
Nanomaterials for Delivering Antibiotics in the Therapy of Pneumonia

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
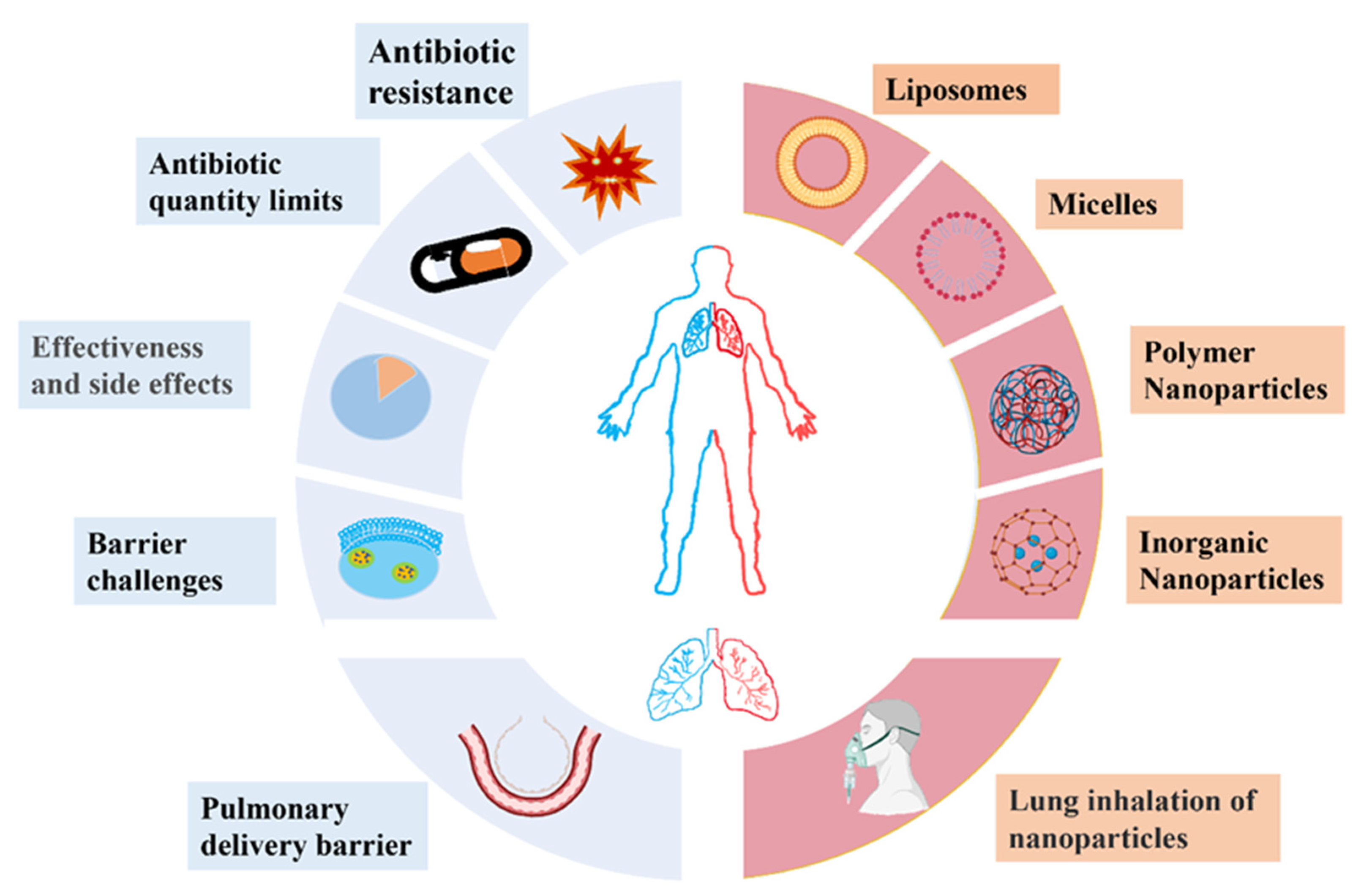
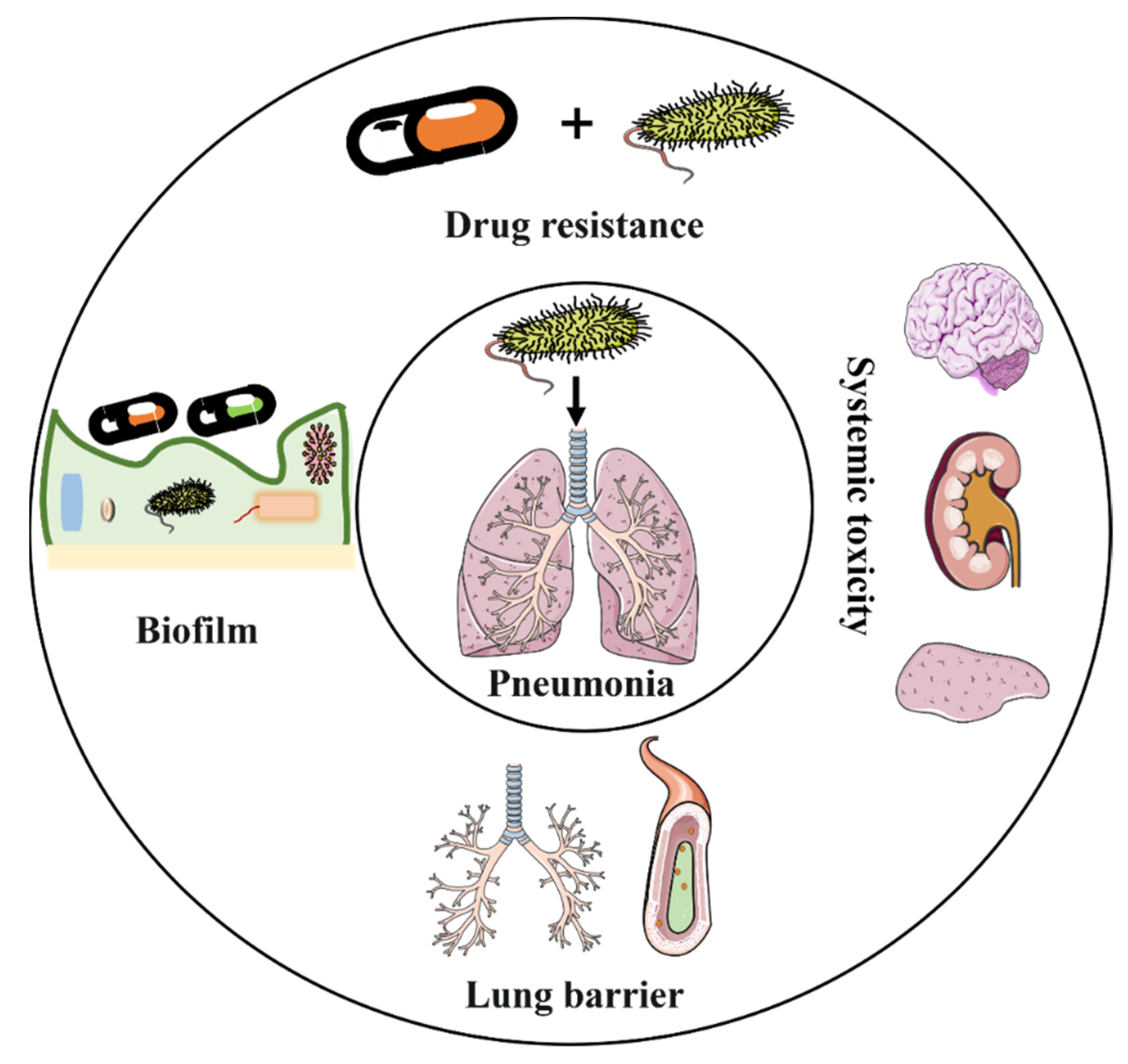
2. Challenges of Traditional Antibiotic Therapy for Bacterial Pneumonia
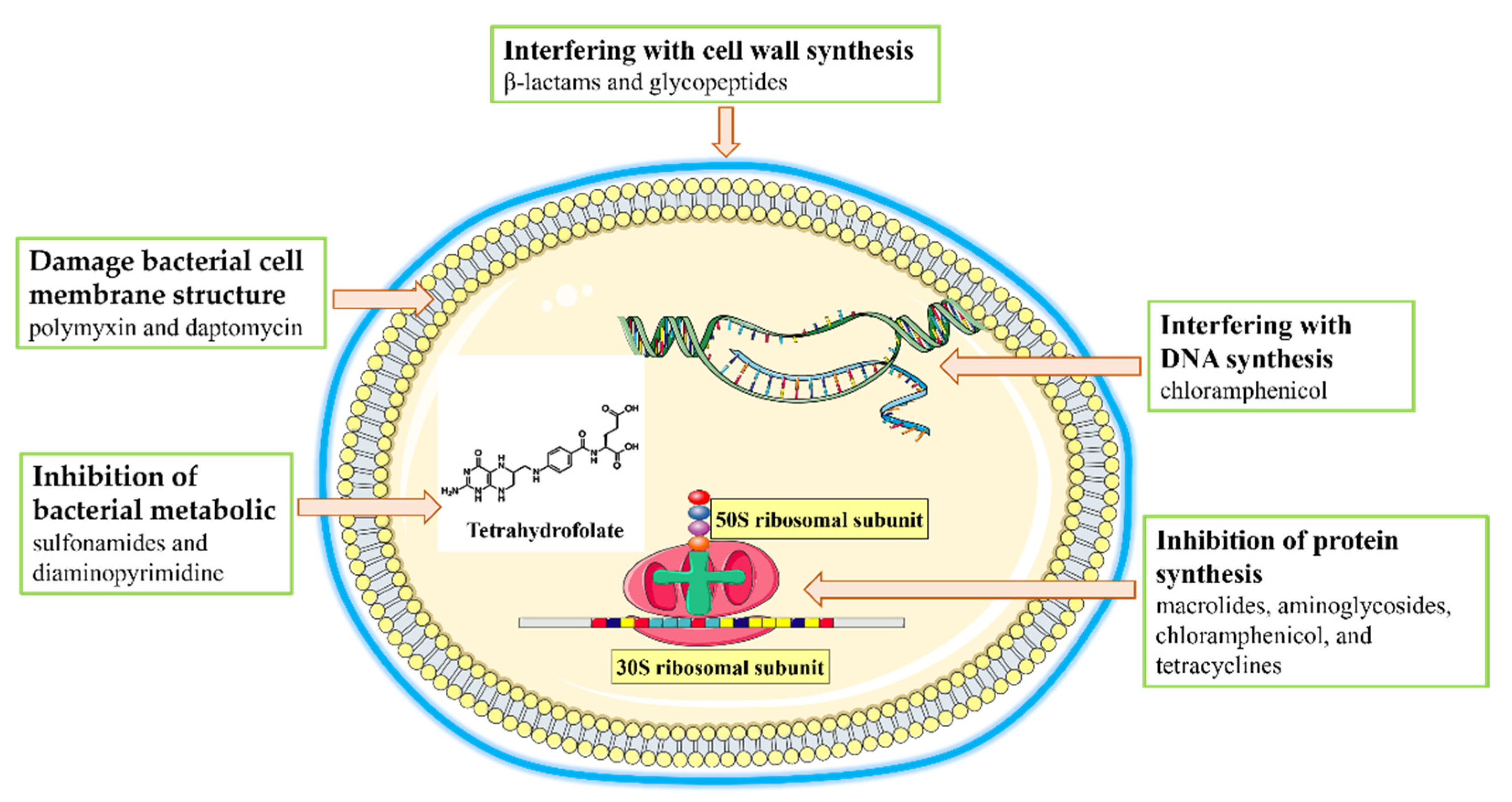
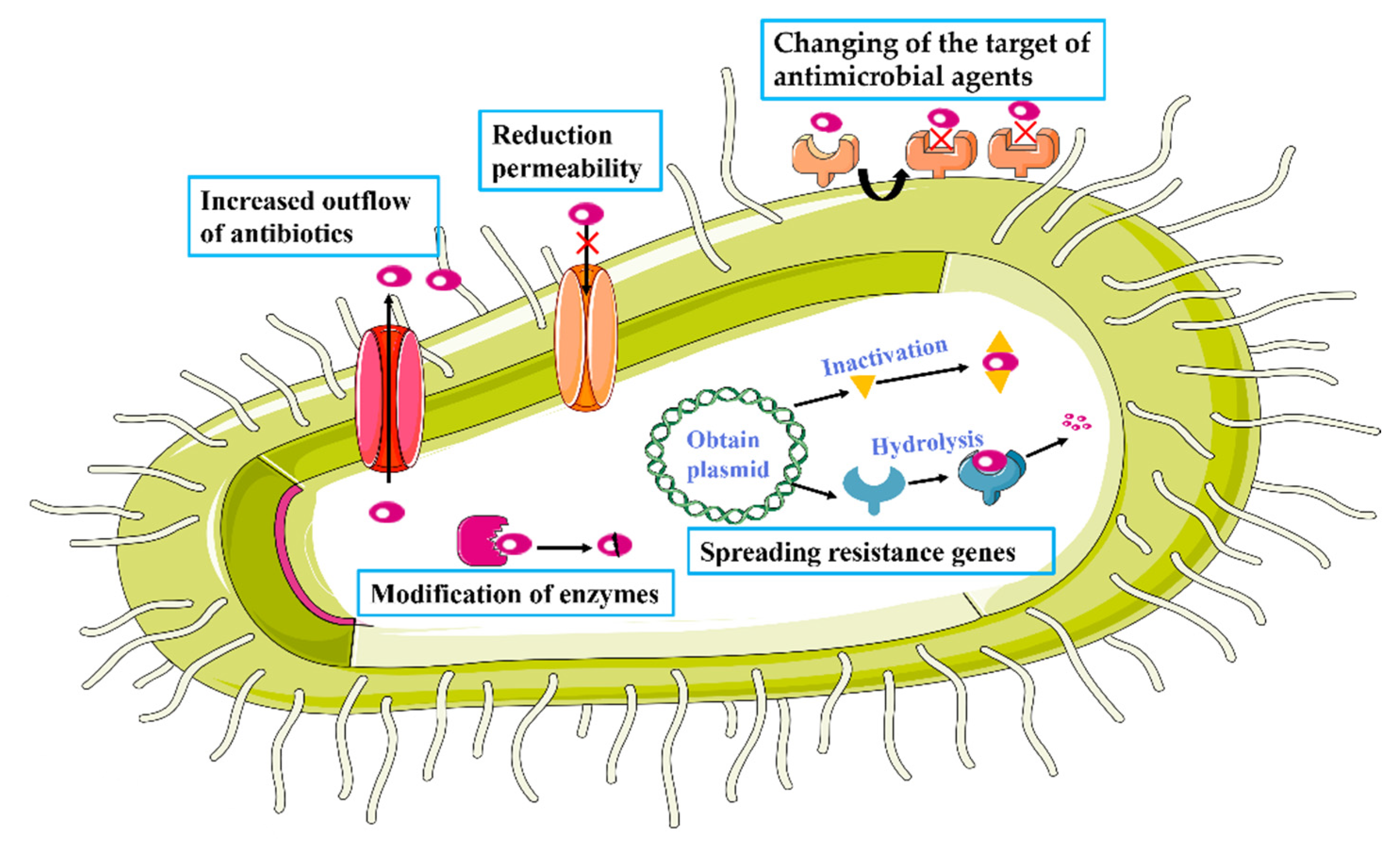
2.1. Antibiotic Resistance
2.2. A Limited Range of Antibiotics
2.3. Low Bioavailability and High Side Effects of Antibiotics
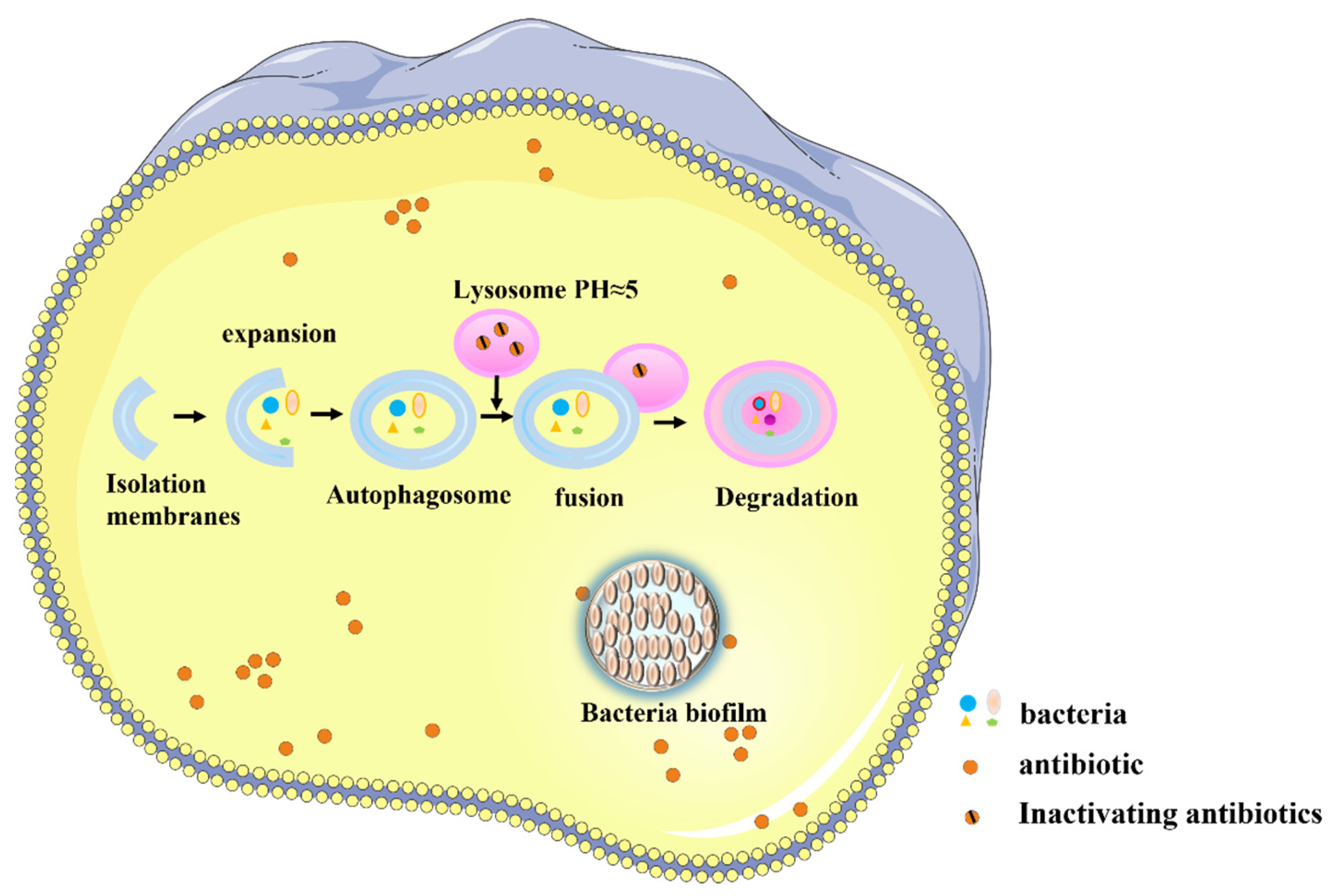
