
Genomic Determinants Potentially Associated with Clinical Manifestations of Human-Pathogenic Tick-Borne Flaviviruses

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
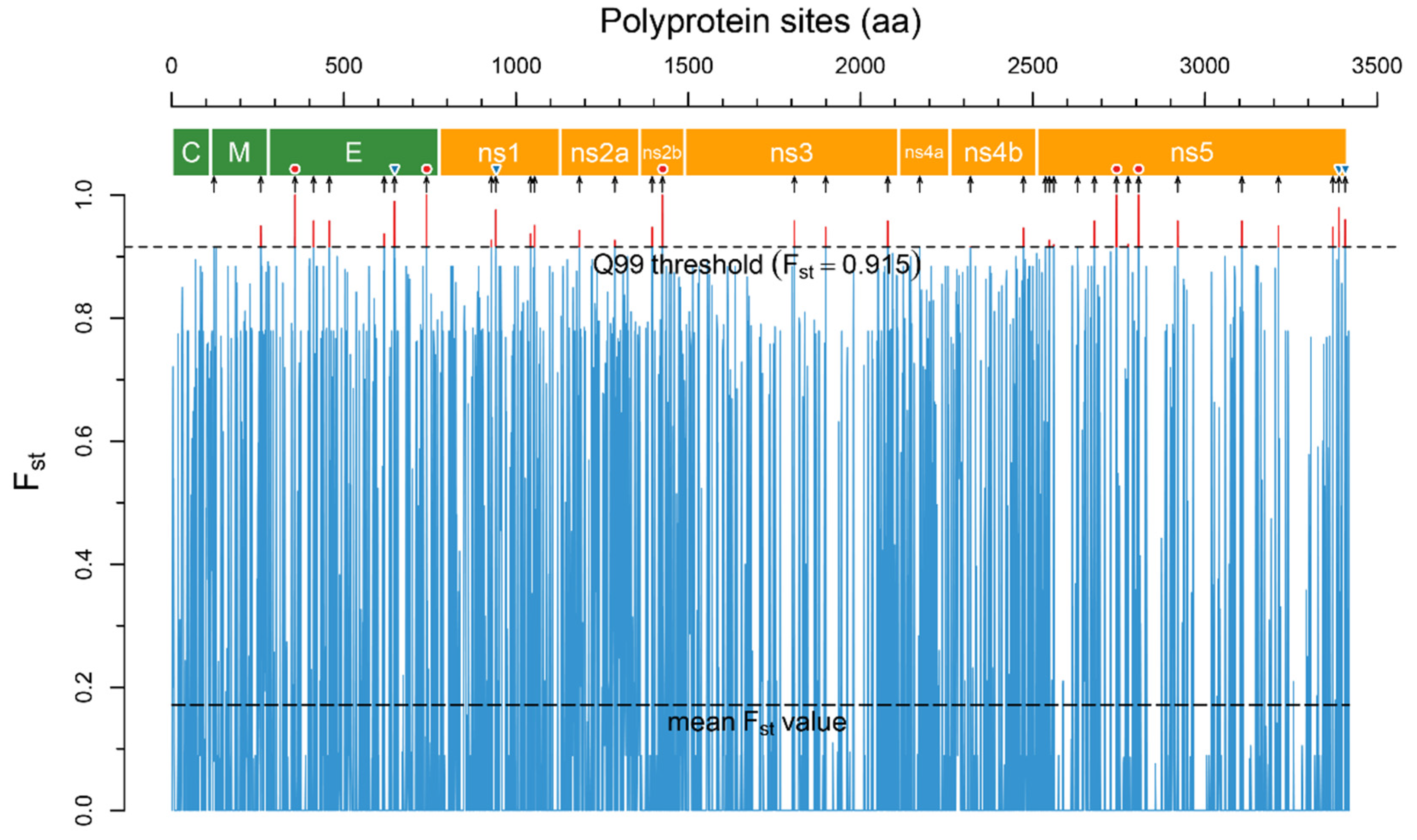
2.1. Molecular Determinants of Clinical Manifestations
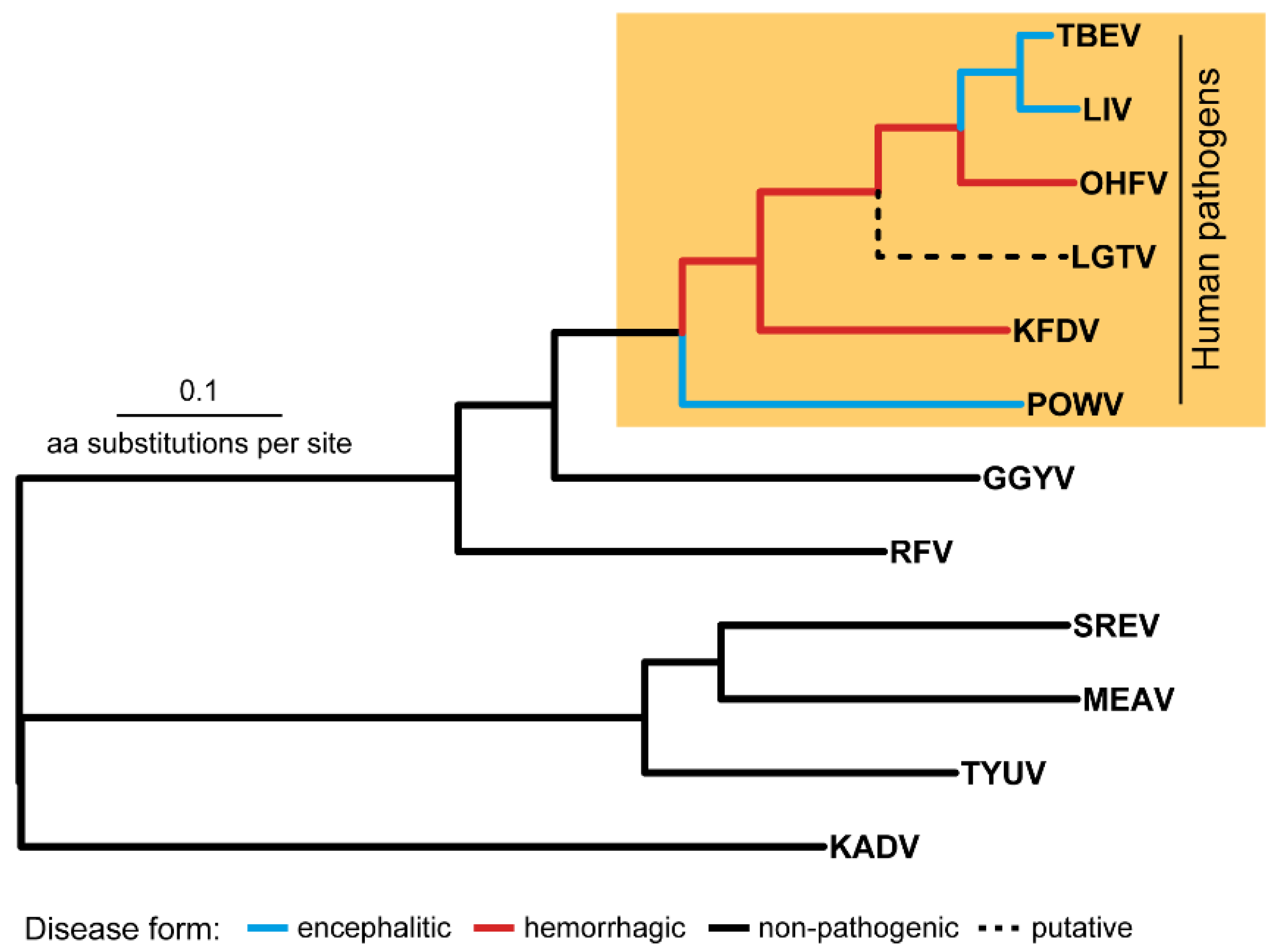
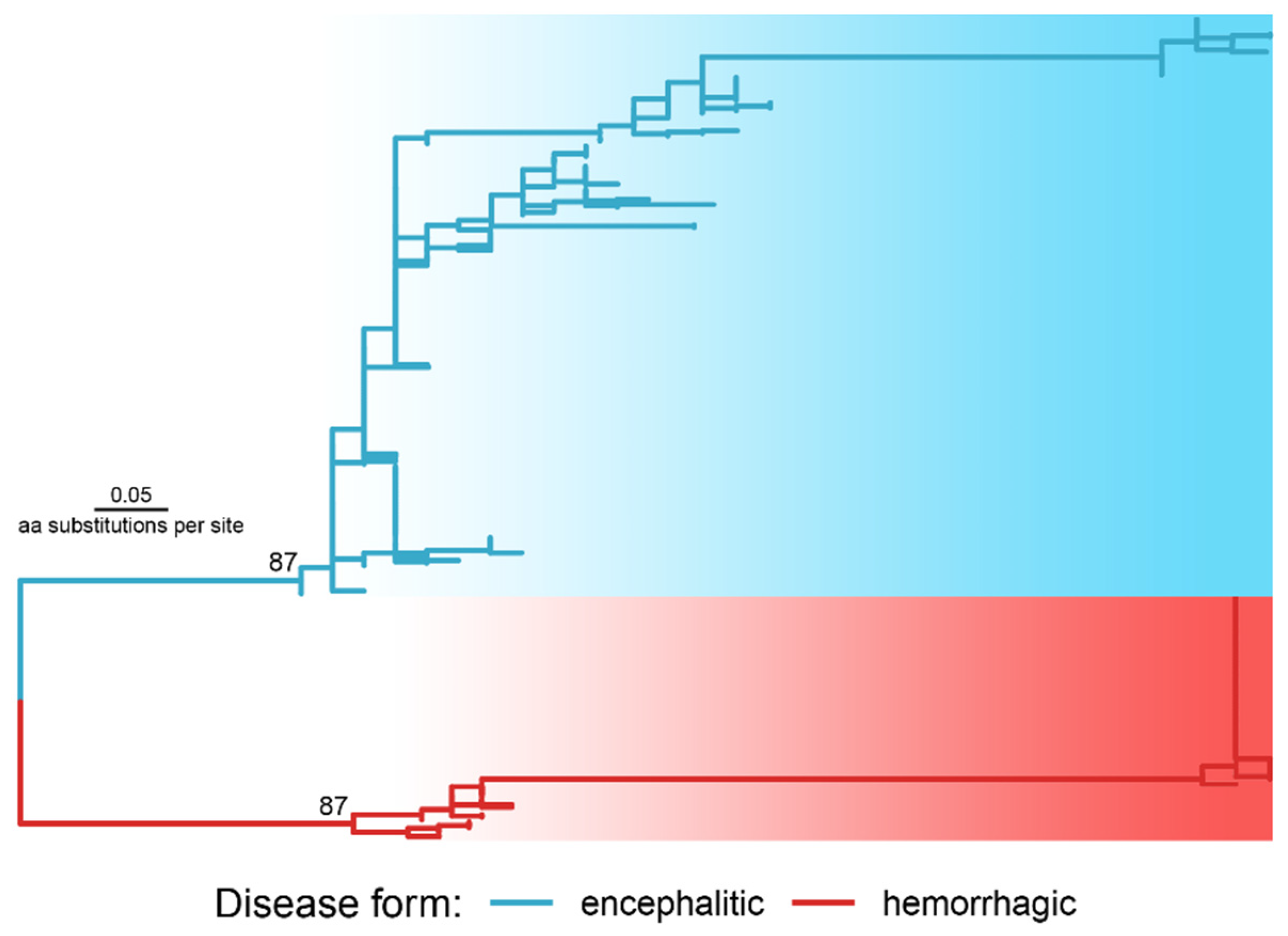
2.2. Phylogenetic Proof
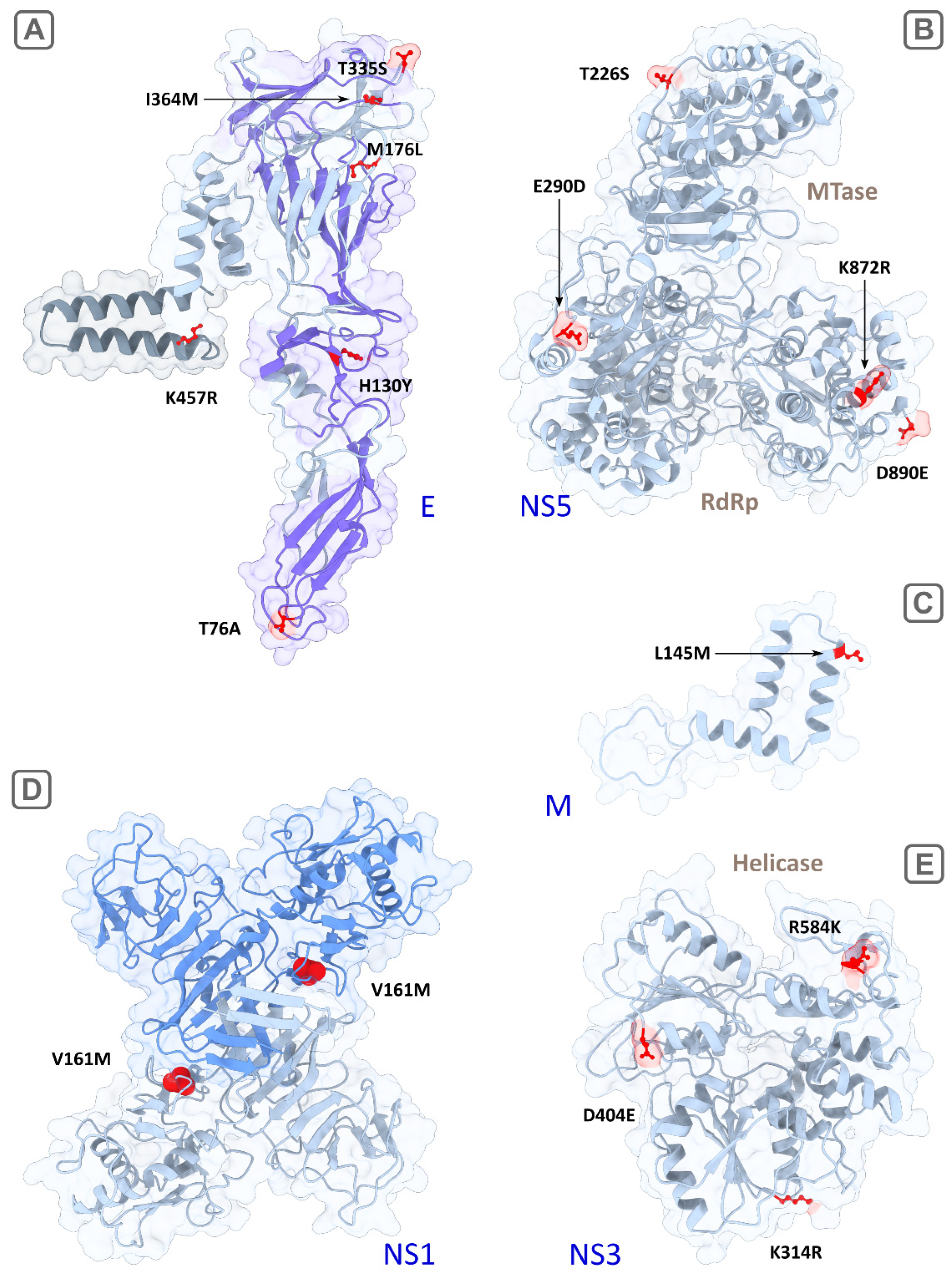
2.3. Reconstruction and Visualisation of Atomic Structures
3. Discussion
3.1. Predicted Determinants in the Reconstructed Structures
3.1.1. M Protein
3.1.2. E Protein
3.1.3. NS1 Protein
3.1.4. NS2b Protein
3.1.5. NS3 Protein
3.1.6. NS5 Protein
3.2. Possible Influence of Vector/Host Specificity
3.3. Absolutely Specific Determinants Indicate LGTV Neurovirulence
3.4. The Role of Point Amino Acid Substitutions and Potential for Further Molecular Dynamics Simulations and Animal Testing
