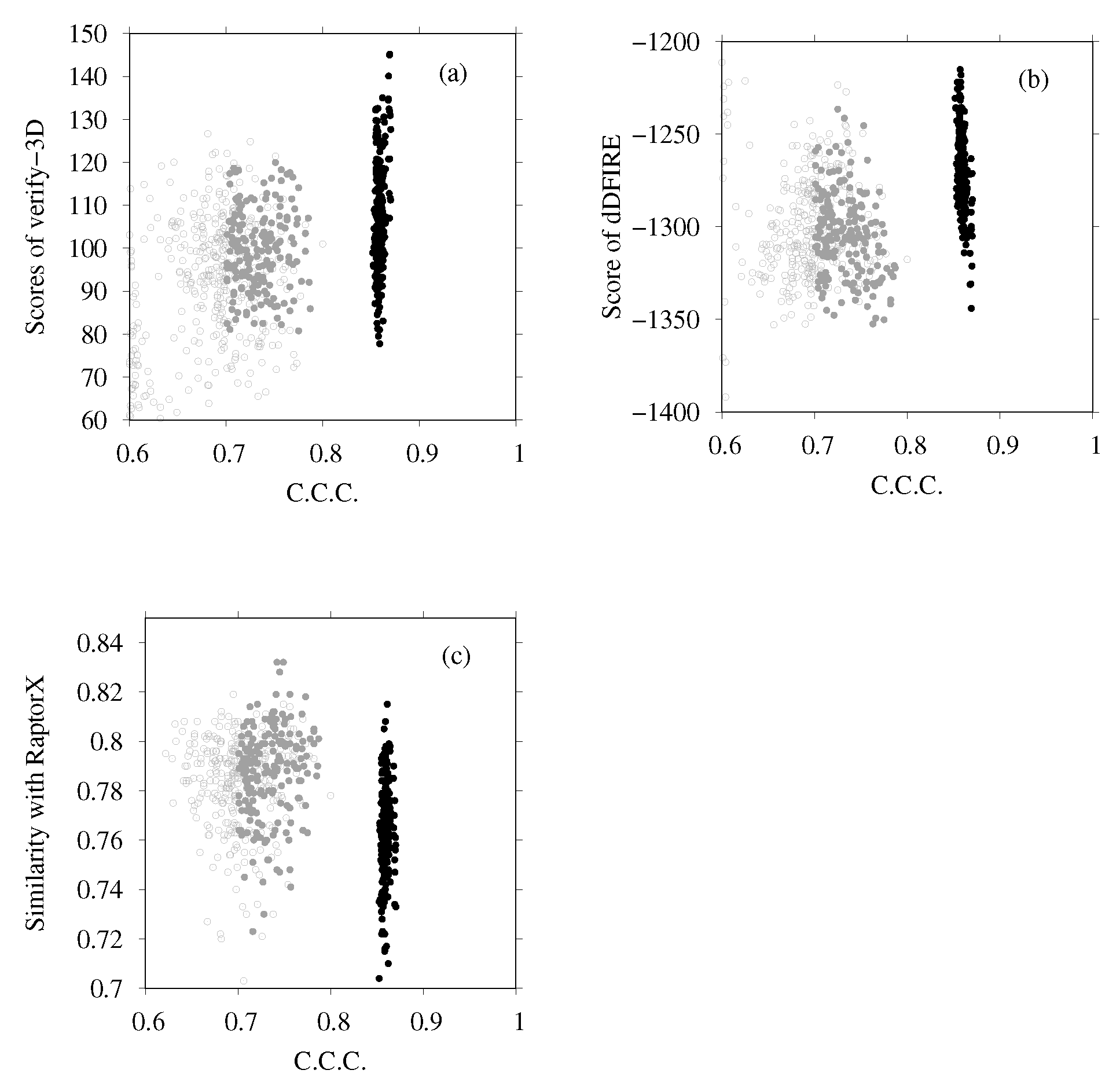
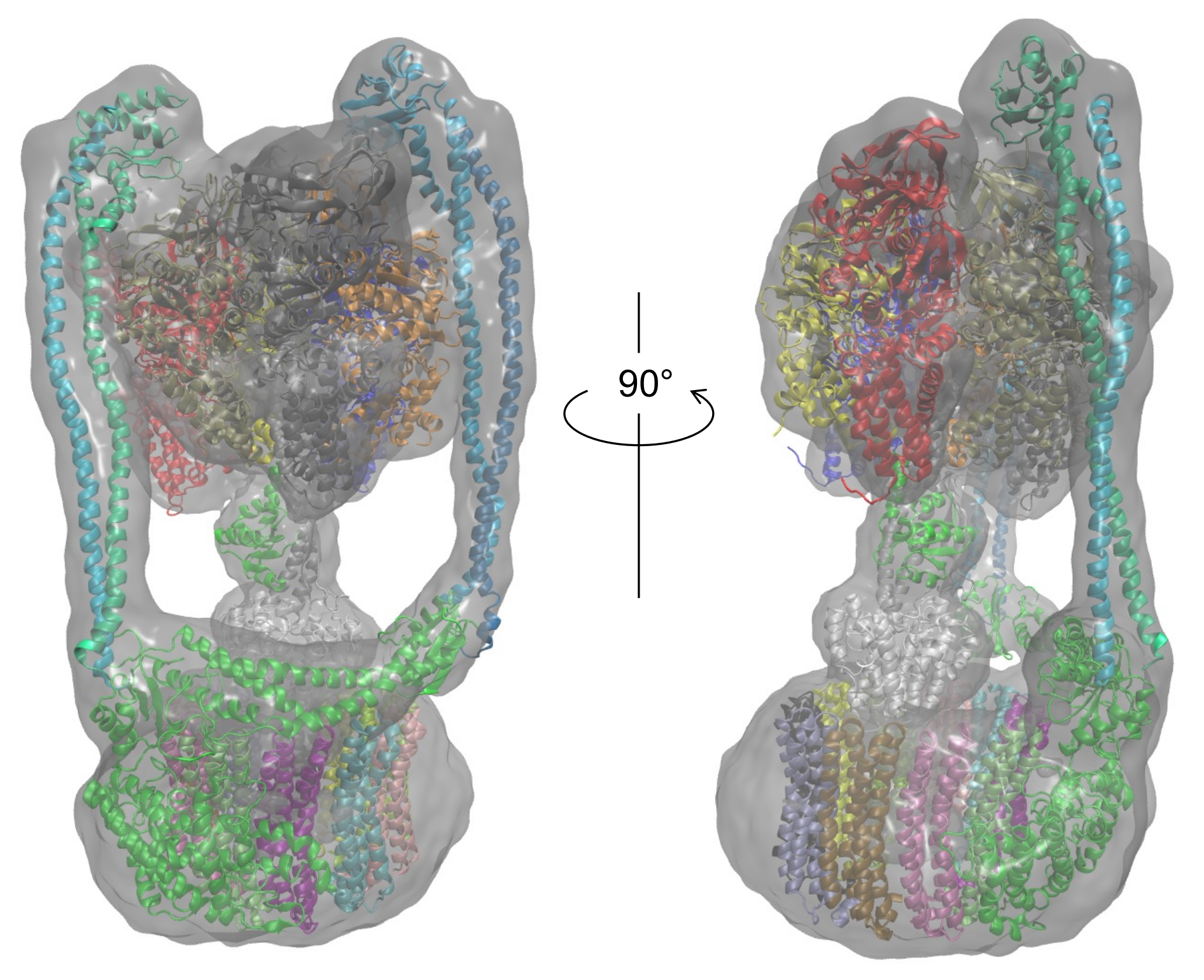
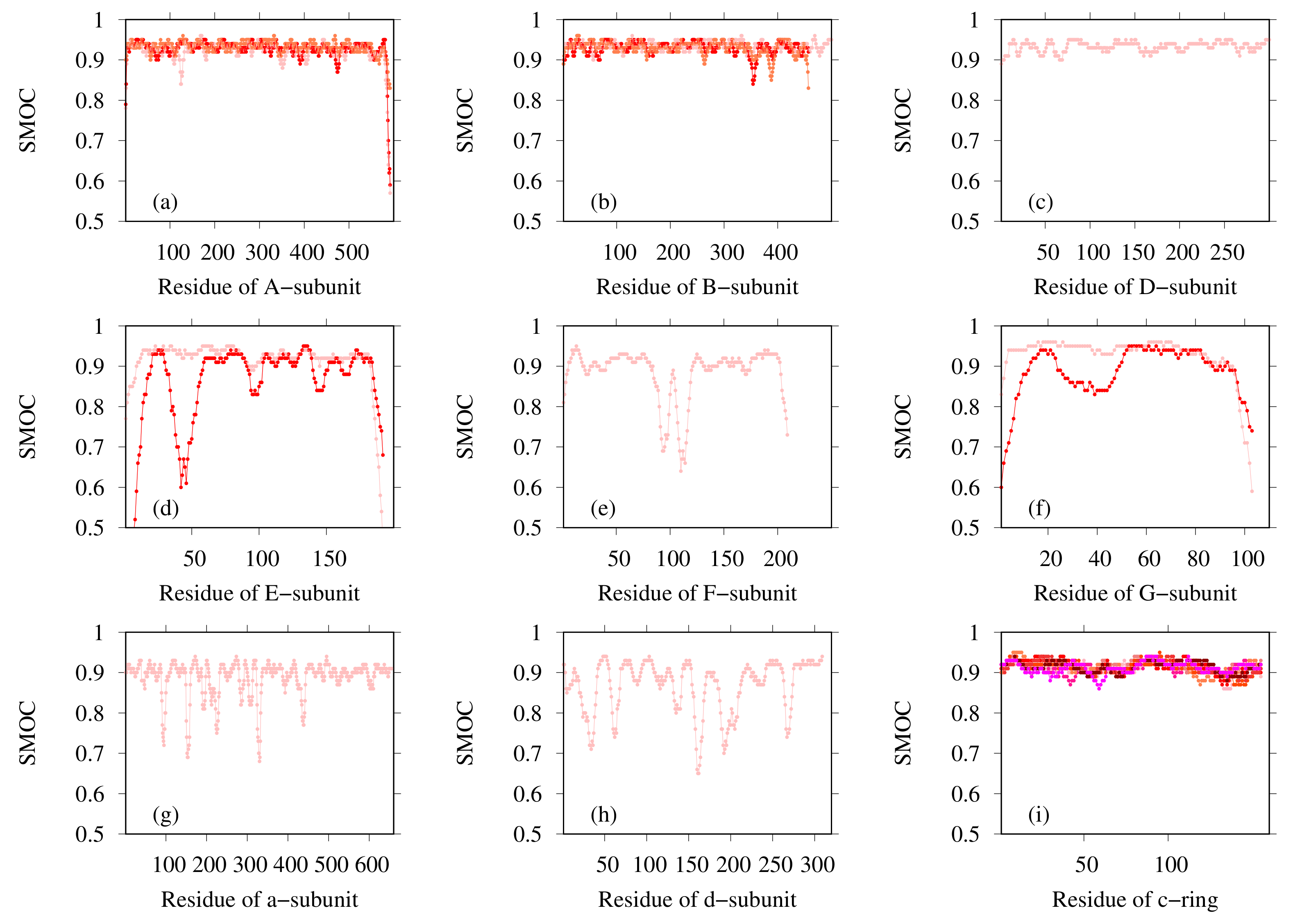
3.2. Modeling of Eh V-ATPase
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kühlbrandt, W. Cryo-EM enters a new era. eLife 2014, 3, e03678. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; Fernandez, I.S.; McMullan, G.; Scheres, S.H.W. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. eLife 2013, 2, e00461. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; McMullan, G.; Scheres, S.H.W. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef]
- Takeda, H.; Tsutsumi, A.; Nishizawa, T.; Lindau, C.; Busto, J.V.; Wenz, L.S.; Ellenrieder, L.; Imai, K.; Straub, S.P.; Mossmann, W.; et al. Mitochondrial sorting and assembly machinery operates by β-barrel switching. Nature 2021, 590, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Fisette, O.; Schröder, G.F.; Schäfer, L.V. Atomistic structure and dynamics of the human MHC-I peptide-loading complex. Proc. Natl. Acad. Sci. USA 2020, 117, 20597–20606. [Google Scholar] [CrossRef]
- Maity, K.; Heumann, J.M.; McGrath, A.P.; Kopcho, N.J.; Hsu, P.K.; Lee, C.W.; Mapes, J.H.; Garza, D.; Krishnan, S.; Morgan, G.P.; et al. Cryo-EM structure of OSCA1.2 from Oryza sativa elucidates the mechanical basis of potential membrane hyperosmolality gating. Proc. Natl. Acad. Sci. USA 2019, 116, 14309–14318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gui, M.; Wang, Z.