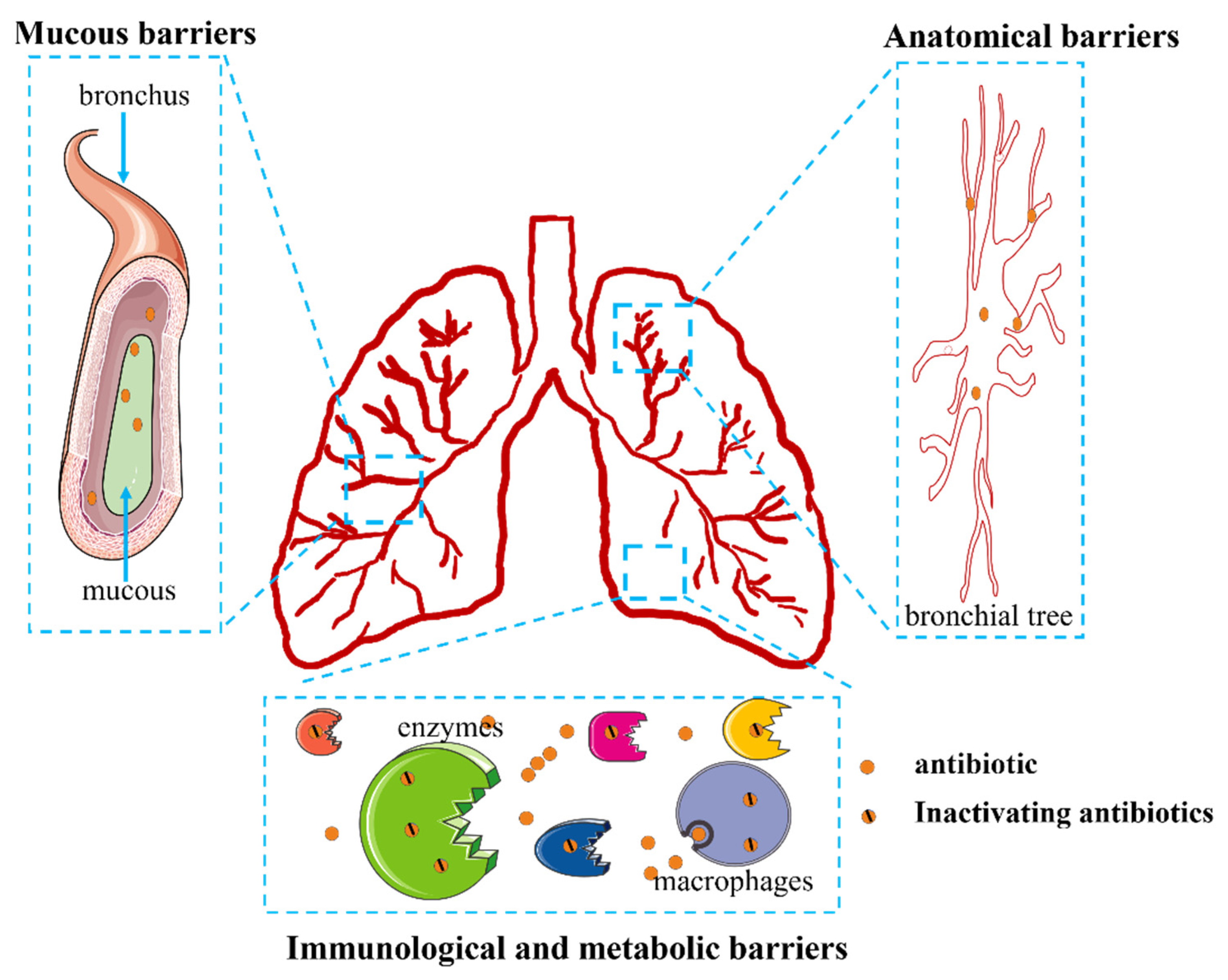
2.4. Barrier Challenges in Pneumonia Treatment
2.4.1. Barrier Challenges for the Systemic Delivery of Antibiotics
2.4.2. Barrier Challenges for the Local Delivery of Antibiotics
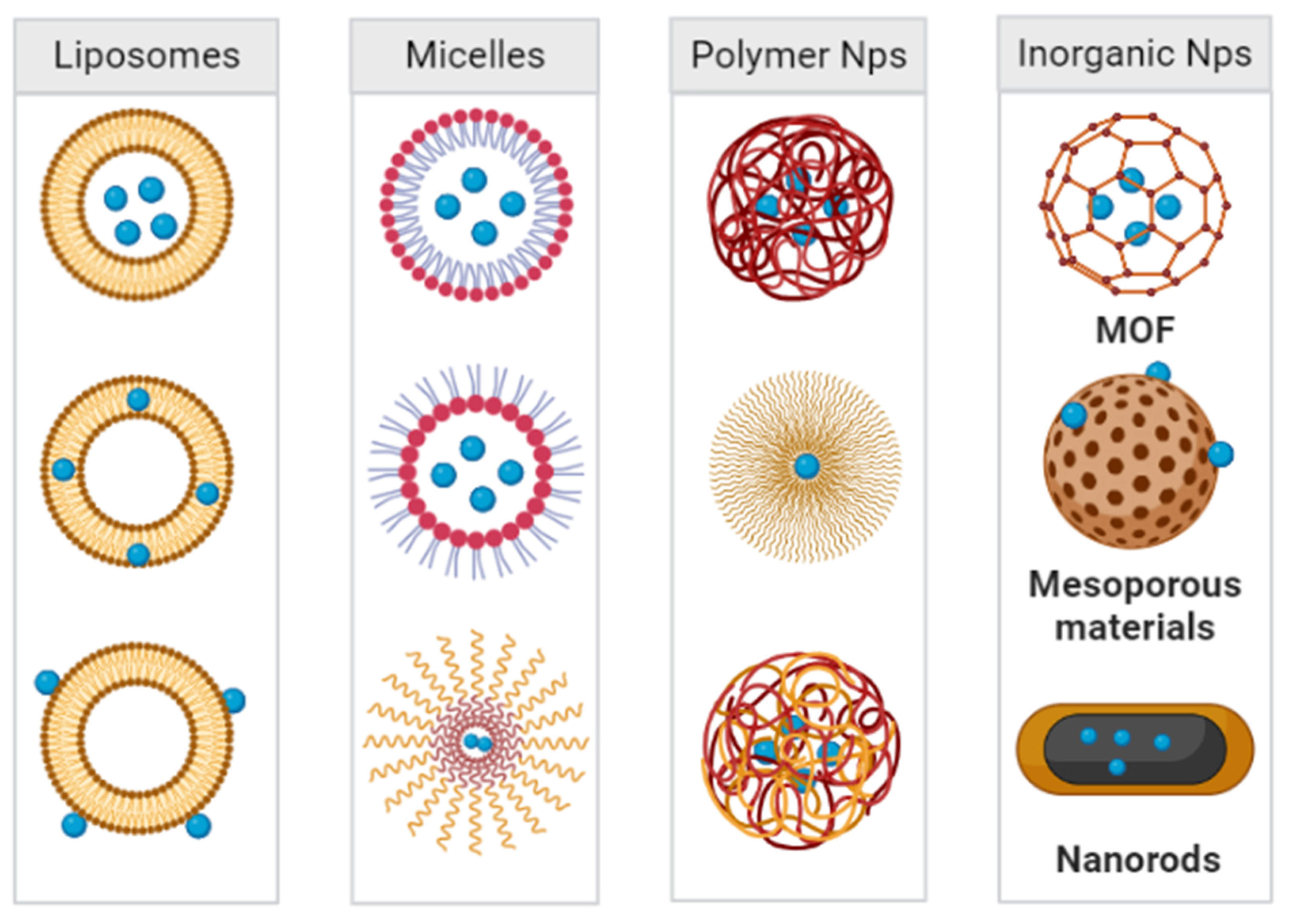
3. Bioactive Nanoparticles for the Treatment of Bacterial Infections in the Lungs
3.1. Liposomes
3.2. Micelles
3.3. Polymer Nanoparticles
3.4. Inorganic Nanoparticles
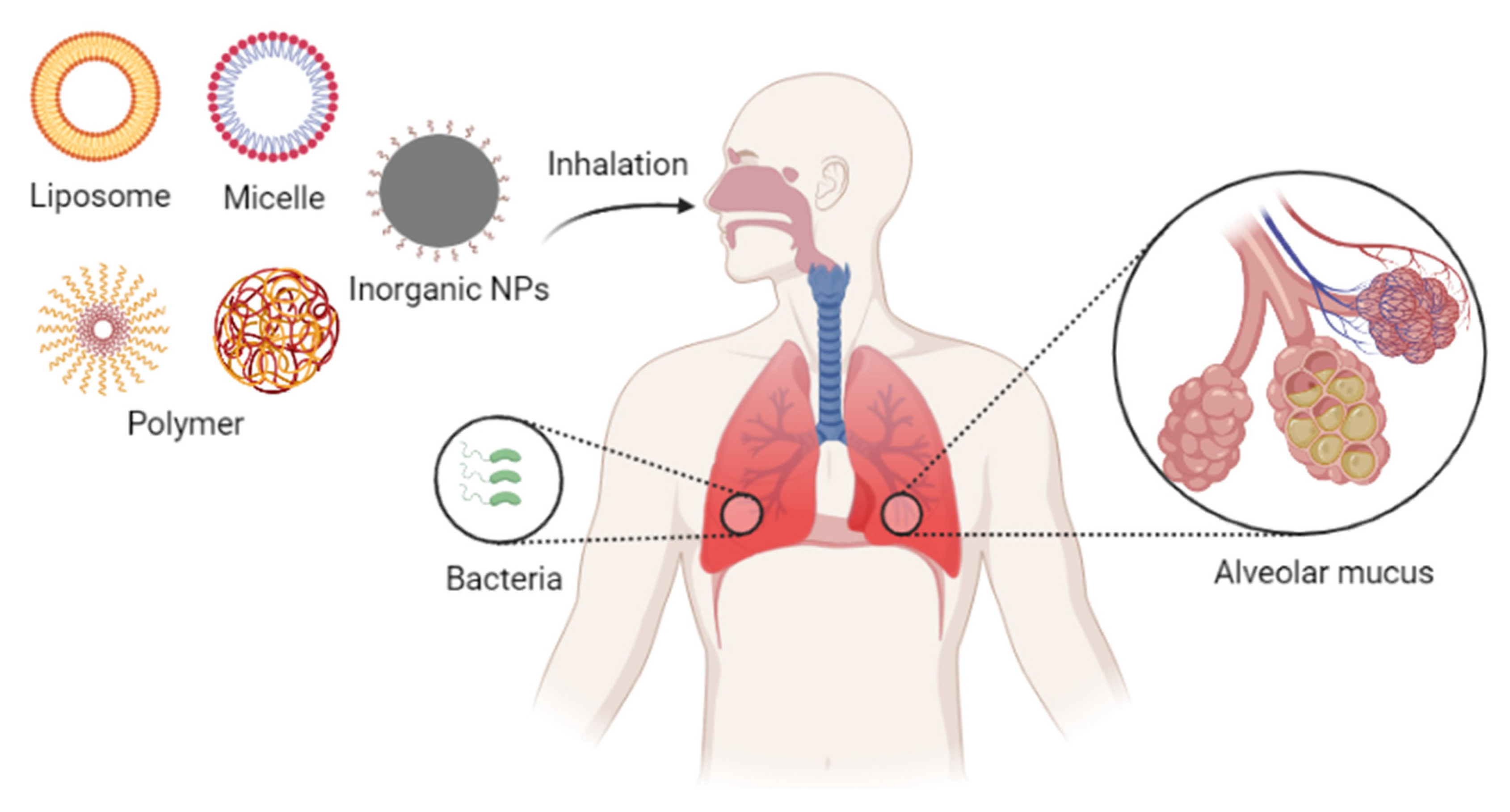
4. Pulmonary Inhalation of Nanoparticles in the Treatment of Pneumonia
4.1. Pulmonary Drug Delivery Barriers
4.2. Nanoparticles for Pulmonary Inhalation
5. Conclusions and Perspectives for Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Chou, C.C.; Shen, C.F.; Chen, S.J.; Chen, H.M.; Wang, Y.C.; Chang, W.S.; Chang, Y.T.; Chen, W.Y.; Huang, C.Y.; Kuo, C.C.; et al. Recommendations and guidelines for the treatment of pneumonia in Taiwan. J. Microbiol. Immunol. Infect. 2019, 52, 172–199. [Google Scholar] [CrossRef] [PubMed]
- WHO. In Pneumonia. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/pneumonia (accessed on 20 August 2022).