4. Materials and Methods
4.1. Protein Sequences

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Number of Sequences | Disease Form 1 | Invertebrate Hosts | Vertebrate Hosts |
|---|---|---|---|---|
| KFDV | 54 | Hem | Haemaphysalis spinigera [1] | Monkeys, small mammals, bats [54] |
| AHFV 2 | 21 | Hem | Ornithodoros savignyi, Hyalomma dromedarii | Sheep [55] |
| OHFV | 21 | Hem | Dermacentor reticulatus [56], Ixodes persulcatus [57] | Microtus gregalis, Ondatra zibethicus [58,59] |
| POWV | 23 | Enc | I. cookei, I. marxi, I. scapularis [43], H. longicornis [41] | Peromyscus leucopus, Odocoileus virginianus, Tamiasciurus hudsonicus [43] |
| LIV | 26 | Enc | I. ricinus | Lagopus lagopus scotica, sheep [44] |
| TBEV 3 | 178 | Enc | Ixodes spp., D. reticulatus [38], H. spp. [39,40] | numerous mammal and bird species [42] (p. 57) |
4.2. Search Algorithm for Genetic Determinants
- Obtaining an aa substitution-rate matrix based on the universal model JTT [61], normalized in the range from 0 to the maximum value. In the original JTT model, substitution weights are changing from −5 (most common substitutions) to 5 (most rare substitutions). Substitutions with a weight of −5 were assigned as 1, substitutions with a weight of 5 were assigned as 10, and rest was converted according to this range of values. Gaps (indels) with the highest weight 11 were additionally added to the weight matrix; Applying of JTT matrix of substitution weights allowed us to estimate differences in substitutions` significance for adaptive transformations due to different physical-chemical properties of residues (mutations which led to significant changes in aa properties is rare).
- For each position in the alignment, a matrix of pairwise evolutionary distances was calculated. If the aa residues in the two compared sequences at a given position matched, then the pairwise distance was 0; if the aa residues did not match, the distance was taken as a weight of the aa substitution from the transformed JTT matrix. Based on the matrix of pairwise evolutionary distances for each position, the average intragroup Hw and intergroup Hb distances (for the “encephalitic” and “hemorrhagic” groups) were calculated. Based on Hw и Hb, the Fst criterion (fixation index) [9] was calculated, showing the degree of intergroup differentiation according to Formula (1):Values Fst range from 0 to 1, values close to 0 indicate the absence of intergroup subdivision, values close to 1-high subdivision. If Hw ≥ Hb or there were no substitutions in a particular position, then Fst was assigned 0.
- 3.