-F.; Gorgulla, C.; Yu, J.J.; Wu, H.; Sun, Z.-Y.J.; Klenk, C.; Merklinger, L.; Morstein, L.; et al. Cryo-EM structure of an activated GPCR–G protein complex in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Malhotra, S.; Monastyrskyy, B.; Cragnolini, T.; Joseph, A.P.; Chiu, W.; Topf, M. Cryo-electron microscopy targets in CASP13: Overview and evaluation of results. Proteins Struct. Funct. Bioinf. 2019, 87, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.R.; Rees, I.; Ludtke, S.J.; Chiu, W.; Baker, M.L. Constructing and validating initial Cα models from subnanometer resolution density maps with pathwalking. Structure 2012, 20, 450–463. [Google Scholar] [CrossRef]
- Chen, M.; Baldwin, P.R.; Ludtke, S.J.; Baker, M.L. De Novo modeling in cryo-EM density maps with Pathwalking. J. Struct. Biol. 2016, 196, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Dimaio, F.; Song, Y.; Li, X.; Brunner, M.J.; Xu, C.; Conticello, V.; Egelman, E.; Marlovits, T.C.; Cheng, Y.; Baker, D. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 2015, 12, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.R.; Kudryashev, M.; Li, X.; Egelman, E.H.; Basler, M.; Cheng, Y.; Baker, D.; Dimaio, F. De novo protein structure determination from near-atomic-resolution cryo-EM maps. Nat. Methods 2015, 12, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Terashi, G.; Kihara, D. De novo main-chain modeling for em maps using MAINMAST. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Gao, H.; Sengupta, J.; Valle, M.; Korostelev, A.; Eswar, N.; Stagg, S.M.; Van Roey, P.; Agrawal, R.K.; Harvey, S.C.; Sali, A.; et al. Study of the structural dynamics of the E. coli 70S ribosome using real-space refinement. Cell 2003, 113, 789–801. [Google Scholar] [CrossRef]
- Velazquez-Muriel, J.A.; Carazo, J.M. Flexible fitting in 3D-EM with incomplete data on superfamily variability. J. Struct. Biol. 2007, 158, 165–181. [Google Scholar] [CrossRef]
- Gorba, C.; Miyashita, O.; Tama, F. Normal-mode flexible fitting of high-resolution structure of biological molecules toward one-dimensional low-resolution data. Biophys. J. 2008, 94, 1589–1599. [Google Scholar] [CrossRef]
- Zhu, J.; Cheng, L.; Fang, Q.; Zhou, Z.H.; Honig, B. Building and Refining Protein Models within Cryo-electron Microscopy Density Maps Based on Homology Modeling and Multiscale Structure Refinement. J. Mol. Biol. 2010, 397, 835–851. [Google Scholar] [CrossRef] [PubMed]
- Kirmizialtin, S.; Loerke, J.; Behrmann, E.; Spahn, C.M.T.; Sanbonmatsu, K.Y. Using molecular simulation to model high-resolution cryo-EM reconstructions. Methods Enzymol. 2015, 558, 497–514. [Google Scholar] [CrossRef]
