
Transcriptional Control of Trpm6 by the Nuclear Receptor FXR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
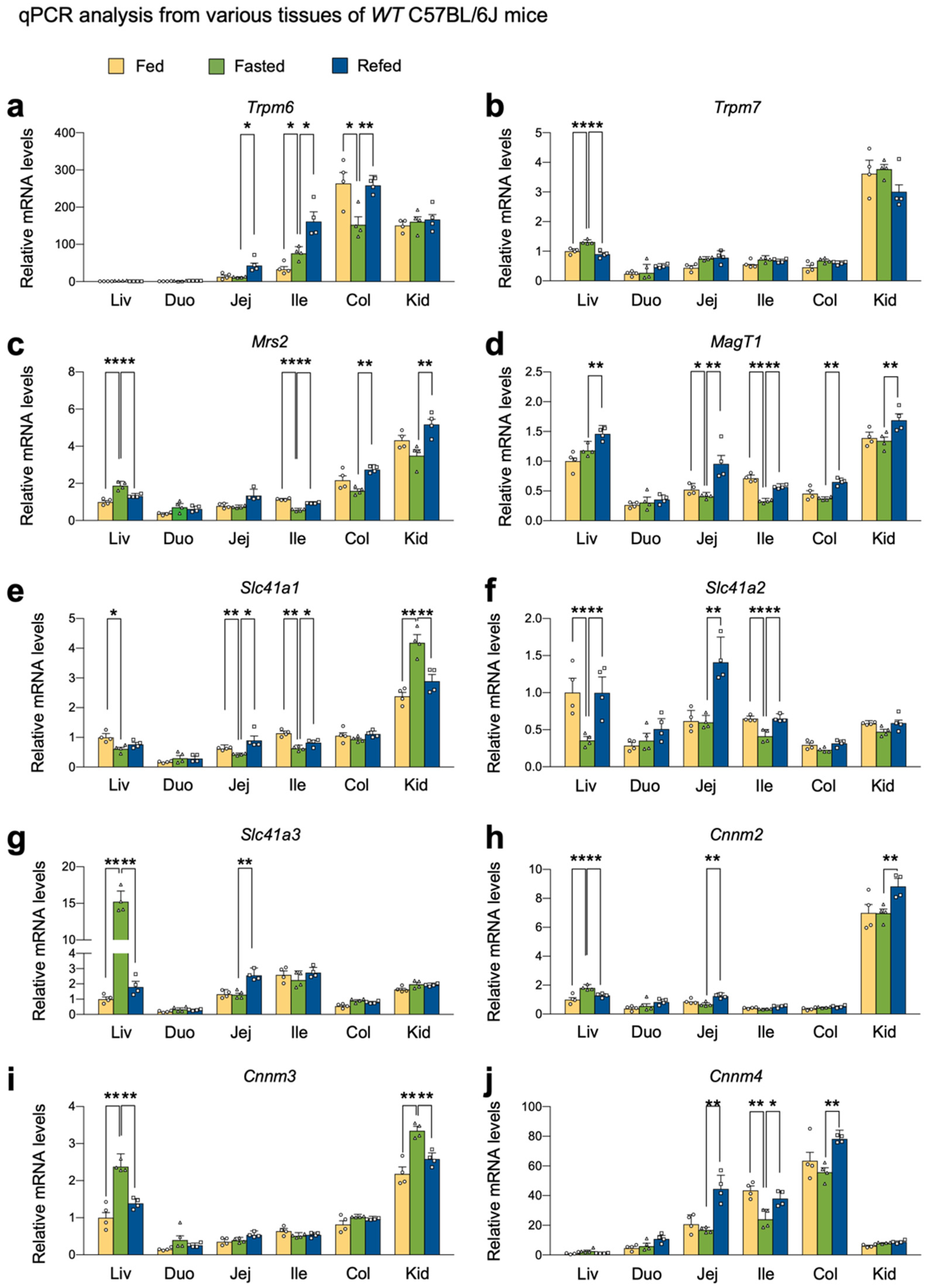
2.1. Nutrient Availability Regulates the Expression of Genes encoding Mg2+ Channels, Exchangers, and Transporters
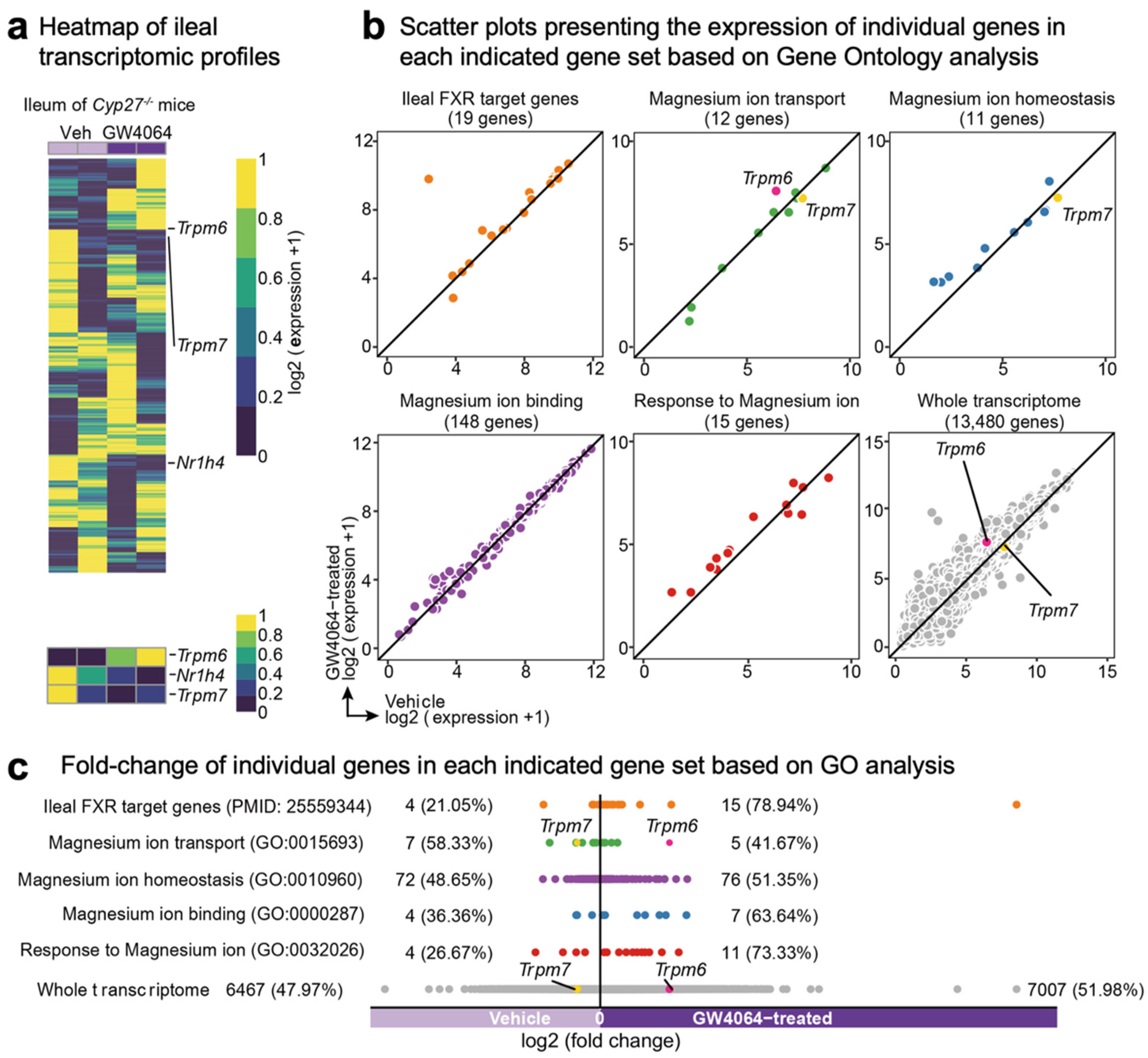
2.2. FXR Agonism Induces Ileal Trpm6 Expression in Cyp27-/- Mice
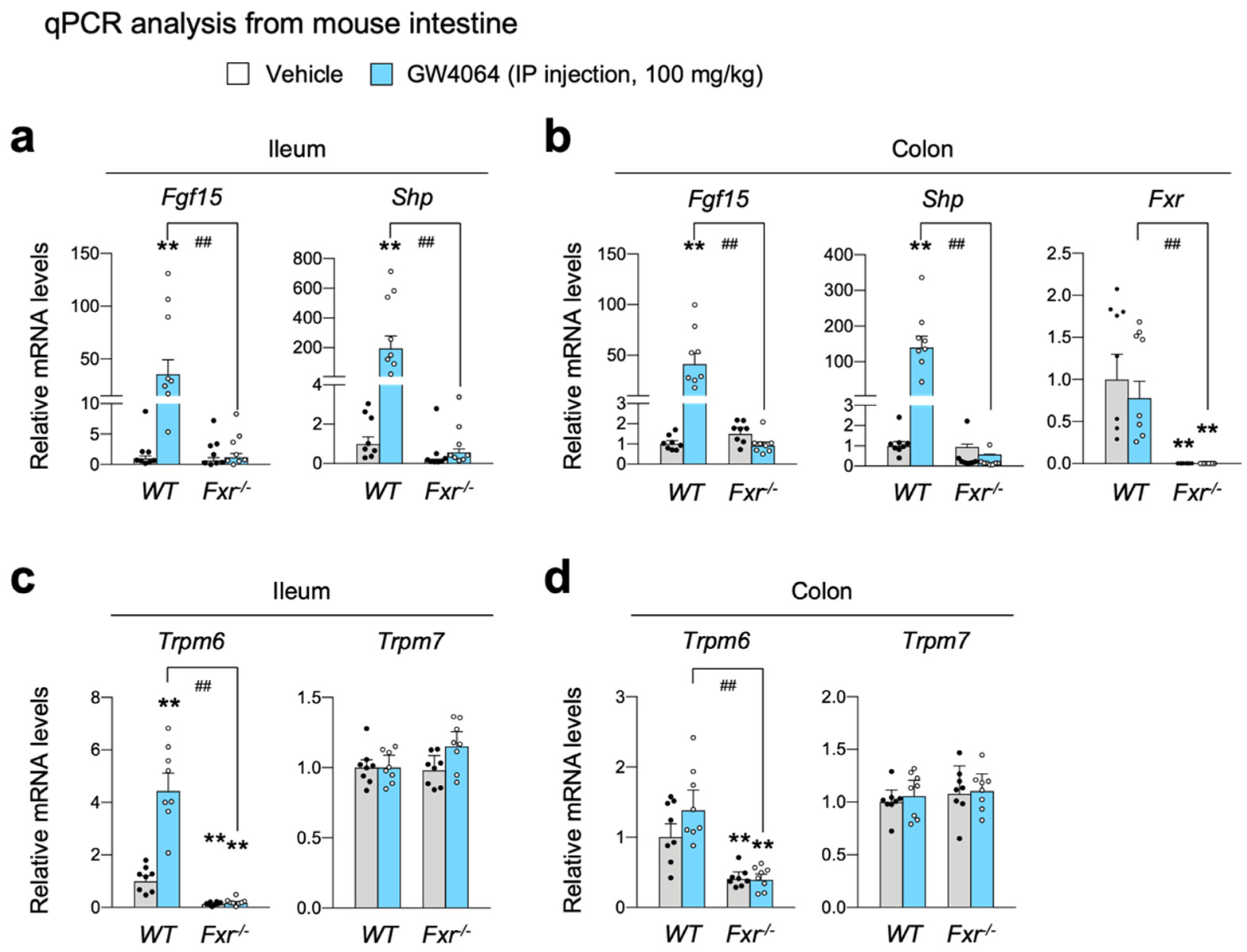
2.3. FXR Is Necessary for Increasing Intestinal Expression of Trpm6 in Response to GW4064
2.4. Cell-Autonomous Activation of FXR Is Required for GW4064-Mediated Induction of Trpm6 in Ileal Epithelial Cells
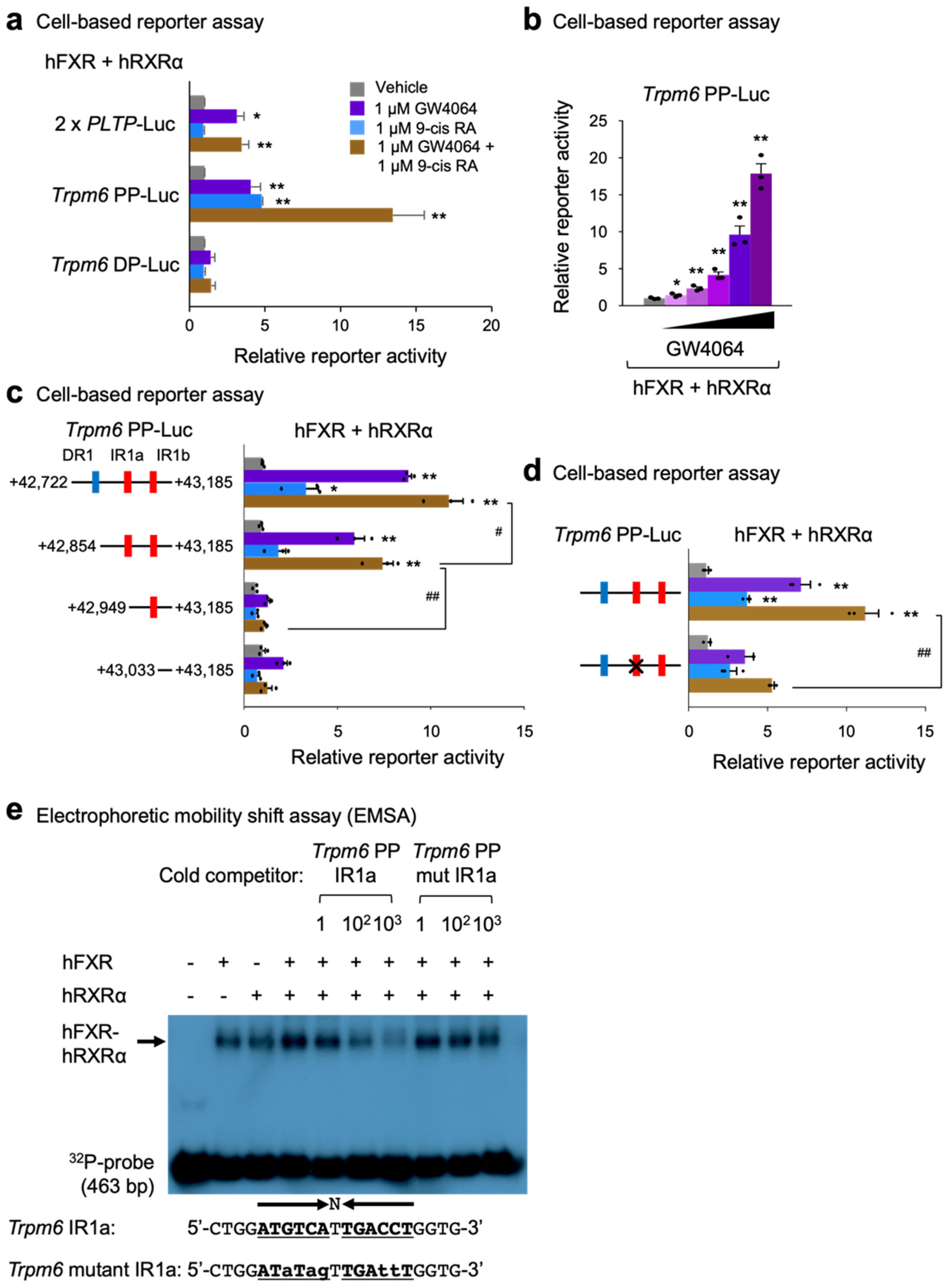
2.5. Identification of Trpm6 as a Direct FXR Target Gene in Mouse Intestine
2.6. Identification of an IR-1 Response Element for FXR Transactivation in Mouse Trpm6 Gene
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animal Experiments
4.3. Transcriptomic Analysis and Visualization
4.4. RNA Purification, cDNA Synthesis, and qPCR Analysis
4.5. Molecular Cloning
4.6. Cell-Based Reporter Assays
4.7. Electrophoretic Mobility Shift Assays
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seol, W.; Choi, H.S.; Moore, D.D. Isolation of proteins that interact specifically with the retinoid X receptor: Two novel orphan receptors. Mol. Endocrinol. 1995, 9, 72–85. [Google Scholar] [PubMed]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Zavacki, A.M.; Lehmann, J.M.; Seol, W.; Willson, T.M.; Kliewer, S.A.; Moore, D.D. Activation of the orphan receptor RIP14 by retinoids. Proc. Natl. Acad. Sci. USA 1997, 94, 7909–7914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous Bile Acids Are Ligands for the Nuclear Receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Oresic, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-beta-muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted Disruption of the Nuclear Receptor FXR/BAR Impairs Bile Acid and Lipid Homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.T.; Makishima, M.; Repa, J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Castellani, L.W.; Sinal, C.J.; Gonzalez, F.J.; Edwards, P.A. Peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1alpha) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes Dev. 2004, 18, 157–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.; Amar, M.J.A.; Guo, G.; Brewer, H.B.; Gonzalez, F.J.; Sinal, C.J. The Farnesoid X-receptor Is an Essential Regulator of Cholesterol Homeostasis. J. Biol. Chem. 2003, 278, 2563–2570. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear Receptor-Dependent Bile Acid Signaling Is Required for Normal Liver Regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef]
- Ryan, K.; Tremaroli, V.; Clemmensen, C.; Kovatcheva-Datchary, P.; Myronovych, A.; Karns, R.; Wilson-Pérez, H.E.; Sandoval, D.A.; Kohli, R.; Bäckhed, F.; et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 2014, 509, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; van Harmelen, K.; Duran-Sandoval, D.; van Dijk, T.H.; Grefhorst, A.; Abdelkarim, M.; Caron, S.; Torpier, G.; Fruchart, J.-C.; Gonzalez, F.J.; et al. The Farnesoid X Receptor Modulates Adiposity and Peripheral Insulin Sensitivity in Mice. J. Biol. Chem. 2006, 281, 11039–11049. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Xie, C.; Lv, Y.; Li, J.; Krausz, K.W.; Shi, J.; Brocker, C.N.; Desai, D.; Amin, S.G.; Bisson, W.H.; et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 2015, 6, 10166. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous Development of Liver Tumors in the Absence of the Bile Acid Receptor Farnesoid X Receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2006, 28, 940–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modica, S.; Murzilli, S.; Salvatore, L.; Schmidt, D.R.; Moschetta, A. Nuclear Bile Acid Receptor FXR Protects against Intestinal Tumorigenesis. Cancer Res. 2008, 68, 9589–9594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Ospina, N.; Potter, C.J.; Xiao, R.; Manickam, K.; Kim, M.-S.; Kim, K.H.; Shneider, B.L.; Picarsic, J.L.; Jacobson, C.J.P.K.M.T.A.; Zhang, J.; et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat. Commun. 