- Henig, O.; Kaye, K.S. Bacterial Pneumonia in Older Adults. Infect. Dis. Clin. N. Am. 2017, 31, 689–713. [Google Scholar] [CrossRef] [PubMed]
- Pakhale, S.; Mulpuru, S.; Verheij, T.J.; Kochen, M.M.; Rohde, G.G.; Bjerre, L.M. Antibiotics for community-acquired pneumonia in adult outpatients. Cochrane Database Syst. Rev. 2014, 10, CD002109. [Google Scholar] [CrossRef] [PubMed]
- File, T.M., Jr.; Marrie, T.J. Burden of community-acquired pneumonia in North American adults. Postgrad. Med. 2010, 122, 130–141. [Google Scholar] [CrossRef]
- Kim, G.L.; Seon, S.H.; Rhee, D.K. Pneumonia and Streptococcus pneumoniae vaccine. Arch. Pharm. Res. 2017, 40, 885–893. [Google Scholar] [CrossRef]
- Musher, D.M.; Abers, M.S.; Bartlett, J.G. Evolving Understanding of the Causes of Pneumonia in Adults, With Special Attention to the Role of Pneumococcus. Clin. Infect. Dis. 2017, 65, 1736–1744. [Google Scholar] [CrossRef]
- Miyashita, N. Atypical pneumonia: Pathophysiology, diagnosis, and treatment. Respir. Investig. 2022, 60, 56–67. [Google Scholar] [CrossRef]
- Adamo, R.; Margarit, I. Fighting Antibiotic-Resistant Klebsiella pneumoniae with “Sweet” Immune Targets. Mbio 2018, 9, e00874–e00918. [Google Scholar] [CrossRef]
- Giske, C.G.; Monnet, D.L.; Cars, O.; Carmeli, Y.; ReAct-Action on Antibiotic. Clinical and economic impact of common multidrug-resistant gram-negative bacilli. Antimicrob Agents Chemother 2008, 52, 813–821. [Google Scholar] [CrossRef]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Peyrani, P.; Mandell, L.; Torres, A.; Tillotson, G.S. The burden of community-acquired bacterial pneumonia in the era of antibiotic resistance. Expert Rev. Respir. Med. 2019, 13, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Abushaheen, M.A.; Fatani, A.J.; Alosaimi, M.; Mansy, W.; George, M.; Acharya, S.; Rathod, S.; Divakar, D.D.; Jhugroo, C.; Vellappally, S.; et al. Antimicrobial resistance, mechanisms and its clinical significance. Dis. Mon. 2020, 66, 100971. [Google Scholar] [CrossRef]
- Xiong, M.H.; Bao, Y.; Yang, X.Z.; Zhu, Y.H.; Wang, J. Delivery of antibiotics with polymeric particles. Adv. Drug Deliv. Rev. 2014, 78, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, I.; Favini-Stabile, S.; Dessen, A. Resistance to antibiotics targeted to the bacterial cell wall. Protein Sci. 2014, 23, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Infect. Control 2006, 34 (Suppl. 1), S3–S10; discussion S64–S73. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. Medchemcomm 2016, 7, 11–27. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef]
- Bertacine Dias, M.V.; Santos, J.C.; Libreros-Zuniga, G.A.; Ribeiro, J.A.; Chavez-Pacheco, S.M. Folate biosynthesis pathway: Mechanisms and insights into drug design for infectious diseases. Future Med. Chem. 2018, 10, 935–959. [Google Scholar] [CrossRef]
- Kalhapure, R.S.; Suleman, N.; Mocktar, C.; Seedat, N.; Govender, T. Nanoengineered drug delivery systems for enhancing antibiotic therapy. J. Pharm. Sci. 2015, 104, 872–905. [Google Scholar] [CrossRef]
- Bailey, M.M.; Berkland, C.J. Nanoparticle formulations in pulmonary drug delivery. Med. Res. Rev. 2009, 29, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, Y.; Tang, W.; Zhao, J.; Jing, L.; McHugh, K.J. Theranostic nanoparticles with disease-specific administration strategies. Nano Today 2022, 42, 101335. [Google Scholar] [CrossRef]
- Morley, V.J.; Kinnear, C.L.; Sim, D.G.; Olson, S.N.; Jackson, L.M.; Hansen, E.; Usher, G.A.; Showalter, S.A.; Pai, M.P.; Woods, R.J.; et al. An adjunctive therapy administered with an antibiotic prevents enrichment of antibiotic-resistant clones of a colonizing opportunistic pathogen. Elife 2020, 9, e58147. [Google Scholar] [CrossRef] [PubMed]
- Dodds, D.R. Antibiotic resistance: A current epilogue. Biochem. Pharmacol. 2017, 134, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.; Waugh, C.; Perry, M.R.; Nair, H. Global disease burden due to antibiotic resistance—State of the evidence. J. Glob. Health 2016, 6, 010306. [Google Scholar] [CrossRef]
- Kritikos, A.; Zanella, M.C.; Huttner, B.; Boillat-Blanco, N. [Side effects of selected antibiotics, not to be missed!]. Rev. Med. Suisse 2020, 16, 719–723. [Google Scholar]
- Aminov, R.I. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef]
- Cole, S.T. Who will develop new antibacterial agents? Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130430. [Google Scholar] [CrossRef]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef]
- Putman, M.; van Veen, H.W.; Konings, W.N. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 2000, 64, 672–693. [Google Scholar] [CrossRef]
- Karaiskos, I.; Giamarellou, H. Multidrug-resistant and extensively drug-resistant Gram-negative pathogens: Current and emerging therapeutic approaches. Expert Opin. Pharmacother. 2014, 15, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.A. Extended-spectrum beta-lactamases in the 21st century: Characterization, epidemiology, and detection of this important resistance threat. Clin. Microbiol. Rev. 2001, 14, 933–951. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 2005, 56, 20–51. [Google Scholar] [CrossRef]
- Redgrave, L.S.; Sutton, S.B.; Webber, M.A.; Piddock, L.J. Fluoroquinolone resistance: Mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014, 22, 438–445. [Google Scholar] [CrossRef]
- Kruger, N.J.; Stingl, K. Two steps away from novelty—Principles of bacterial DNA uptake. Mol. Microbiol. 2011, 80, 860–867. [Google Scholar] [CrossRef]
- Novick, R.P.; Christie, G.E.; Penades, J.R. The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 2010, 8, 541–551. [Google Scholar] [CrossRef]
- Nolivos, S.; Cayron, J.; Dedieu, A.; Page, A.; Delolme, F.; Lesterlin, C. Role of AcrAB-TolC multidrug efflux pump in drug-resistance acquisition by plasmid transfer. Science 2019, 364, 778–782. [Google Scholar] [CrossRef]
- Mandell, L.A.; Wunderink, R.G.; Anzueto, A.; Bartlett, J.G.; Campbell, G.D.; Dean, N.C.; Dowell, S.F.; File, T.M., Jr.; Musher, D.M.; Niederman, M.S.; et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin. Infect. Dis. 2007, 44 (Suppl. 2), S27–S72. [Google Scholar] [CrossRef]
- Lim, W.S.; Baudouin, S.V.; George, R.C.; Hill, A.T.; Jamieson, C.; Le Jeune, I.; Macfarlane, J.T.; Read, R.C.; Roberts, H.J.; Levy, M.L.; et al. BTS guidelines for the management of community acquired pneumonia in adults: Update 2009. Thorax 2009, 64 (Suppl. 3), iii1–iii55. [Google Scholar] [CrossRef]