- Singharoy, A.; Teo, I.; McGreevy, R.; Stone, J.E.; Zhao, J.; Schulten, K. Molecular dynamics-based refinement and validation for sub-5 Å cryo-electron microscopy maps. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.N.; Moriarty, N.W.; Kirmizialtin, S.; Afonine, P.V.; Poon, B.; Sobolev, O.V.; Adams, P.D.; Sanbonmatsu, K. Cryo-fit: Democratization of flexible fitting for cryo-EM. J. Struct. Biol. 2019, 208, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Poon, B.K.; Read, R.J.; Sobolev, O.V.; Terwilliger, T.C.; Urzhumtsev, A.; Adams, P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 2018, 74, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Trabuco, L.G.; Villa, E.; Mitra, K.; Frank, J.; Schulten, K. Flexible Fitting of Atomic Structures into Electron Microscopy Maps Using Molecular Dynamics. Structure 2008, 16, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Trabuco, L.G.; Villa, E.; Schreiner, E.; Harrison, C.B.; Schulten, K. Molecular dynamics flexible fitting: A practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods 2009, 49, 174–180. [Google Scholar] [CrossRef] [PubMed]
- phenix.cryo_fit2 webpage. Available online: https://www.phenix-online.org/documentation/reference/dock_in_map.html (accessed on 10 January 2022).