2016, 7, 10713. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Seok, S.; Fu, T.; Choi, S.-E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR–CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Panzitt, K.; Jungwirth, E.; Krones, E.; Lee, J.M.; Pollheimer, M.; Thallinger, G.G.; Kolb-Lenz, D.; Xiao, R.; Thorell, A.; Trauner, M.; et al. FXR-dependent Rubicon induction impairs autophagy in models of human cholestasis. J. Hepatol. 2020, 72, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- Maloney, P.R.; Parks, D.J.; Haffner, C.D.; Fivush, A.M.; Chandra, G.; Plunket, K.D.; Creech, K.L.; Moore, L.B.; Wilson, J.G.; Lewis, M.C.; et al. Identification of a Chemical Tool for the Orphan Nuclear Receptor FXR. J. Med. Chem. 2000, 43, 2971–2974. [Google Scholar] [CrossRef]
- Downes, M.; Verdecia, M.A.; Roecker, A.; Hughes, R.; Hogenesch, J.B.; Kast-Woelbern, H.R.; Bowman, M.E.; Ferrer, J.-L.; Anisfeld, A.M.; Edwards, P.A.; et al. A Chemical, Genetic, and Structural Analysis of the Nuclear Bile Acid Receptor FXR. Mol. Cell 2003, 11, 1079–1092. [Google Scholar] [CrossRef]
- Fang, S.; Suh, J.M.; Reilly, S.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urizar, N.L.; Liverman, A.B.; Dodds, D.T.; Silva, F.V.; Ordentlich, P.; Yan, Y.; Gonzalez, F.J.; Heyman, R.A.; Mangelsdorf, D.J.; Moore, D.D. A Natural Product That Lowers Cholesterol As an Antagonist Ligand for FXR. Science 2002, 296, 1703–1706. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6α-Ethyl-Chenodeoxycholic Acid (6-ECDCA), a Potent and Selective FXR Agonist Endowed with Anticholestatic Activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Rinella, M.E.; Dufour, J.-F.; Anstee, Q.M.; Goodman, Z.; Younossi, Z.; Harrison, S.A.; Loomba, R.; Sanyal, A.J.; Bonacci, M.; Trylesinski, A.; et al. Non-invasive evaluation of response to obeticholic acid in patients with NASH: Results from the REGENERATE study. J. Hepatol. 2021. [Google Scholar] [CrossRef]