- A bootstrap analysis was used to verify estimated Fst, according to the following scheme: from each group (“encephalitic” and “hemorrhagic”) of polyprotein sequences, a replica was selected from 96 random sequences with a return (according to the smallest sample size of viruses that cause hemorrhagic fevers). For each position of each replica, Fst was calculated. The procedure was repeated 2000 times. Thus, 2000 Fst values were obtained for each aligned position. The probability of the null hypothesis-no differentiation was calculated using the formula:where P–the probability of the null hypothesis (p-value), n–the number of replicas with Fst = 0. If p > 0.05 then Fst value was replaced by 0 (no differentiation). For further analysis, the average value of Fst from 2000 bootstrap replicas was taken for each position.
- 4.
- Finally, Fst values for all positions were ranged in the ascending order from 0 to 1 with a step of 0.01. The quantile (Q) of the largest Fst values (excluding Q0) was selected with the formation of new datasets (subsets) from the alignment, with the highest Fst values. From 100 obtained subsets, each next subset (ascending) contained fewer alignment positions, but with higher Fst values and increasing mean differentiation between groups (encephalitis and hemorrhagic). For each of 100 subsets, a phylogenetic tree was constructed using the UPGMA method using the JTT distance matrix. The structure of each tree was analyzed visually. The subset with the minimum quantile of the ranked Fst was selected, in which the tree was divided into two monophyletic clusters, one of which included only species that cause hemorrhagic fevers, and the other cluster included only encephalitis.
- 5.
- Thus, selected subset of data was considered the candidate dataset to search determinants of different clinical manifestations of virus manifestation. For the statistical assessment of the tree topology, we performed additional phylogenetic analysis in IQTREE v.1.6.12 [62] with the ultrafast bootstrap support [63] and model selection using ModelFinder [64] implemented in IQTREE.
4.3. Reconstruction, Visualization, and Analysis of 3D Models of Protein Molecules
5. Conclusions
- the E protein where the most of determinants lie on the front sheet of the virion surface and one–in the transmembrane region. These sites take part in virus budding and membrane fusion which in total can affect cell tropism;
- non-structural proteins NS1, NS3 and NS5 which provide intracellular persistence of viruses [18] while mutations in them facilitate changes in a tropism to various tissues at the intracellular level and immune response;
- the NS5 protein with determinants located on the inter domain interface and at the regions near active sites.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, J.; Hu, Z.; Deng, F.; Shen, S. Tick-Borne Viruses. Virol. Sin. 2018, 33, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Gritsun, T.S.; Lashkevich, V.A.; Gould, E.A. Tick-borne encephalitis. Antivir. Res. 2003, 57, 129–146. [Google Scholar] [CrossRef]
- Grard, G.; Moureau, G.; Charrel, R.N.; Lemasson, J.J.; Gonzalez, J.P.; Gallian, P.; Gritsun, T.S.; Holmes, E.C.; Gould, E.A.; de Lamballerie, X. Genetic characterization of tick-borne flaviviruses: New insights into evolution, pathogenetic determinants and taxonomy. Virology 2007, 361, 80–92. [Google Scholar] [CrossRef]
- Bondaryuk, A.N.; Andaev, E.I.; Dzhioev, Y.P.; Zlobin, V.I.; Tkachev, S.E.; Kozlova, I.V.; Bukin, Y.S. Delimitation of the tick-borne flaviviruses. Resolving the tick-borne encephalitis virus and louping-ill virus paraphyletic taxa. Mol. Phylogenet. Evol. 2022, 169, 107411. [Google Scholar] [CrossRef]
- Heinze, D.M.; Gould, E.A.; Forrester, N.L. Revisiting the clinal concept of evolution and dispersal for the tick-borne flaviviruses by using phylogenetic and biogeographic analyses. J. Virol. 2012, 86, 8663–8671. [Google Scholar] [CrossRef]