- Ratje, A.H.; Loerke, J.; Mikolajka, A.; Brünner, M.; Hildebrand, P.W.; Starosta, A.L.; Dönhöfer, A.; Connell, S.R.; Fucini, P.; Mielke, T.; et al. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature 2010, 468, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins Struct. Funct. Bioinf. 2021, 89, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Anishchenko, I.; Park, H.; Humphreys, I.R.; Baker, D. Protein oligomer modeling guided by predicted interchain contacts in CASP14. Proteins Struct. Funct. Bioinf. 2021, 89, 1824–1833. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Brysbaert, G.; Mauri, T.; Nadzirin, N.; Velankar, S.; Chaleil, R.A.G.; Clarence, T.; Bates, P.A.; Kong, R.; Liu, B.; et al. Prediction of protein assemblies, the next frontier: The CASP14-CAPRI experiment. Proteins Struct. Funct. Bioinf. 2021, 89, 1800–1823. [Google Scholar] [CrossRef]
- Dapkūnas, J.; Olechnovič, K.; Česlovas, V. Modeling of protein complexes in CASP14 with emphasis on the interaction interface prediction. Proteins Struct. Funct. Bioinf. 2021, 89, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Woo, H.; Yang, J.; Kwon, S.; Won, J.; Seok, C. Protein oligomer structure prediction using GALAXY in CASP14. Proteins Struct. Funct. Bioinf. 2021, 89, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ghani, U.; Desta, I.; Jindal, A.; Khan, O.; Jones, G.; Kotelnikov, S.; Padhorny, D.; Vajda, S.; Kozakov, D. Improved Docking of Protein Models by a Combination of Alphafold2 and ClusPro. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2 and extended multiple-sequence alignments. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Moult, J.; Albrecht, R.; Chang, G.A.; Chao, K.; Fraser, A.; Greenfield, J.; Hartmann, M.D.; Herzberg, O.; Josts, I.; et al. Computational models in the service of X-ray and cryo-electron microscopy structure determination. Proteins Struct. Funct. Bioinf. 2021, 89, 1633–1646. [Google Scholar] [CrossRef]
- Mosalaganti, S.; Obarska-Kosinska, A.; Siggel, M.; Turonova, B.; Zimmerli, C.E.; Buczak, K.; Schmidt, F.H.; Margiotta, E.; Mackmull, M.T.; Hagen, W.; et al. Artificial intelligence reveals nuclear pore complexity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Nakamura, T.; Oda, T.; Fukasawa, Y.; Tomii, K. Template-based quaternary structure prediction of proteins using enhanced profile–profile alignments. Proteins Struct. Funct. Bioinf. 2018, 86, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Tomii, K.; Akiyama, Y. FORTE: A profile-profile comparison tool for protein fold recognition. Bioinformatics 2004, 20, 594–595. [Google Scholar] [CrossRef][Green Version]