- Woodhead, M.; Blasi, F.; Ewig, S.; Garau, J.; Huchon, G.; Ieven, M.; Ortqvist, A.; Schaberg, T.; Torres, A.; van der Heijden, G.; et al. Guidelines for the management of adult lower respiratory tract infections—Full version. Clin. Microbiol. Infect. 2011, 17 (Suppl. 6), E1–E59. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.A.; Villegas, M.V.; Quinn, J.P. Current concepts in antibiotic-resistant gram-negative bacteria. Expert Rev. Anti-Infect. Ther. 2007, 5, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S.; Rahman, N.A.A.; Peile, E.; Rahman, M.; Sartelli, M.; Hassali, M.A.; Islam, T.; Islam, S.; Haque, M. Microbial Resistance Movements: An Overview of Global Public Health Threats Posed by Antimicrobial Resistance, and How Best to Counter. Front. Public. Health 2020, 8, 535668. [Google Scholar] [CrossRef] [PubMed]
- Shlaes, D.M.; Sahm, D.; Opiela, C.; Spellberg, B. The FDA reboot of antibiotic development. Antimicrob. Agents Chemother. 2013, 57, 4605–4607. [Google Scholar] [CrossRef]
- Aulner, N.; Danckaert, A.; Ihm, J.; Shum, D.; Shorte, S.L. Next-Generation Phenotypic Screening in Early Drug Discovery for Infectious Diseases. Trends Parasitol. 2019, 35, 559–570. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef]
- Wright, G.D. Something old, something new: Revisiting natural products in antibiotic drug discovery. Can. J. Microbiol. 2014, 60, 147–154. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Benjamin, D.K., Jr.; Bradley, J.; Guidos, R.J.; Jones, R.N.; Murray, B.E.; Bonomo, R.A.; Gilbert, D. x ‘20 Progress—Development of new drugs active against gram-negative bacilli: An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2013, 56, 1685–1694. [Google Scholar] [CrossRef]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut 2016, 65, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Couce, A.; Blazquez, J. Side effects of antibiotics on genetic variability. FEMS Microbiol. Rev. 2009, 33, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.S.; Harding, J.; Molloy, R.; Land, L.; Longcroft-Neal, K.; Moore, D.; Ross, J.D.C. Adverse effects of a single dose of gentamicin in adults: A systematic review. Br. J. Clin. Pharmacol. 2018, 84, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Pamer, E.G. Resurrecting the intestinal microbiota to combat antibiotic-resistant pathogens. Science 2016, 352, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Zogg, H.; Wei, L.; Bartlett, A.; Ghoshal, U.C.; Rajender, S.; Ro, S. Gut Microbial Dysbiosis in the Pathogenesis of Gastrointestinal Dysmotility and Metabolic Disorders. J. Neurogastroenterol. Motil. 2021, 27, 19–34. [Google Scholar] [CrossRef]
- Rashid, M.U.; Zaura, E.; Buijs, M.J.; Keijser, B.J.; Crielaard, W.; Nord, C.E.; Weintraub, A. Determining the Long-term Effect of Antibiotic Administration on the Human Normal Intestinal Microbiota Using Culture and Pyrosequencing Methods. Clin. Infect. Dis. 2015, 60 (Suppl. 2), S77–S84. [Google Scholar] [CrossRef]
- Pinto-Alphandary, H.; Andremont, A.; Couvreur, P. Targeted delivery of antibiotics using liposomes and nanoparticles: Research and applications. Int. J. Antimicrob. Agents 2000, 13, 155–168. [Google Scholar] [CrossRef]
- Couvreur, P.; Fattal, E.; Andremont, A. Liposomes and nanoparticles in the treatment of intracellular bacterial infections. Pharm. Res. 1991, 8, 1079–1086. [Google Scholar] [CrossRef]
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004, 36, 2445–2462. [Google Scholar] [CrossRef]
- Das, D.; Saha, S.S.; Bishayi, B. Intracellular survival of Staphylococcus aureus: Correlating production of catalase and superoxide dismutase with levels of inflammatory cytokines. Inflamm. Res. 2008, 57, 340–349. [Google Scholar] [CrossRef]
- Carryn, S.; Chanteux, H.; Seral, C.; Mingeot-Leclercq, M.P.; Van Bambeke, F.; Tulkens, P.M. Intracellular pharmacodynamics of antibiotics. Infect. Dis. Clin. N. Am. 2003, 17, 615–634. [Google Scholar] [CrossRef] [PubMed]
- Onyeji, C.O.; Nightingale, C.H.; Nicolau, D.P.; Quintiliani, R. Efficacies of liposome-encapsulated clarithromycin and ofloxacin against Mycobacterium avium-M. intracellulare complex in human macrophages. Antimicrob. Agents Chemother. 1994, 38, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Baldry, S. Attack of the clones. Nat. Rev. Microbiol. 2010, 8, 390. [Google Scholar] [CrossRef]
- Mah, T.F.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends. Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Okuda, T.; Lu, X.Y.; Chan, H.K. Amorphous powders for inhalation drug delivery. Adv. Drug Deliv. Rev. 2016, 100, 102–115. [Google Scholar] [CrossRef]
- Courrier, H.M.; Butz, N.; Vandamme, T.F. Pulmonary drug delivery systems: Recent developments and prospects. Crit. Rev. Ther. Drug Carr. Syst. 2002, 19, 425–498. [Google Scholar] [CrossRef]
- Patton, J.S.; Byron, P.R. Inhaling medicines: Delivering drugs to the body through the lungs. Nat. Rev. Drug Discov. 2007, 6, 67–74. [Google Scholar] [CrossRef]
- Thakur, A.K.; Chellappan, D.K.; Dua, K.; Mehta, M.; Satija, S.; Singh, I. Patented therapeutic drug delivery strategies for targeting pulmonary diseases. Expert Opin. Ther. Pat. 2020, 30, 375–387. [Google Scholar] [CrossRef]
- Newman, S.P. Drug delivery to the lungs: Challenges and opportunities. Ther. Deliv. 2017, 8, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, D. Will pulmonary drug delivery for systemic application ever fulfill its rich promise? Expert Opin. Drug Deliv. 2016, 13, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Meng, J.; Li, Y.; Yang, C.; Hou, Y.; Tang, W.; McHugh, K.J.; Jing, L. Nanotechnology-enhanced immunotherapy for metastatic cancer. Innovation 2021, 2, 100174. [Google Scholar] [CrossRef] [PubMed]
- Flume, P.A.; VanDevanter, D.R. Clinical applications of pulmonary delivery of antibiotics. Adv. Drug Deliv. Rev. 2015, 85, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Luepke, K.H.; Suda, K.J.; Boucher, H.; Russo, R.L.; Bonney, M.W.; Hunt, T.D.; Mohr, J.F., 3rd. Past, Present, and Future of Antibacterial Economics: Increasing Bacterial Resistance, Limited Antibiotic Pipeline, and Societal Implications. Pharmacotherapy 2017, 37, 71–84. [Google Scholar] [CrossRef]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control Release Off. J. Control Release Soc. 2011, 156, 128–145. [Google Scholar] [CrossRef]
- Gupta, A.; Mumtaz, S.; Li, C.H.; Hussain, I.; Rotello, V.M. Combatting antibiotic-resistant bacteria using nanomaterials. Chem. Soc. Rev. 2019, 48, 415–427. [Google Scholar] [CrossRef]
- Dua, K.; Shukla, S.D.; Tekade, R.K.; Hansbro, P.M. Whether a novel drug delivery system can overcome the problem of biofilms in respiratory diseases? Drug Deliv. Transl. Res. 2017, 7, 179–187. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, L.; Su, L.; van der Mei, H.C.; Jutte, P.C.; Ren, Y.; Busscher, H.J. Nanotechnology-based antimicrobials and delivery systems for biofilm-infection control. Chem. Soc. Rev. 2019, 48, 428–446. [Google Scholar] [CrossRef]