- Moureau, G.; Cook, S.; Lemey, P.; Nougairede, A.; Forrester, N.L.; Khasnatinov, M.; Charrel, R.N.; Firth, A.E.; Gould, E.A.; de Lamballerie, X. New insights into flavivirus evolution, taxonomy and biogeographic history, extended by analysis of canonical and alternative coding sequences. PLoS ONE 2015, 10, e0117849. [Google Scholar] [CrossRef]
- Halliburton, R. Introduction to Population Genetics; Pearson/Prentice Hall: Upper Saddle River, NJ, USA, 2004. [Google Scholar]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of levels of gene flow from DNA sequence data. Genetics 1992, 132, 583–589. [Google Scholar] [CrossRef]
- Arenas, M. Trends in substitution models of molecular evolution. Front. Genet. 2015, 6, 319. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef]
- Luo, D.; Wei, N.; Doan, D.N.; Paradkar, P.N.; Chong, Y.; Davidson, A.D.; Kotaka, M.; Lescar, J.; Vasudevan, S.G. Flexibility between the protease and helicase domains of the dengue virus NS3 protein conferred by the linker region and its functional implications. J. Biol. Chem. 2010, 285, 18817–18827. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Gong, P. Crystal Structure of the full-length Japanese encephalitis virus NS5 reveals a conserved methyltransferase-polymerase interface. PLoS Pathog. 2013, 9, e1003549. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Song, H.; Qi, J.; Liu, Y.; Wang, H.; Su, C.; Shi, Y.; Gao, G.F. Contribution of intertwined loop to membrane association revealed by Zika virus full-length NS1 structure. EMBO J. 2016, 35, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Li, L.; Dick, D.; Shope, R.E.; Feldmann, H.; Barrett, A.D.T.; Holbrook, M.R. Analysis of the complete genome of the tick-borne flavivirus Omsk hemorrhagic fever virus. Virology 2003, 313, 81–90. [Google Scholar] [CrossRef]
- Růžek, D.; Yoshii, K.; Bloom, M.E.; Gould, E.A. Virology. In The TBE Book, 5th ed.; Dobler, G., Erber, W., Bröker, M., Schmitt, H.J., Eds.; Global Health Press: Singapore, 2022. [Google Scholar]
- Pangerl, K.; Heinz, F.X.; Stiasny, K. Mutational analysis of the zippering reaction during flavivirus membrane fusion. J. Virol. 2011, 85, 8495–8501. [Google Scholar] [CrossRef]
- Barnard, T.R.; Abram, Q.H.; Lin, Q.F.; Wang, A.B.; Sagan, S.M. Molecular Determinants of Flavivirus Virion Assembly. Trends Biochem. Sci. 2021, 46, 378–390. [Google Scholar] [CrossRef]
- Kaufmann, B.; Rossmann, M.G. Molecular mechanisms involved in the early steps of flavivirus cell entry. Microbes Infect. 2011, 13, 1–9. [Google Scholar] [CrossRef]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef]
- Trowbridge, J.M.; Gallo, R.L. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology 2002, 12, 117R–125R. [Google Scholar] [CrossRef]
- Khoo, U.S.; Chan, K.Y.; Chan, V.S.; Lin, C.L. DC-SIGN and L-SIGN: The SIGNs for infection. J. Mol. Med. 2008, 86, 861–874. [Google Scholar] [CrossRef]
- Kim, S.Y.; Li, B.; Linhardt, R.J. Pathogenesis and Inhibition of Flaviviruses from a Carbohydrate Perspective. Pharmaceuticals 2017, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Westlake, D.; Bielefeldt-Ohmann, H.; Prow, N.A.A.; Hall, R.A.A. Novel Flavivirus Attenuation Markers Identified in the Envelope Protein of Alfuy Virus. Viruses 2021, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zheng, H.; Tong, W.; Li, G.; Wang, T.; Li, L.; Gao, F.; Shan, T.; Yu, H.; Zhou, Y.; et al. Acidity/Alkalinity of Japanese Encephalitis Virus E Protein Residue 138 Alters Neurovirulence in Mice. J. Virol. 2018, 92, e00108-18. [Google Scholar] [CrossRef] [PubMed]
- Mandl, C.W.; Kroschewski, H.; Allison, S.L.; Kofler, R.; Holzmann, H.; Meixner, T.; Heinz, F.X. Adaptation of tick-borne encephalitis virus to BHK-21 cells results in the formation of multiple heparan sulfate binding sites in the envelope protein and attenuation in vivo. J. Virol. 2001, 75, 5627–5637. [Google Scholar] [CrossRef]
- Lee, E.; Lobigs, M. Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray Valley encephalitis virus. J. Virol. 2002, 76, 4901–4911. [Google Scholar] [CrossRef]
- Carbaugh, D.L.; Lazear, H.M. Flavivirus Envelope Protein Glycosylation: Impacts on Viral Infection and Pathogenesis. J. Virol. 2020, 94, e00104-20. [Google Scholar] [CrossRef]