- Tomii, K.; Hirokawa, T.; Motono, C. Protein structure prediction using a variety of profile libraries and 3D verification. Proteins Struct. Funct. Bioinf. 2005, 61, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Velankar, S.; Kryshtafovych, A.; Huang, S.Y.; Schneidman-Duhovny, D.; Sali, A.; Segura, J.; Fernandez-Fuentes, N.; Viswanath, S.; Elber, R.; et al. Prediction of homoprotein and heteroprotein complexes by protein docking and template-based modeling: A CASP-CAPRI experiment. Proteins Struct. Funct. Bioinf. 2016, 84, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Velankar, S.; Baek, M.; Heo, L.; Seok, C.; Wodak, S.J. The challenge of modeling protein assemblies: The CASP12-CAPRI experiment. Proteins Struct. Funct. Bioinf. 2018, 86, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Shiota, T.; Imai, K.; Qiu, J.; Hewitt, V.L.; Tan, K.; Shen, H.H.; Sakiyama, N.; Fukasawa, Y.; Hayat, S.; Kamiya, M.; et al. Molecular architecture of the active mitochondrial protein gate. Science 2015, 349, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Moult, J.; Billings, W.M.; Corte, D.D.; Fidelis, K.; Kwon, S.; Olechnovič, K.; Seok, C.; Česlovas, V.; Won, J.; et al. Modeling SARS-CoV-2 proteins in the CASP-commons experiment. Proteins Struct. Funct. Bioinf. 2021, 89, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- She, J.; Zeng, W.; Guo, J.; Chen, Q.; Bai, X.C.; Jiang, Y. Structural mechanisms of phospholipid activation of the human TPC2 channel. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, J.; Song, C.; Imai, F.L.; Takagi, J.; Ueno, H.; Murata, T.; Iino, R.; Murata, K. Off-axis rotor in Enterococcus hirae V-ATPase visualized by Zernike phase plate single-particle cryo-electron microscopy. Sci. Rep. 2018, 8, 15632. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Pearson, W. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol. Biol. 2000, 132, 185–219. [Google Scholar] [CrossRef]
- Yamada, K.; Tomii, K. Revisiting amino acid substitution matrices for identifying distantly related proteins. Bioinformatics 2014, 30, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Lim, K.; Tomii, K. Simple adjustment of the sequence weight algorithm remarkably enhances PSI-BLAST performance. BMC Bioinform. 2017, 18, 288. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; von den Driesch, L.; Galiez, C.; Martin, M.J.; Söding, J.; Steinegger, M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2016, 45, D170–D176. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Y. Ab initio folding of terminal segments with secondary structures reveals the fine difference between two closely related all-atom statistical energy functions. Protein Sci. 2008, 17, 1212–1219. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Y. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins Struct. Funct. Bioinf. 2008, 72, 793–803. [Google Scholar] [CrossRef]
- Wang, S.; Li, W.; Liu, S.; Xu, J. RaptorX-Property: A web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef] [PubMed]