- Chu, L.; Gao, H.; Cheng, T.; Zhang, Y.; Liu, J.; Huang, F.; Yang, C.; Shi, L.; Liu, J. A charge-adaptive nanosystem for prolonged and enhanced in vivo antibiotic delivery. Chem. Commun. 2016, 52, 6265–6268. [Google Scholar] [CrossRef]
- Davoodi, P.; Lee, L.Y.; Xu, Q.; Sunil, V.; Sun, Y.; Soh, S.; Wang, C.-H. Drug delivery systems for programmed and on-demand release. Adv. Drug Deliv. Rev. 2018, 132, 104–138. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.S.; Figueiredo, C.; Azevedo, N.F.; Braeckmans, K.; De Smedt, S.C. Nanomaterials and molecular transporters to overcome the bacterial envelope barrier: Towards advanced delivery of antibiotics. Adv. Drug Deliv. Rev. 2018, 136–137, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Sen Karaman, D.; Ercan, U.; Bakay, E.; Topaloglu, N.; Rosenholm, J. Evolving Technologies and Strategies for Combating Antibacterial Resistance in the Advent of the Postantibiotic Era. Adv. Funct. Mater. 2020, 30, 1908783. [Google Scholar] [CrossRef]
- Bao, C.; Liu, B.; Li, B.; Chai, J.; Zhang, L.; Jiao, L.; Li, D.; Yu, Z.; Ren, F.; Shi, X.; et al. Enhanced Transport of Shape and Rigidity-Tuned α-Lactalbumin Nanotubes across Intestinal Mucus and Cellular Barriers. Nano Lett. 2020, 20, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.; Del Fiol, F.S.; Balcão, V.M. Prospects for the Use of New Technologies to Combat Multidrug-Resistant Bacteria. Front. Pharmacol. 2019, 10, 692. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Song, H.; Qu, X.; Chang, J.; Yang, B.; Lu, S. Carbon dots as a new class of nanomedicines: Opportunities and challenges. Coord. Chem. Rev. 2021, 442, 214010. [Google Scholar] [CrossRef]
- Gadekar, V.; Borade, Y.; Kannaujia, S.; Rajpoot, K.; Anup, N.; Tambe, V.; Kalia, K.; Tekade, R.K. Nanomedicines accessible in the market for clinical interventions. J. Control Release 2021, 330, 372–397. [Google Scholar] [CrossRef]
- Singh, A.; Amod, A.; Pandey, P.; Bose, P.; Pingali, M.S.; Shivalkar, S.; Varadwaj, P.K.; Sahoo, A.K.; Samanta, S.K. Bacterial biofilm infections, their resistance to antibiotics therapy and current treatment strategies. Biomed. Mater. 2022, 17, 022003. [Google Scholar] [CrossRef]
- Hallaj-Nezhadi, S.; Hassan, M. Nanoliposome-based antibacterial drug delivery. Drug Deliv. 2015, 22, 581–589. [Google Scholar] [CrossRef]
- Meng, J.; Zhang, P.; Chen, Q.; Wang, Z.; Gu, Y.; Ma, J.; Li, W.; Yang, C.; Qiao, Y.; Hou, Y.; et al. Two-Pronged Intracellular Co-Delivery of Antigen and Adjuvant for Synergistic Cancer Immunotherapy. Adv. Mater. 2022, 34, 2202168. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A.; Gubernator, J.; Gula, G.; Bocer, T.; Doroszkiewicz, W. The interaction between Pseudomonas aeruginosa cells and cationic PC:Chol:DOTAP liposomal vesicles versus outer-membrane structure and envelope properties of bacterial cell. Int. J. Pharm. 2009, 367, 211–219. [Google Scholar] [CrossRef]
- Su, F.Y.; Chen, J.; Son, H.N.; Kelly, A.M.; Convertine, A.J.; West, T.E.; Skerrett, S.J.; Ratner, D.M.; Stayton, P.S. Polymer-augmented liposomes enhancing antibiotic delivery against intracellular infections. Biomater. Sci. 2018, 6, 1976–1985. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, K.; Patel, U.; Pham, C.; McAlpin, A.; Budisalich, T.; Jayawardena, S.N. Targeted Delivery of Antibiotic Therapy to Inhibit Pseudomonas aeruginosa Using Lipid-Coated Mesoporous Silica Core-Shell Nanoassembly. ACS Appl. Bio Mater. 2020, 3, 6708–6721. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Sung, C.T.; Aljuffali, I.A.; Chen, C.H.; Hu, K.Y.; Fang, J.Y. Intravenous anti-MRSA phosphatiosomes mediate enhanced affinity to pulmonary surfactants for effective treatment of infectious pneumonia. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Allemailem, K.S.; Almatroudi, A.; Alrumaihi, F.; Aljaghwani, A.; Alnuqaydan, A.M.; Khalilullah, H.; Younus, H.; El-Kady, A.M.; Aldakheel, F.M.; Khan, A.A.; et al. Antimicrobial, Immunomodulatory and Anti-Inflammatory Potential of Liposomal Thymoquinone: Implications in the Treatment of Bacterial Pneumonia in Immunocompromised Mice. Biomedicines 2021, 9, 1673. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.; Park, H.; Yu, S.; Zhou, F.; Li, J.; Xu, Q.; Zhou, Q.T. Evaluation of co-delivery of colistin and ciprofloxacin in liposomes using an in vitro human lung epithelial cell model. Int. J. Pharm. 2019, 569, 118616. [Google Scholar] [CrossRef] [PubMed]
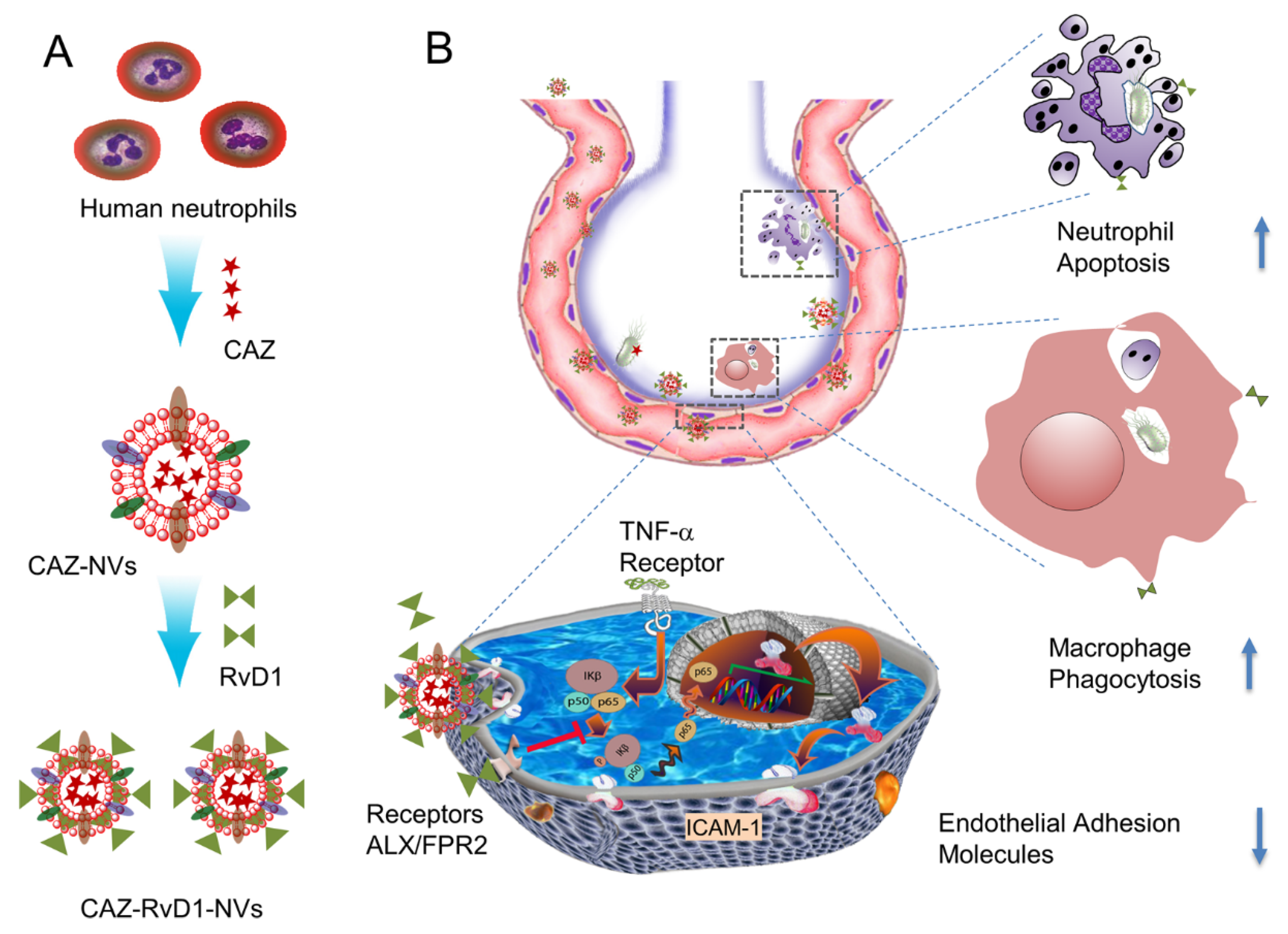
- Gao, J.; Wang, S.; Dong, X.; Leanse, L.G.; Dai, T.; Wang, Z. Co-delivery of resolvin D1 and antibiotics with nanovesicles to lungs resolves inflammation and clears bacteria in mice. Commun. Biol. 2020, 3, 680. [Google Scholar] [CrossRef]
- Rao, Y.; Sun, Y.; Li, P.; Xu, M.; Chen, X.; Wang, Y.; Chen, Y.; Deng, X.; Yu, S.; Hu, H. Hypoxia-sensitive adjuvant loaded liposomes enhance the antimicrobial activity of azithromycin via phospholipase-triggered releasing for Pseudomonas aeruginosa biofilms eradication. Int. J. Pharm. 2022, 623, 121910. [Google Scholar] [CrossRef]
- Pushparaj Selvadoss, P.; Nellore, J.; Balaraman Ravindrran, M.; Sekar, U. Novel pyochelin-based PEGylated liposomes for enhanced delivery of antibiotics against resistant clinical isolates of Pseudomonas aeruginosa. Artif. Cells Nanomed. Biotechnol. 2018, 46, 2043–2053. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, X.; Wang, B.; Zhang, G.; Liu, M.; Geng, S.; Sun, L.; An, J.; Zhang, Z.; Zhang, H. A soft anti-virulence liposome realizing the explosive release of antibiotics at an infectious site to improve antimicrobial therapy. J. Mater. Chem. B 2021, 9, 147–158. [Google Scholar] [CrossRef]
- Gaspar, D.P.; Gaspar, M.M.; Eleutério, C.V.; Grenha, A.; Blanco, M.; Gonçalves, L.M.D.; Taboada, P.; Almeida, A.J.; Remuñán-López, C. Microencapsulated Solid Lipid Nanoparticles as a Hybrid Platform for Pulmonary Antibiotic Delivery. Mol. Pharm. 2017, 14, 2977–2990. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wu, M.; Ye, W.; Huang, Z.; Ma, X.; Wang, W.; Wang, W.; Huang, Y.; Pan, X.; Wu, C. Inhalable solid lipid nanoparticles for intracellular tuberculosis infection therapy: Macrophage-targeting and pH-sensitive properties. Drug Deliv. Transl. Res. 2021, 11, 1218–1235. [Google Scholar] [CrossRef] [PubMed]
- Brandelli, A. Nanostructures as promising tools for delivery of antimicrobial peptides. Mini Rev. Med. Chem. 2012, 12, 731–741. [Google Scholar] [CrossRef]