- Hu, T.; Wu, Z.; Wu, S.; Chen, S.; Cheng, A. The key amino acids of E protein involved in early flavivirus infection: Viral entry. Virol. J. 2021, 18, 136. [Google Scholar] [CrossRef]
- Op De Beeck, A.; Molenkamp, R.; Caron, M.; Ben Younes, A.; Bredenbeek, P.; Dubuisson, J. Role of the transmembrane domains of prM and E proteins in the formation of yellow fever virus envelope. J. Virol. 2003, 77, 813–820. [Google Scholar] [CrossRef]
- Muller, D.A.; Young, P.R. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antivir. Res. 2013, 98, 192–208. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Dutta, S.; Konwerski, J.; Jose, J.; Jurkiw, T.J.; DelProposto, J.; Ogata, C.M.; Skiniotis, G.; Kuhn, R.J.; et al. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 2014, 343, 881–885. [Google Scholar] [CrossRef]
- Edeling, M.A.; Diamond, M.S.; Fremont, D.H. Structural basis of Flavivirus NS1 assembly and antibody recognition. Proc. Natl. Acad. Sci. USA 2014, 111, 4285–4290. [Google Scholar] [CrossRef] [PubMed]
- Potapova, U.V.; Feranchuk, S.I.; Potapov, V.V.; Kulakova, N.V.; Kondratov, I.G.; Leonova, G.N.; Belikov, S.I. NS2B/NS3 protease: Allosteric effect of mutations associated with the pathogenicity of tick-borne encephalitis virus. J. Biomol. Struct. Dyn. 2012, 30, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Thurmond, S.; Zhou, K.; Sanchez-Aparicio, M.T.; Fang, J.; Lu, J.; Gao, L.; Ren, W.; Cui, Y.; Veit, E.C.; et al. Structural basis for STAT2 suppression by flavivirus NS5. Nat. Struct. Mol. Biol. 2020, 27, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Ciota, A.T.; Kramer, L.D. Insights into arbovirus evolution and adaptation from experimental studies. Viruses 2010, 2, 2594–2617. [Google Scholar] [CrossRef]
- Lickova, M.; Fumacova Havlikova, S.; Slavikova, M.; Slovak, M.; Drexler, J.F.; Klempa, B. Dermacentor reticulatus is a vector of tick-borne encephalitis virus. Ticks Tick Borne Dis. 2020, 11, 101414. [Google Scholar] [CrossRef]
- Abdiyeva, K.; Turebekov, N.; Yegemberdiyeva, R.; Dmitrovskiy, A.; Yeraliyeva, L.; Shapiyeva, Z.; Nurmakhanov, T.; Sansyzbayev, Y.; Froeschl, G.; Hoelscher, M.; et al. Vectors, molecular epidemiology and phylogeny of TBEV in Kazakhstan and central Asia. Parasit. Vectors 2020, 13, 504. [Google Scholar] [CrossRef]
- Yun, S.M.; Song, B.G.; Choi, W.; Park, W.I.; Kim, S.Y.; Roh, J.Y.; Ryou, J.; Ju, Y.R.; Park, C.; Shin, E.H. Prevalence of tick-borne encephalitis virus in ixodid ticks collected from the republic of Korea during 2011–2012. Osong Public Health Res. Perspect. 2012, 3, 213–221. [Google Scholar] [CrossRef]
- L’vov, D.K.; Al’khovskiĭ, S.V.; Shchelkanov, M.; Deriabin, P.G.; Gitel’man, A.K.; Botikov, A.G.; Aristova, V.A. Genetic characterisation of Powassan virus (POWV) isolated from Haemophysalis longicornis ticks in Primorye and two strains of Tick-borne encephalitis virus (TBEV) (Flaviviridae, Flavivirus): Alma-Arasan virus (AAV) isolated from Ixodes persulcatus ticks in Kazakhstan and Malyshevo virus isolated from Aedes vexans nipponii mosquitoes in Khabarovsk kray. Vopr. Virusol. 2014, 59, 18–22. [Google Scholar]
- Chitimia-Dobler, L.; Mackenstedt, U.; Kahl, O. Transmission/natural cycle. In The TBE Book, 5th ed.; Dobler, G., Erber, W., Bröker, M., Schmitt, H.J., Eds.; Global Health Press: Singapore, 2022. [Google Scholar]
- Hermance, M.E.; Thangamani, S. Powassan Virus: An Emerging Arbovirus of Public Health Concern in North America. Vector Borne Zoonotic Dis. 2017, 17, 453–462. [Google Scholar] [CrossRef]
- Gilbert, L. Louping ill virus in the UK: A review of the hosts, transmission and ecological consequences of control. Exp. Appl. Acarol. 2016, 68, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Pletnev, A.G.; Men, R. Attenuation of the Langat tick-borne flavivirus by chimerization with mosquito-borne flavivirus dengue type 4. Proc. Natl. Acad. Sci. USA 1998, 95, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Thind, I.S.; Price, W.H. A chick embryo attenuated strain (TP21 E5) of Langat virus. II. Stability after passage in various laboratory animals and tissue cultures. Am. J. Epidemiol. 1966, 84, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Laurini, E.; Marson, D.; Aulic, S.; Fermeglia, A.; Pricl, S. Computational Mutagenesis at the SARS-CoV-2 Spike Protein/Angiotensin-Converting Enzyme 2 Binding Interface: Comparison with Experimental Evidence. ACS Nano 2021, 15, 6929–6948. [Google Scholar] [CrossRef]