- De Baaij, J.H.F.; Hoenderop, J.G.J.; Bindels, R.J. Magnesium in Man: Implications for Health and Disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef]
- Gommers, L.M.M.; Hoenderop, J.G.J.; Bindels, R.J.M.; De Baaij, J.H.F. Hypomagnesemia in Type 2 Diabetes: A Vicious Circle? Diabetes 2016, 65, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Dudley, S.C., Jr. Magnesium, Oxidative Stress, Inflammation, and Cardiovascular Disease. Antioxidants 2020, 9, 907. [Google Scholar] [CrossRef]
- Banach, W.; Nitschke, K.; Krajewska, N.; Mongiałło, W.; Matuszak, O.; Muszyński, J.; Skrypnik, D. The Association between Excess Body Mass and Disturbances in Somatic Mineral Levels. Int. J. Mol. Sci. 2020, 21, 7306. [Google Scholar] [CrossRef]
- Bosman, W.; Hoenderop, J.G.J.; de Baaij, J.H.F. Genetic and drug-induced hypomagnesemia: Different cause, same mechanism. Proc. Nutr. Soc. 2021, 80, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Schlingmann, K.P.; Weber, S.; Peters, M.; Nejsum, L.N.; Vitzthum, H.; Klingel, K.; Kratz, M.; Haddad, E.; Ristoff, E.; Dinour, D.; et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat. Genet. 2002, 31, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Walder, R.Y.; Landau, D.; Meyer, P.; Shalev, H.; Tsolia, M.; Borochowitz, Z.; Boettger, M.B.; Beck, G.E.; Englehardt, R.K.; Carmi, R.; et al. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat. Genet. 2002, 31, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Stuiver, M.; Lainez, S.; Will, C.; Terryn, S.; Günzel, D.; Debaix, H.; Sommer, K.; Kopplin, K.; Thumfart, J.; Kampik, N.B.; et al. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am. J. Hum. Genet. 2011, 88, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chubanov, V.; Schlingmann, K.P.; Wäring, J.; Heinzinger, J.; Kaske, S.; Waldegger, S.; Schnitzler, M.M.Y.; Gudermann, T. Hypomagnesemia with Secondary Hypocalcemia due to a Missense Mutation in the Putative Pore-forming Region of TRPM6. J. Biol. Chem. 2007, 282, 7656–7667. [Google Scholar] [CrossRef] [Green Version]