- Feng, W.; Li, G.; Kang, X.; Wang, R.; Liu, F.; Zhao, D.; Li, H.; Bu, F.; Yu, Y.; Moriarty, T.F.; et al. Cascade-Targeting Poly(amino acid) Nanoparticles Eliminate Intracellular Bacteria via On-Site Antibiotic Delivery. Adv. Mater. 2022, 34, 2109789. [Google Scholar] [CrossRef]
- Chen, M.; Xie, S.; Wei, J.; Song, X.; Ding, Z.; Li, X. Antibacterial Micelles with Vancomycin-Mediated Targeting and pH/Lipase-Triggered Release of Antibiotics. ACS Appl. Mater. Interfaces 2018, 10, 36814–36823. [Google Scholar] [CrossRef] [PubMed]
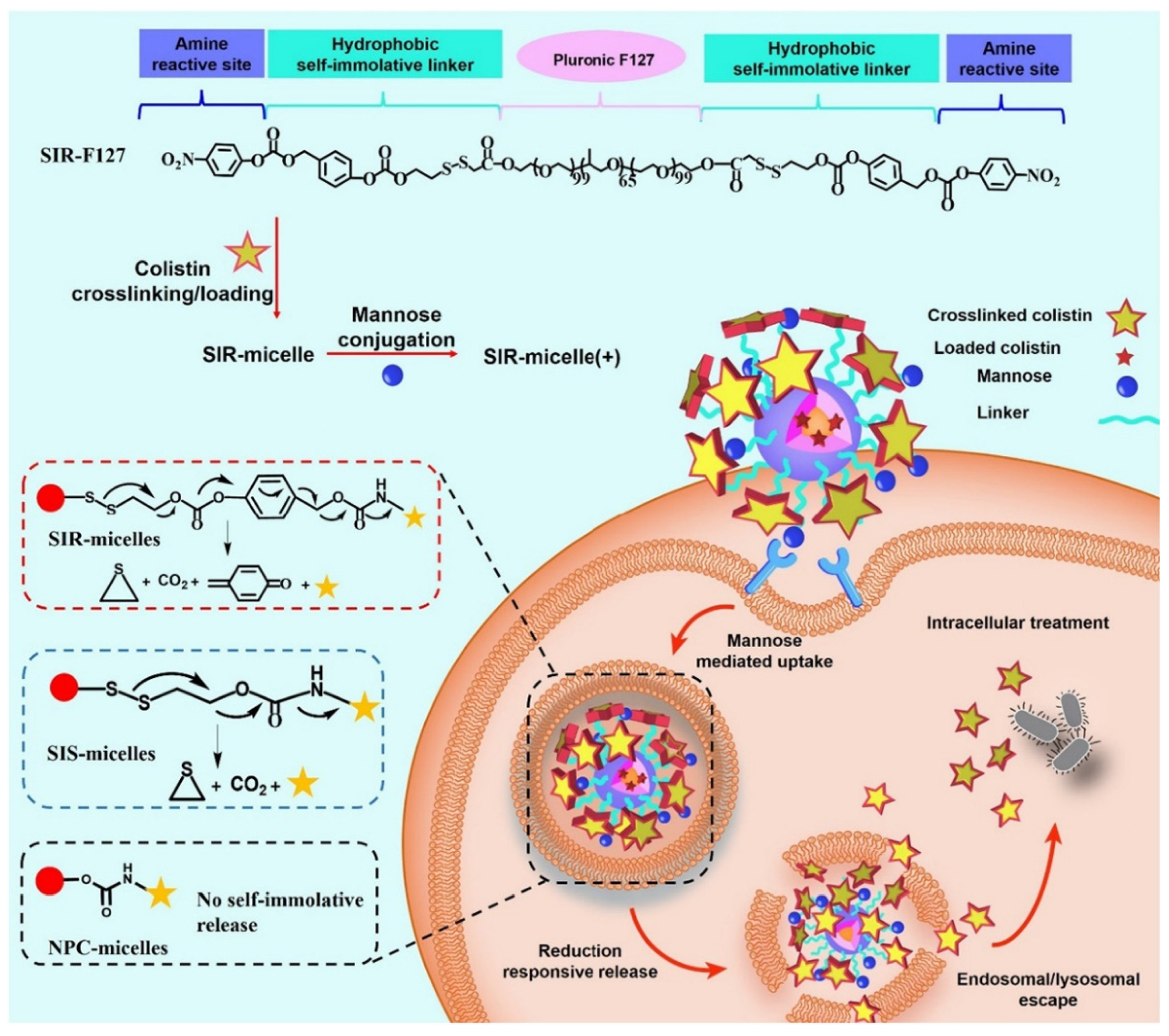
- Yang, X.; Qiu, Q.; Liu, G.; Ren, H.; Wang, X.; Lovell, J.F.; Zhang, Y. Traceless antibiotic-crosslinked micelles for rapid clearance of intracellular bacteria. J. Control Release Off. J. Control Release Soc. 2022, 341, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Wang, C.; Guo, R.; Jiang, X.; Li, W.; Chen, X.; Li, K.; Hong, W. Step-by-step dual stimuli-responsive nanoparticles for efficient bacterial biofilm eradication. Biomater. Sci. 2021, 9, 6889–6902. [Google Scholar] [CrossRef]
- Lu, C.; Xiao, Y.; Liu, Y.; Sun, F.; Qiu, Y.; Mu, H.; Duan, J. Hyaluronic acid-based levofloxacin nanomicelles for nitric oxide-triggered drug delivery to treat bacterial infections. Carbohydr. Polym. 2020, 229, 115479. [Google Scholar] [CrossRef]
- Park, S.C.; Ko, C.; Hyeon, H.; Jang, M.K.; Lee, D. Imaging and Targeted Antibacterial Therapy Using Chimeric Antimicrobial Peptide Micelles. ACS Appl. Mater. Interfaces 2020, 12, 54306–54315. [Google Scholar] [CrossRef]
- Dinarello, C.A. Anti-inflammatory Agents: Present and Future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Gao, J.; Wang, Z. Bioresponsive Nanoparticles Targeted to Infectious Microenvironments for Sepsis Management. Adv. Mater. 2018, 30, e1803618. [Google Scholar] [CrossRef] [PubMed]
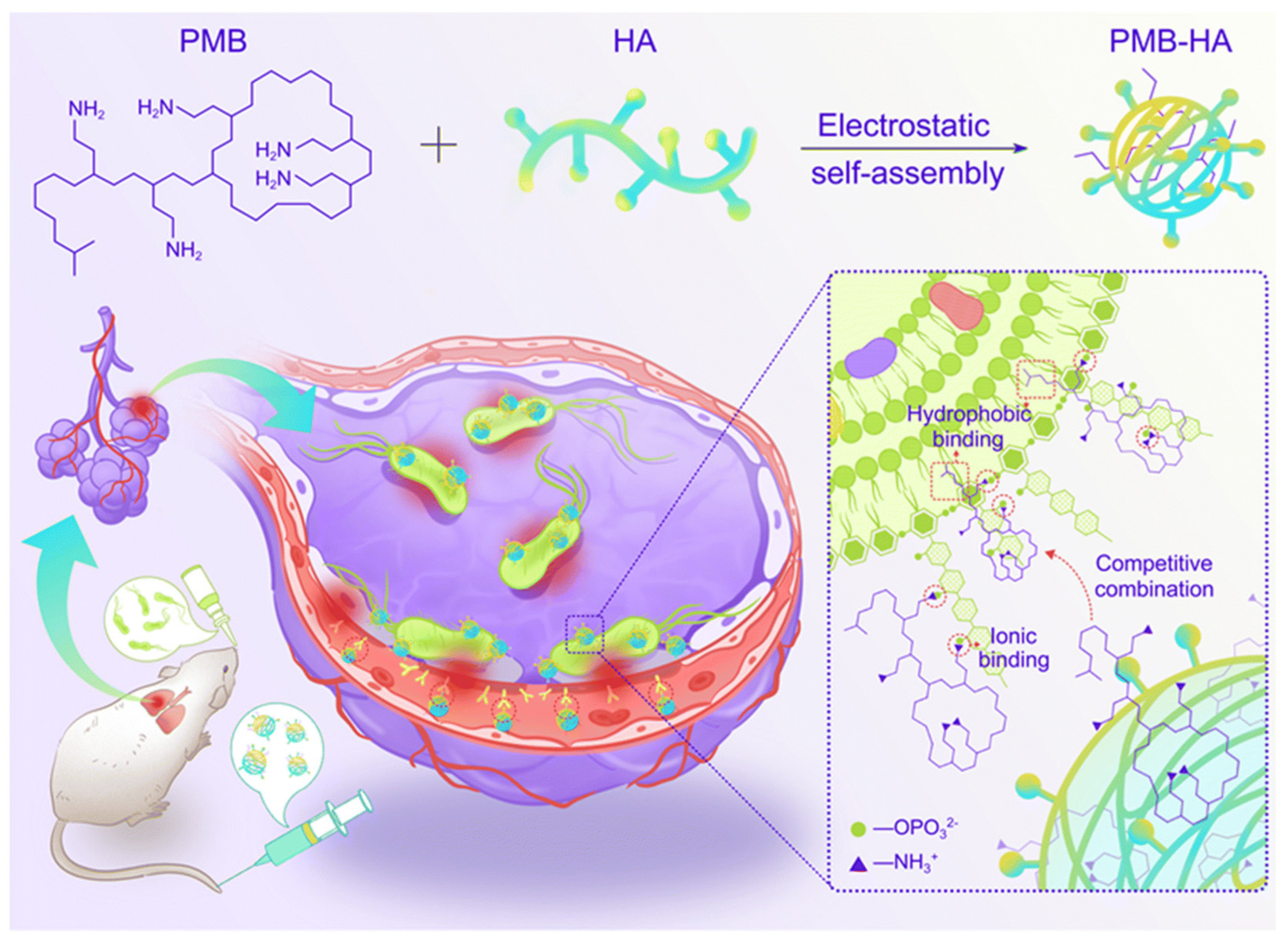
- Chai, M.; Gao, Y.; Liu, J.; Deng, Y.; Hu, D.; Jin, Q.; Ji, J. Polymyxin B-Polysaccharide Polyion Nanocomplex with Improved Biocompatibility and Unaffected Antibacterial Activity for Acute Lung Infection Management. Adv. Healthc. Mater. 2020, 9, e1901542. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Kuo, S.C.; Yao, B.Y.; Fang, Z.S.; Lee, Y.T.; Chang, Y.C.; Chen, T.L.; Hu, C.J. Colistin nanoparticle assembly by coacervate complexation with polyanionic peptides for treating drug-resistant gram-negative bacteria. Acta Biomater. 2018, 82, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ouyang, Q.; Zhai, T.; Sun, J.; Wu, J.; Qin, F.; Zhang, N.; Yue, S.; Yang, X.; Zhang, H.; et al. An inflammation-targeted nanoparticle with bacteria forced release of polymyxin B for pneumonia therapy. Nanoscale 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Lin, W.; Gao, J.; Shi, X.; Davaritouchaee, M.; Nielsen, A.E.; Mancini, R.J.; Wang, Z. pH-Responsive Nanoparticles Targeted to Lungs for Improved Therapy of Acute Lung Inflammation/Injury. ACS Appl. Mater. Interfaces 2019, 11, 16380–16390. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Li, J.; Zhang, Y.; Zheng, Y.; Zhang, Y.; Meng, H.; Wu, G.; Hu, Y.; Gao, Y.; Huang, S.; et al. Synergy between Clinical Microenvironment Targeted Nanoplatform and Near-Infrared Light Irradiation for Managing Pseudomonas aeruginosa Infections. ACS Appl. Mater. Interfaces 2021, 13, 38979–38989. [Google Scholar] [CrossRef]
- Du, X.; Xue, J.; Jiang, M.; Lin, S.; Huang, Y.; Deng, K.; Shu, L.; Xu, H.; Li, Z.; Yao, J.; et al. A Multiepitope Peptide, rOmp22, Encapsulated in Chitosan-PLGA Nanoparticles as a Candidate Vaccine Against Acinetobacter baumannii Infection. Int. J. Nanomed. 2021, 16, 1819–1836. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, Y.; Fan, B.; Wang, Y.; Mao, Z.; Wang, W.; Wu, J. Inflammation-targeting polymeric nanoparticles deliver sparfloxacin and tacrolimus for combating acute lung sepsis. J. Control Release 2020, 321, 463–474. [Google Scholar] [CrossRef]
- Hussain, S.; Joo, J.; Kang, J.; Kim, B.; Braun, G.B.; She, Z.-G.; Kim, D.; Mann, A.P.; Mölder, T.; Teesalu, T.; et al. Antibiotic-loaded nanoparticles targeted to the site of infection enhance antibacterial efficacy. Nat. Biomed. Eng. 2018, 2, 95–103. [Google Scholar] [CrossRef]
- Chin, W.; Zhong, G.; Pu, Q.; Yang, C.; Lou, W.; De Sessions, P.F.; Periaswamy, B.; Lee, A.; Liang, Z.C.; Ding, X.; et al. A macromolecular approach to eradicate multidrug resistant bacterial infections while mitigating drug resistance onset. Nat. Commun. 2018, 9, 917. [Google Scholar] [CrossRef]
- Lou, W.; Venkataraman, S.; Zhong, G.; Ding, B.; Tan, J.P.K.; Xu, L.; Fan, W.; Yang, Y.Y. Antimicrobial polymers as therapeutics for treatment of multidrug-resistant Klebsiella pneumoniae lung infection. Acta Biomater. 2018, 78, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lou, W.; Zhong, G.; Lee, A.; Leong, J.; Chin, W.; Ding, B.; Bao, C.; Tan, J.P.K.; Pu, Q.; et al. Degradable antimicrobial polycarbonates with unexpected activity and selectivity for treating multidrug-resistant Klebsiella pneumoniae lung infection in mice. Acta Biomater. 2019, 94, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Zhu, Y.; Wang, Y.; Qu, S.; Huang, Y.; Zheng, L.; Duan, S.; Yu, B.; Qin, M.; Xu, F.-J. Cationic Polysaccharide Conjugates as Antibiotic Adjuvants Resensitize Multidrug-Resistant Bacteria and Prevent Resistance. Adv. Mater. 2022, 34, 2204065. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, X.; Zhang, S.; Chen, N.; Sun, Y.; Ma, K.; Hong, D.; Li, L.; Du, Y.; Lu, X.; et al. TPGS-based and S-thanatin functionalized nanorods for overcoming drug resistance in Klebsiella pneumonia. Nat. Commun. 2022, 13, 3731. [Google Scholar] [CrossRef]
- Pan, Y.; Zheng, H.; Li, G.; Li, Y.; Jiang, J.; Chen, J.; Xie, Q.; Wu, D.; Ma, R.; Liu, X.; et al. Antibiotic-Like Activity of Atomic Layer Boron Nitride for Combating Resistant Bacteria. ACS Nano 2022, 16, 7674–7688. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Zou, S.; Lu, C.; Xiao, Y.; Bai, H.; Zhang, X.; Mu, H.; Zhang, X.; Duan, J. Hyaluronic acid-coated ZIF-8 for the treatment of pneumonia caused by methicillin-resistant Staphylococcus aureus. Int. J. Biol. Macromol. 2020, 155, 103–109. [Google Scholar] [CrossRef]
- Shen, X.; Ma, R.; Huang, Y.; Chen, L.; Xu, Z.; Li, D.; Meng, X.; Fan, K.; Xi, J.; Yan, X.; et al. Nano-decocted ferrous polysulfide coordinates ferroptosis-like death in bacteria for anti-infection therapy. Nano Today 2020, 35, 100981. [Google Scholar] [CrossRef]
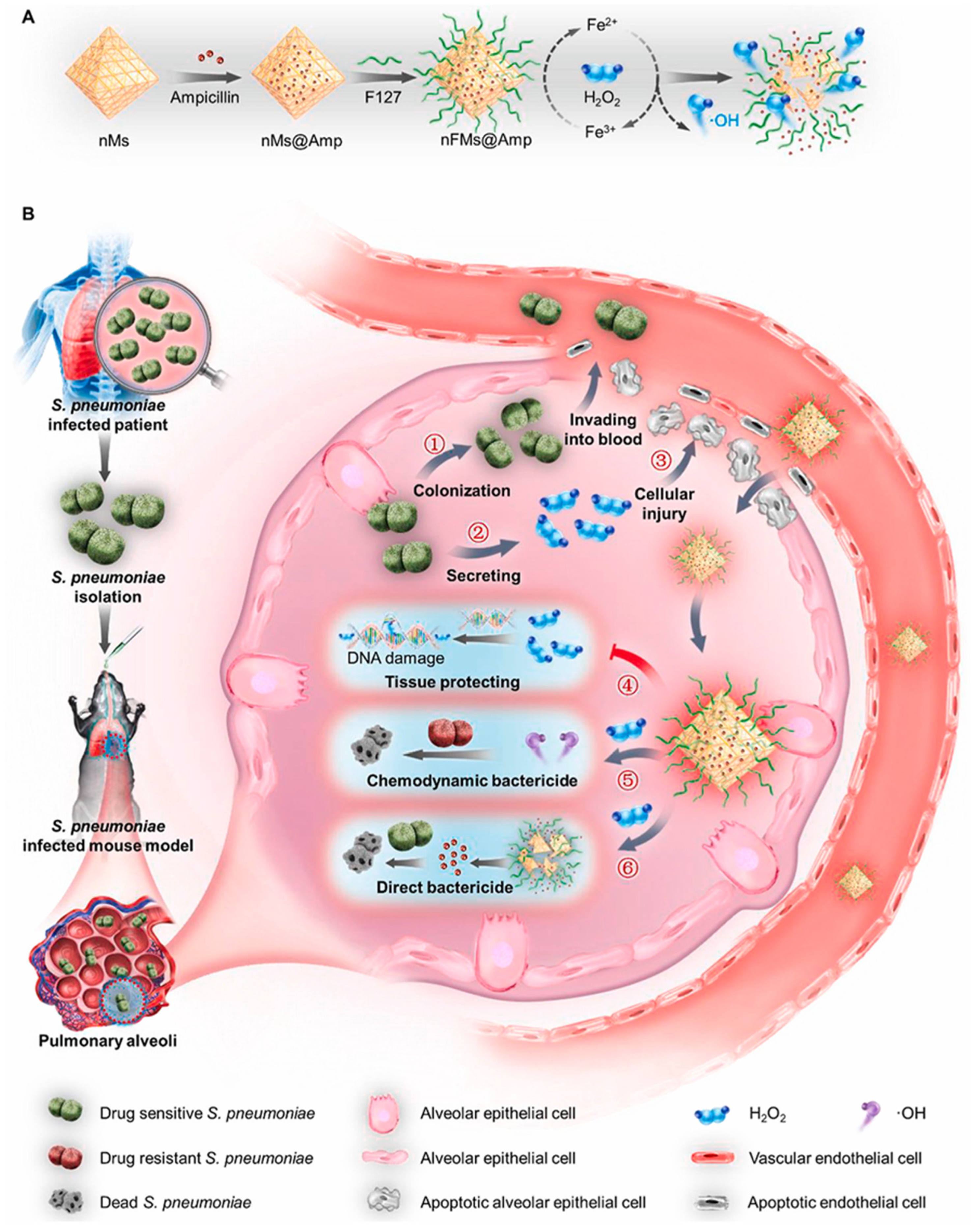