- Wriggers, W. Conventions and workflows for using Situs. Acta Crystallogr. D 2012, 68, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Klaholz, B.P.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D.; Urzhumtsev, A. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. D 2018, 74, 814–840. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Micros. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Farabella, I.; Vasishtan, D.; Joseph, A.P.; Pandurangan, A.P.; Sahota, H.; Topf, M. TEMPy: A Python library for assessment of three-dimensional electron microscopy density fits. J. Appl. Crystallogr. 2015, 48, 1314–1323. [Google Scholar] [CrossRef]
- She, J.; Guo, J.; Chen, Q.; Zeng, W.; Jiang, Y.; Bai, X.C. Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 2018, 556, 130–134. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinf. 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Murata, T.; Yamato, I.; Kakinuma, Y.; Leslie, A.G.W.; Walker, J.E. Structure of the rotor of the V-type Na+-ATPase from Enterococcus hirae. Science 2005, 308, 654–659. [Google Scholar] [CrossRef]
- Saijo, S.; Arai, S.; Hossain, K.M.M.; Yamato, I.; Suzuki, K.; Kakinuma, Y.; Ishizuka-Katsura, Y.; Ohsawa, N.; Terada, T.; Shirouzu, M.; et al. Crystal structure of the central axis DF complex of the prokaryotic V-ATPase. Proc. Natl. Acad. Sci. USA 2011, 108, 19955–19960. [Google Scholar] [CrossRef]
- Arai, S.; Saijo, S.; Suzuki, K.; Mizutani, K.; Kakinuma, Y.; Ishizuka-Katsura, Y.; Ohsawa, N.; Terada, T.; Shirouzu, M.; Yokoyama, S.; et al. Rotation mechanism of Enterococcus hirae v 1-ATPase based on asymmetric crystal structures. Nature 2013, 493, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kimura, T.; Shinoda, H.; Bai, G.; Daniels, M.J.; Arai, Y.; Nakano, M.; Nagai, T. Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Pintilie, G.D.; Zhang, J.; Goddard, T.D.; Chiu, W.; Gossard, D.C. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 2010, 170, 427–438. [Google Scholar] [CrossRef]
- Nakanishi, A.; Kishikawa, J.; Tamakoshi, M.; Mitsuoka, K.; Yokoyama, K. Cryo EM structure of intact rotary H+-ATPase/synthase from Thermus thermophilus. Nat. Commun. 2018, 9, 89. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Mukherjee, S.; Zhang, Y. MM-align: A quick algorithm for aligning multiple-chain protein complex structures using iterative dynamic programming. Nucleic Acids Res. 2009, 37, e83. [Google Scholar] [CrossRef]
- Mizutani, K.; Yamamoto, M.; Suzuki, K.; Yamato, I.; Kakinuma, Y.; Shirouzu, M.; Walker, J.E.; Yokoyama, S.; Iwata, S.; Murata, T. Structure of the rotor ring modified with N,N’-dicyclohexylcarbodiimide of the Na+-transporting vacuolar ATPase. Proc. Natl. Acad. Sci. USA 2011, 108, 13474–13479. [Google Scholar] [CrossRef]
- Schep, D.G.; Zhao, J.; Rubinstein, J.L. Models for the a subunits of the Thermus thermophilus V/A-ATPase and Saccharomyces cerevisiae V-ATPase enzymes by cryo-EM and evolutionary covariance. Proc. Natl. Acad. Sci. USA 2016, 113, 3245–3250. [Google Scholar] [CrossRef]
- Balakrishna, A.M.; Hunke, C.; Grüber, G. The Structure of Subunit E of the Pyrococcus horikoshii OT3 A-ATP Synthase Gives Insight into the Elasticity of the Peripheral Stalk. J. Mol. Biol. 2012, 420, 155–163. [Google Scholar] [CrossRef]