- Voets, T.; Nilius, B.; Hoefs, S.; van der Kemp, A.W.C.M.; Droogmans, G.; Bindels, R.J.M.; Hoenderop, J.G.J. TRPM6 Forms the Mg2+ Influx Channel Involved in Intestinal and Renal Mg2+ Absorption. J. Biol. Chem. 2004, 279, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Groenestege, W.M.T.; Hoenderop, J.G.; Heuvel, L.V.D.; Knoers, N.; Bindels, R.J. The Epithelial Mg2+ Channel Transient Receptor Potential Melastatin 6 Is Regulated by Dietary Mg2+Content and Estrogens. J. Am. Soc. Nephrol. 2006, 17, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.G.; Rios, F.J.; Montezano, A.C.; Touyz, R.M. TRPM7, Magnesium, and Signaling. Int. J. Mol. Sci. 2019, 20, 1877. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Jiang, J.; Yue, L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yu, H.; Huang, J.; Faouzi, M.; Schmitz, C.; Penner, R.; Fleig, A. The TRPM6 kinase domain determines the Mg.ATP sensitivity of TRPM7/M6 heteromeric ion channels. J. Biol. Chem. 2014, 289, 5217–5227. [Google Scholar] [CrossRef] [Green Version]
- Takashina, Y.; Manabe, A.; Tabuchi, Y.; Ikari, A. Cyanidin Increases the Expression of Mg2+ Transport Carriers Mediated by the Activation of PPARalpha in Colonic Epithelial MCE301 Cells. Nutrients 2019, 11, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, A.V.; Hocher, B.; Verkaart, S.; van Zeeland, F.; Pfab, T.; Slowinski, T.; Chen, Y.-P.; Schlingmann, K.P.; Schaller, A.; Gallati, S.; et al. Loss of insulin-induced activation of TRPM6 magnesium channels results in impaired glucose tolerance during pregnancy. Proc. Natl. Acad. Sci. USA 2012, 109, 11324–11329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thebault, S.; Alexander, R.T.; Groenestege, W.M.T.; Hoenderop, J.G.; Bindels, R.J. EGF Increases TRPM6 Activity and Surface Expression. J. Am. Soc. Nephrol. 2008, 20, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.M.; Hart, S.; Kong, B.; Fang, J.; Zhong, X.-B.; Guo, G.L. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 2009, 51, 1410–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chubanov, V.; Ferioli, S.; Wisnowsky, A.; Simmons, D.G.; Leitzinger, C.; Einer, C.; Jonas, W.; Shymkiv, Y.; Bartsch, H.; Braun, A.; et al. Epithelial magnesium transport by TRPM6 is essential for prenatal development and adult survival. eLife 2016, 5, e20914. [Google Scholar] [CrossRef]