- Wu, Y.; Jiang, W.; Huo, S.; Li, S.; Xu, Y.; Ding, S.; Zhou, J.; Liu, H.; Lv, W.; Wang, Y. Nano-metal-organic-frameworks for treating H(2)O(2)-Secreting bacteria alleviate pulmonary injury and prevent systemic sepsis. Biomaterials 2021, 279, 121237. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, K.; Liu, Z.; Zhang, Y.; Chen, Z.; Sun, H.; Ren, J.; Qu, X. Corrigendum to “Activation of biologically relevant levels of reactive oxygen species by Au/g-C(3)N(4) hybrid nanozyme for bacteria killing and wound disinfection” [Biomaterials, 113(2017)145-157]. Biomaterials 2020, 233, 119754. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, X.; Jia, Z.; Huo, D.; Liu, Y.; Liu, J. Cationic chitosan@Ruthenium dioxide hybrid nanozymes for photothermal therapy enhancing ROS-mediated eradicating multidrug resistant bacterial infection. J. Colloid Interface Sci. 2021, 603, 615–632. [Google Scholar] [CrossRef]
- Guo, Q.; Luo, Y.; Guo, H.; Lan, T.; Wang, S.; Geng, K.; Lu, X.; Tao, L.; Shen, X. A photo-thermal nanocomposite capable of relieving inflammatory response to compete multidrug-resistant pseudomonas aeruginosa infection. Chem. Eng. J. 2022, 446, 137173. [Google Scholar] [CrossRef]
- Biagi, M.; Butler, D.; Tan, X.; Qasmieh, S.; Wenzler, E. A Breath of Fresh Air in the Fog of Antimicrobial Resistance: Inhaled Polymyxins for Gram-Negative Pneumonia. Antibiotics 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L. Inhaled antibiotic therapy: What drug? What dose? What regimen? What formulation? J. Cyst. Fibros. 2002, 1 (Suppl. 2), 189–193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Quon, B.S.; Goss, C.H.; Ramsey, B.W. Inhaled antibiotics for lower airway infections. Ann. Am. Thorac. Soc. 2014, 11, 425–434. [Google Scholar] [CrossRef]
- Ye, M.; Zhao, Y.; Wang, Y.; Yodsanit, N.; Xie, R.; Gong, S. pH-Responsive Polymer–Drug Conjugate: An Effective Strategy to Combat the Antimicrobial Resistance. Adv. Funct. Mater. 2020, 30, 2002655. [Google Scholar] [CrossRef]
- Champion, J.A.; Walker, A.; Mitragotri, S. Role of particle size in phagocytosis of polymeric microspheres. Pharm. Res. 2008, 25, 1815–1821. [Google Scholar] [CrossRef]
- Usmani, O.S.; Biddiscombe, M.F.; Underwood, S.R.; Barnes, P.J. Characterization of the generation of radiolabeled monodisperse albuterol particles using the spinning-top aerosol generator. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2004, 45, 69–73. [Google Scholar]
- Sibille, Y.; Reynolds, H.Y. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am. Rev. Respir. Dis. 1990, 141, 471–501. [Google Scholar] [CrossRef]
- van der Weide, H.; Cossío, U.; Gracia, R.; Te Welscher, Y.M.; Ten Kate, M.T.; van der Meijden, A.; Marradi, M.; Ritsema, J.A.S.; Vermeulen-de Jongh, D.M.C.; Storm, G.; et al. Therapeutic Efficacy of Novel Antimicrobial Peptide AA139-Nanomedicines in a Multidrug-Resistant Klebsiella pneumoniae Pneumonia-Septicemia Model in Rats. Antimicrob. Agents Chemother. 2020, 64, e00517–e00620. [Google Scholar] [CrossRef]
- Falciani, C.; Zevolini, F.; Brunetti, J.; Riolo, G.; Gracia, R.; Marradi, M.; Loinaz, I.; Ziemann, C.; Cossío, U.; Llop, J.; et al. Antimicrobial Peptide-Loaded Nanoparticles as Inhalation Therapy for Pseudomonas aeruginosa Infections. Int. J. Nanomed. 2020, 15, 1117–1128. [Google Scholar] [CrossRef]
- Kwon, E.; Skalak, M.; Bertucci, A.; Braun, G.; Ricci, F.; Ruoslahti, E.; Sailor, M.; Bhatia, S. Porous Silicon Nanoparticle Delivery of Tandem Peptide Anti-Infectives for the Treatment of Pseudomonas aeruginosa Lung Infections. Adv. Mater. 2017, 29, 1701527. [Google Scholar] [CrossRef] [PubMed]
- Su, F.Y.; Srinivasan, S.; Lee, B.; Chen, J.; Convertine, A.J.; West, T.E.; Ratner, D.M.; Skerrett, S.J.; Stayton, P.S. Macrophage-targeted drugamers with enzyme-cleavable linkers deliver high intracellular drug dosing and sustained drug pharmacokinetics against alveolar pulmonary infections. J. Control Release 2018, 287, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Casciaro, B.; d’Angelo, I.; Zhang, X.; Loffredo, M.R.; Conte, G.; Cappiello, F.; Quaglia, F.; Di, Y.P.; Ungaro, F.; Mangoni, M.L. Poly(lactide-co-glycolide) Nanoparticles for Prolonged Therapeutic Efficacy of Esculentin-1a-Derived Antimicrobial Peptides against Pseudomonas aeruginosa Lung Infection: In Vitro and in Vivo Studies. Biomacromolecules 2019, 20, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
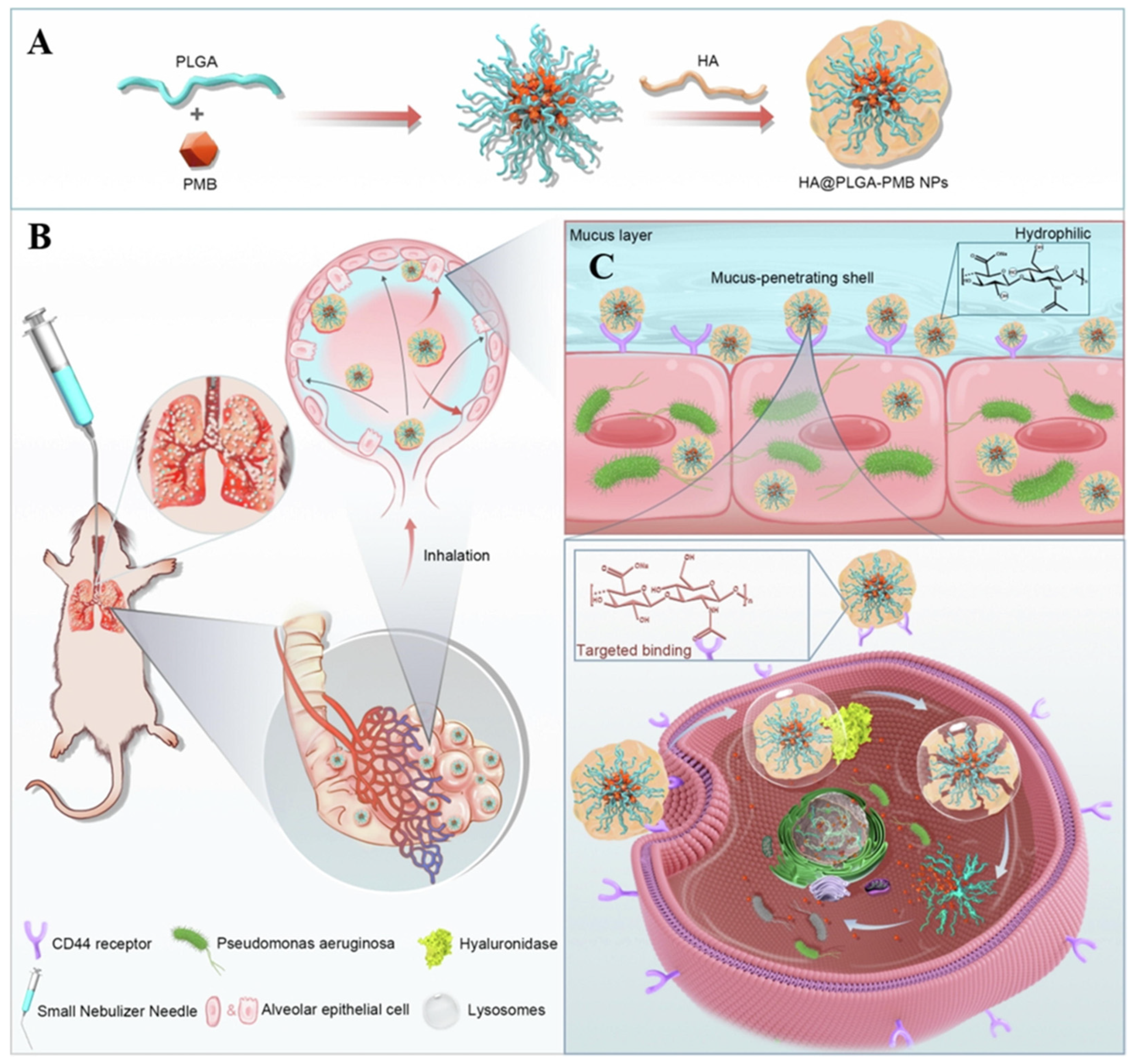
- Wu, J.; Zhai, T.; Sun, J.; Yu, Q.; Feng, Y.; Li, R.; Wang, H.; Ouyang, Q.; Yang, T.; Zhan, Q.; et al. Mucus-permeable polymyxin B-hyaluronic acid/ poly (lactic-co-glycolic acid) nanoparticle platform for the nebulized treatment of lung infections. J. Colloid Interface Sci. 2022, 624, 307–319. [Google Scholar] [CrossRef]
- Ren, H.-M.; Han, L.; Zhang, L.; Zhao, Y.-Q.; Lei, C.; Xiu, Z.; Zhao, N.; Yu, B.; Zhou, F.; Duan, S.; et al. Inhalable responsive polysaccharide-based antibiotic delivery nanoparticles to overcome mucus barrier for lung infection treatment. Nano Today 2022, 44, 101489. [Google Scholar] [CrossRef]
- Yang, J.; Duan, S.; Ye, H.; Ge, C.; Piao, C.; Chen, Y.; Lee, M.; Yin, L. Pro-Peptide-Reinforced, Mucus-Penetrating Pulmonary siRNA Delivery Mitigates Cytokine Storm in Pneumonia. Adv. Funct. Mater. 2021, 31, 2008960. [Google Scholar] [CrossRef]
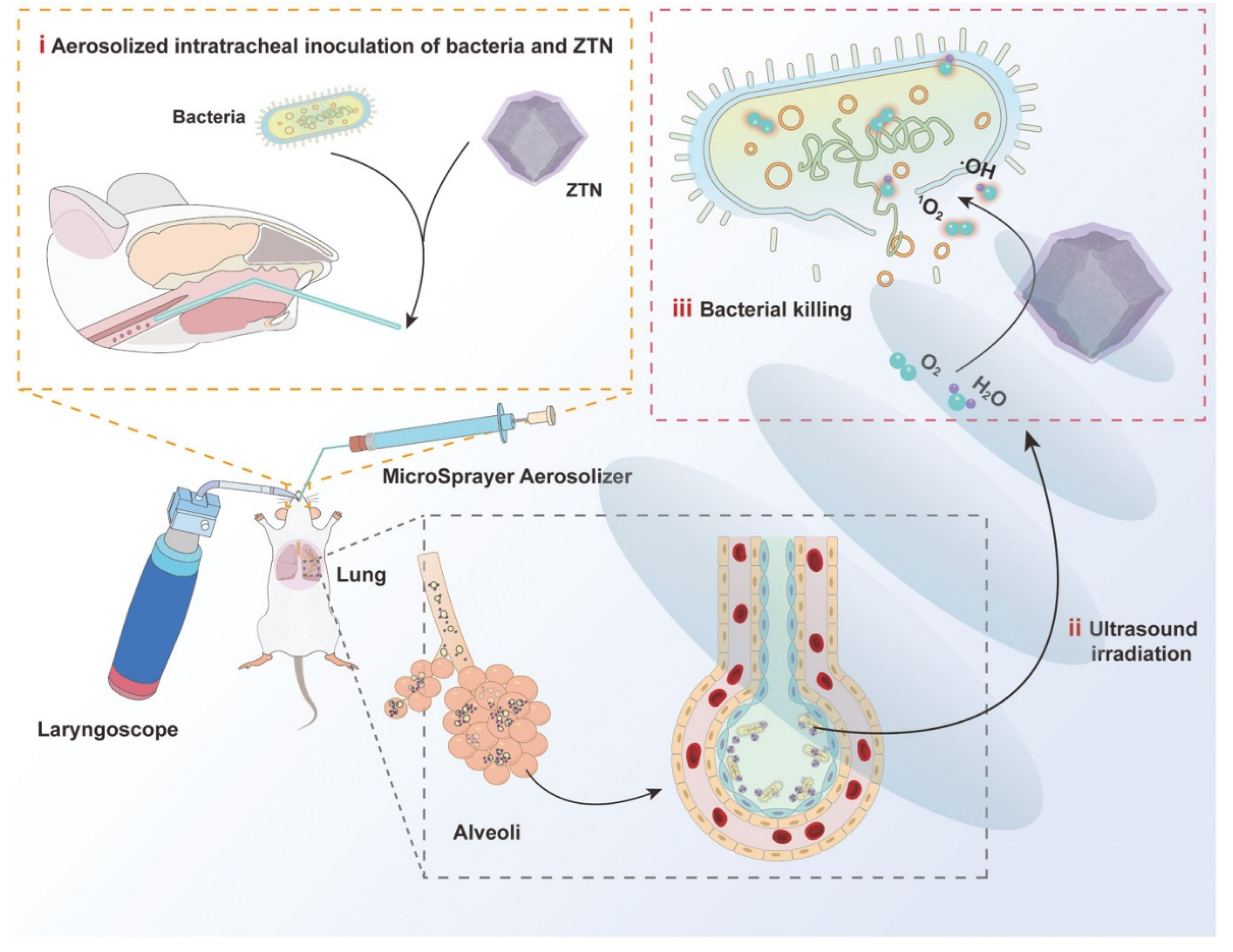
- Pan, X.; Wu, N.; Tian, S.; Guo, J.; Wang, C.; Sun, Y.; Huang, Z.; Chen, F.; Wu, Q.; Jing, Y.; et al. Inhalable MOF-Derived Nanoparticles for Sonodynamic Therapy of Bacterial Pneumonia. Adv. Funct. Mater. 2022, 32, 2112145. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Ouyang, Q.; Li, Y.; Zhang, P.; Jin, W.; Qu, S.; Yang, F.; He, Z.; Qin, M. Nanomaterials for Delivering Antibiotics in the Therapy of Pneumonia. Int. J. Mol. Sci. 2022, 23, 15738. https://doi.org/10.3390/ijms232415738
Tang J, Ouyang Q, Li Y, Zhang P, Jin W, Qu S, Yang F, He Z, Qin M. Nanomaterials for Delivering Antibiotics in the Therapy of Pneumonia. International Journal of Molecular Sciences. 2022; 23(24):15738. https://doi.org/10.3390/ijms232415738
Chicago/Turabian StyleTang, Jie, Qiuhong Ouyang, Yanyan Li, Peisen Zhang, Weihua Jin, Shuang Qu, Fengmei Yang, Zhanlong He, and Meng Qin. 2022. "Nanomaterials for Delivering Antibiotics in the Therapy of Pneumonia" International Journal of Molecular Sciences 23, no. 24: 15738. https://doi.org/10.3390/ijms232415738
APA StyleTang, J., Ouyang, Q., Li, Y., Zhang, P., Jin, W., Qu, S., Yang, F., He, Z., & Qin, M. (2022). Nanomaterials for Delivering Antibiotics in the Therapy of Pneumonia. International Journal of Molecular Sciences, 23(24), 15738. https://doi.org/10.3390/ijms232415738