- Macheboeuf, P.; Buffalo, C.; Fu, C.Y.; Zinkernagel, A.S.; Cole, J.N.; Johnson, J.E.; Ghosh, V.N.P. Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature 2011, 472, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sazanov, L.A. Structure and conformational plasticity of the intact Thermus thermophilus V/A-type ATPase. Science 2019, 365, eaaw9144. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. Multi-State Modeling of G-protein Coupled Receptors at Experimental Accuracy. bioRxiv 2021. [Google Scholar] [CrossRef]
- del Alamo, D.; Sala, D.; Mchaourab, H.S.; Meiler, J. Sampling the conformational landscapes of transporters and receptors with AlphaFold2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
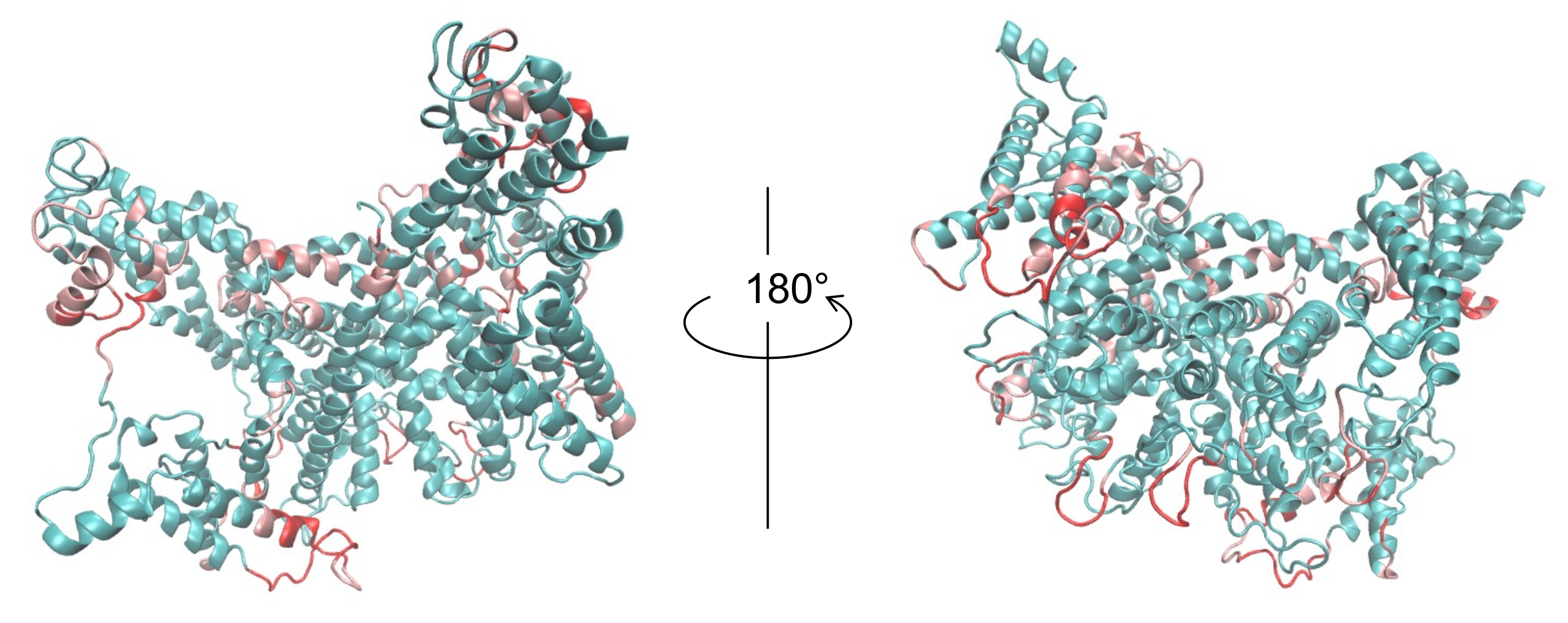{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | |
|---|---|
| (1) | SSEARCH(nr,MIQS) + MAFFT + PSI-BLASTexB(nr,3) |
| (2) | DELTA-BLAST(CDD) |
| (3) | HHblits(Uniclust) + PSI-BLASTexB(nr,1) |
| 6NQ1 | Our Model | |
|---|---|---|
| CC | 0.823 | 0.755 |
| CC | 0.794 | 0.713 |
| CC | 0.516 | 0.468 |
| CC | 0.585 | 0.557 |
| CCC | 0.593 | 0.566 |
| Correlation (fit_in_map) | 0.858 | 0.829 |
| SMOC (avg.) | 0.850 | 0.811 |
| TM-score | 1.0 | 0.930 |
| Subunit | Template (PDB ID(s)) | Method (Resolution) |
|---|---|---|
| a-subunit | 5Y5X | EM (5.0 Å) |
| c-ring | 2BL2 | X-ray (2.1 Å) |
| d-subunit | 1R5Z | X-ray (2.05 Å) |
| E-subunit (Stalk A) | 4DT0 | X-ray (3.65 Å) |
| G-subunit (Stalk A) | 3DHR | X-ray (2.0 Å) |
| E-subunit (Stalk B) | 4DT0 | X-ray (3.65 Å) |
| G-subunit (Stalk B) | 2XNX | X-ray (3.3 Å) |
| A3B3-complex | 5KND | X-ray (2.89 Å) |
| DF-complex | 5KND/3AON | X-ray (2.89 Å/2.0 Å) |
| Our Model | |
|---|---|
| CC | 0.652 |
| CC | 0.534 |
| CC | 0.352 |
| CC | 0.892 |
| CCC | 0.386 |
| Correlation (fit_in_map) | 0.901 |
| SMOC (avg.) | 0.908 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamori, Y.; Tomii, K. Application of Homology Modeling by Enhanced Profile–Profile Alignment and Flexible-Fitting Simulation to Cryo-EM Based Structure Determination. Int. J. Mol. Sci. 2022, 23, 1977. https://doi.org/10.3390/ijms23041977
Yamamori Y, Tomii K. Application of Homology Modeling by Enhanced Profile–Profile Alignment and Flexible-Fitting Simulation to Cryo-EM Based Structure Determination. International Journal of Molecular Sciences. 2022; 23(4):1977. https://doi.org/10.3390/ijms23041977
Chicago/Turabian StyleYamamori, Yu, and Kentaro Tomii. 2022. "Application of Homology Modeling by Enhanced Profile–Profile Alignment and Flexible-Fitting Simulation to Cryo-EM Based Structure Determination" International Journal of Molecular Sciences 23, no. 4: 1977. https://doi.org/10.3390/ijms23041977
APA StyleYamamori, Y., & Tomii, K. (2022). Application of Homology Modeling by Enhanced Profile–Profile Alignment and Flexible-Fitting Simulation to Cryo-EM Based Structure Determination. International Journal of Molecular Sciences, 23(4), 1977. https://doi.org/10.3390/ijms23041977