- Zsurka, G.; Gregan, J.; Schweyen, R.J. The Human Mitochondrial Mrs2 Protein Functionally Substitutes for Its Yeast Homologue, A Candidate Magnesium Transporter. Genomics 2001, 72, 158–168. [Google Scholar] [CrossRef]
- Shindo, Y.; Fujii, T.; Komatsu, H.; Citterio, D.; Hotta, K.; Suzuki, K.; Oka, K. Newly Developed Mg2+–Selective Fluorescent Probe Enables Visualization of Mg2+ Dynamics in Mitochondria. PLoS ONE 2011, 6, e23684. [Google Scholar] [CrossRef] [Green Version]
- Chaigne-Delalande, B.; Li, F.-Y.; O’Connor, G.M.; Lukacs, M.J.; Jiang, P.; Zheng, L.; Shatzer, A.; Biancalana, M.; Pittaluga, S.; Matthews, H.F.; et al. Mg2+ Regulates Cytotoxic Functions of NK and CD8 T Cells in Chronic EBV Infection Through NKG2D. Science 2013, 341, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Li, F.-Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef]
- Kolisek, M.; Launay, P.; Beck, A.; Sponder, G.; Serafini, N.; Brenkus, M.; Froschauer, E.M.; Martens, H.; Fleig, A.; Schweigel, M. SLC41A1 Is a Novel Mammalian Mg2+ Carrier. J. Biol. Chem. 2008, 283, 16235–16247. [Google Scholar] [CrossRef] [Green Version]
- Kolisek, M.; Nestler, A.; Vormann, J.; Schweigel-Röntgen, M. Human gene SLC41A1 encodes for the Na+/Mg2+ exchanger. Am. J. Physiol.-Cell Physiol. 2012, 302, C318–C326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahni, J.; Nelson, B.; Scharenberg, A.M. SLC41A2 encodes a plasma-membrane Mg2+ transporter. Biochem. J. 2006, 401, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Baaij, J.H.; Stuiver, M.; Meij, I.C.; Lainez, S.; Kopplin, K.; Venselaar, H.; Müller, D.; Bindels, R.J.M.; Hoenderop, J.G.J. Membrane topology and intracellular processing of cyclin M2 (CNNM2). J. Biol. Chem. 2012, 287, 13644–13655. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, H.; Reshef, A.; Maeda, N.; Lippoldt, A.; Shpizen, S.; Triger, L.; Eggertsen, G.; Björkhem, I.; Leitersdorf, E. Markedly Reduced Bile Acid Synthesis but Maintained Levels of Cholesterol and Vitamin D Metabolites in Mice with Disrupted Sterol 27-Hydroxylase Gene. J. Biol. Chem. 1998, 273, 14805–14812. [Google Scholar] [CrossRef] [Green Version]
- Bijsmans, I.T.; Milona, A.; Ijssennagger, N.; Willemsen, E.C.; Pittol, J.M.R.; Jonker, J.W.; Lange, K.; Hooiveld, G.J.; van Mil, S.W. Characterization of stem cell-derived liver and intestinal organoids as a model system to study nuclear receptor biology. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 687–700. [Google Scholar] [CrossRef]
- Sandelin, A.; Wasserman, W. Prediction of Nuclear Hormone Receptor Response Elements. Mol. Endocrinol. 2005, 19, 595–606. [Google Scholar] [CrossRef]
- Claudel, T.; Inoue, Y.; Barbier, O.; Duran-Sandoval, D.; Kosykh, V.; Fruchart, J.; Fruchart, J.-C.; Gonzalez, F.J.; Staels, B. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology 2003, 125, 544–555. [Google Scholar] [CrossRef]
- Chennamsetty, I.; Claudel, T.; Kostner, K.M.; Baghdasaryan, A.; Kratky, D.; Levak-Frank, S.; Frank, S.; Gonzalez, F.J.; Trauner, M.; Kostner, G.M. Farnesoid X receptor represses hepatic human APOA gene expression. J. Clin. Investig. 2011, 121, 3724–3734. [Google Scholar] [CrossRef] [Green Version]
- Urizar, N.L.; Dowhan, D.H.; Moore, D.D. The Farnesoid X-activated Receptor Mediates Bile Acid Activation of Phospholipid Transfer Protein Gene Expression. J. Biol. Chem. 2000, 275, 39313–39317. [Google Scholar] [CrossRef] [Green Version]
- Preidis, G.A.; Kim, K.H.; Moore, D.D. Nutrient-sensing nuclear receptors PPARalpha and FXR control liver energy balance. J. Clin. Investig. 2017, 127, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Moore, D.D. Regulation of Liver Energy Balance by the Nuclear Receptors Farnesoid X Receptor and Peroxisome Proliferator Activated Receptor α. Dig. Dis. 2017, 35, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M. Transcriptional coordination of hepatic autophagy by nutrient-sensing nuclear receptor PPARalpha and FXR. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walder, R.Y.; Yang, B.; Stokes, J.B.; Kirby, P.A.; Cao, X.; Shi, P.; Searby, C.C.; Husted, R.F.; Sheffield, V.C. Mice defective in Trpm6 show embryonic mortality and neural tube defects. Hum. Mol. Genet. 2009, 18, 4367–4375. [Google Scholar] [CrossRef] [Green Version]
- Woudenberg-Vrenken, T.E.; Sukinta, A.; Van Der Kemp, A.W.; Bindels, R.J.; Hoenderop, J.G. Transient Receptor Potential Melastatin 6 Knockout Mice Are Lethal whereas Heterozygous Deletion Results in Mild Hypomagnesemia. Nephron Physiol. 2011, 117, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Milona, A.; Owen, B.M.; van Mil, S.; Dormann, D.; Mataki, C.; Boudjelal, M.; Cairns, W.; Schoonjans, K.; Milligan, S.; Parker, M.; et al. The normal mechanisms of pregnancy-induced liver growth are not maintained in mice lacking the bile acid sensor Fxr. Am. J. Physiol. Liver Physiol. 2010, 298, G151–G158. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Jo, Y.; Ryu, D.; Jeong, C.; Choe, S.; Lee, J. Artificial-intelligence-driven discovery of prognostic biomarker for sarcopenia. J. Cachex Sarcopenia Muscle 2021, 12, 2220–2230. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.Y.; Lee, J.M. Transcriptional Control of Trpm6 by the Nuclear Receptor FXR. Int. J. Mol. Sci. 2022, 23, 1980. https://doi.org/10.3390/ijms23041980
Kim EY, Lee JM. Transcriptional Control of Trpm6 by the Nuclear Receptor FXR. International Journal of Molecular Sciences. 2022; 23(4):1980. https://doi.org/10.3390/ijms23041980
Chicago/Turabian StyleKim, Eun Young, and Jae Man Lee. 2022. "Transcriptional Control of Trpm6 by the Nuclear Receptor FXR" International Journal of Molecular Sciences 23, no. 4: 1980. https://doi.org/10.3390/ijms23041980
APA StyleKim, E. Y., & Lee, J. M. (2022). Transcriptional Control of Trpm6 by the Nuclear Receptor FXR. International Journal of Molecular Sciences, 23(4), 1980. https://doi.org/10.3390/ijms23041980