- Diaz-Valle, A.; Falcon-Gonzalez, J.M.; Carrillo-Tripp, M. Hot Spots and Their Contribution to the Self-Assembly of the Viral Capsid: In Silico Prediction and Analysis. Int. J. Mol. Sci. 2019, 20, 5966. [Google Scholar] [CrossRef]
- Upfold, N.; Ross, C.; Tastan Bishop, O.; Knox, C. The In Silico Prediction of Hotspot Residues that Contribute to the Structural Stability of Subunit Interfaces of a Picornavirus Capsid. Viruses 2020, 12, 387. [Google Scholar] [CrossRef]
- Zech, F.; Schniertshauer, D.; Jung, C.; Herrmann, A.; Cordsmeier, A.; Xie, Q.; Nchioua, R.; Prelli Bozzo, C.; Volcic, M.; Koepke, L.; et al. Spike residue 403 affects binding of coronavirus spikes to human ACE2. Nat. Commun. 2021, 12, 6855. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef]
- Pattnaik, P. Kyasanur forest disease: An epidemiological view in India. Rev. Med. Virol. 2006, 16, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Abdulhaq, A.A.; Hershan, A.A.; Karunamoorthi, K.; Al-Mekhlafi, H.M. Human Alkhumra hemorrhagic Fever: Emergence, history and epidemiological and clinical profiles. Saudi J. Biol. Sci. 2022, 29, 1900–1910. [Google Scholar] [CrossRef] [PubMed]
- Gritsun, T.S.; Nuttall, P.A.; Gould, E.A. Tick-borne flaviviruses. Adv. Virus Res. 2003, 61, 317–371. [Google Scholar] [PubMed]
- Wagner, E.; Shin, A.; Tukhanova, N.; Turebekov, N.; Nurmakhanov, T.; Sutyagin, V.; Berdibekov, A.; Maikanov, N.; Lezdinsh, I.; Shapiyeva, Z.; et al. First Indications of Omsk Haemorrhagic Fever Virus beyond Russia. Viruses 2022, 14, 754. [Google Scholar] [CrossRef] [PubMed]
- Rudakov, N.V.; Yastrebov, V.K.; Yakimenko, V.V. Epidemiology of Omsk Haemorragic Fever. Epidemiol. Vaccine Prev. 2015, 14, 39–48. [Google Scholar] [CrossRef]
- Růžek, D.; Holbrook, M.R.; Yakimenko, V.V.; Karan, L.S.; Tkachev, S.E. Omsk Hemorrhagic Fever Virus. In Manual of Security Sensitive Microbes and Toxins; Liu, D., Ed.; CRC Press: Boca Raton, FL, USA, 2014; p. 884. [Google Scholar]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–232. [Google Scholar]
- Pele, J.; Becu, J.M.; Abdi, H.; Chabbert, M. Bios2mds: An R package for comparing orthologous protein families by metric multidimensional scaling. BMC Bioinform. 2012, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Mathura, V.S.; Kolippakkam, D. APDbase: Amino acid Physico-chemical properties Database. Bioinformation 2005, 1, 2–4. [Google Scholar] [CrossRef]




| Protein | Position 1 | Residue 2 | Mean Fst | Domain | Note | |
|---|---|---|---|---|---|---|
| Enc(enc/hem,%) | Hem(hem/enc,%) | |||||
| M | 9 | K(87/22) | R(78/13) | 0.916 | N-terminus | |
| 145 | L(98/0) | M(98/2) | 0.950 | transmembrane region | ||
| E | 76 3 | T(100/0) | A(100/0) | 1.000 | bc loop, domain II | front sheet 4 |
| 130 | H(88/16) | Y(84/12) | 0.958 | e strand, domain II | front sheet | |
| 176 | M(78/22) | L(78/22) | 0.958 | G0H0 loop, domain I | back sheet | |
| 335 | T(77/22) | S(78/22) | 0.937 | BCx loop, domain III | front sheet | |
| 364 | I(100/1) | M(99/0) | 0.989 | DxE loop, domain III | front sheet | |
| 457 | K(100/0) | R(100/0) | 1.000 | transmembrane region | ||
| NS1 | 148 | R(92/0) | K(100/8) | 0.926 | “wing” domain | |
| 161 | V(99/0) | M(99/0) | 0.976 | “wing” domain | ||
| 262 | S(84/22) | A(78/16) | 0.937 | C-terminal domain | antibody binding region | |
| 274 | I(80/22) | L(78/19) | 0.950 | C-terminal domain | ||
| NS2a | 52 | R(62/0) | T(100/0) | 0.943 | ||
| 155 | L(90/17) | Y(78/0) | 0.926 | |||
| NS2b | 33 | V(89/8) | A(92/0) | 0.947 | ||
| 63 | E(99.4/0) | D(100/0) | 0.99 | |||
| NS3 | 314 | K(89/15) | R(85/11) | 0.958 | helicase domain | motif III |
| 404 | D(77/22) | E(78/22) | 0.947 | helicase domain | motif V | |
| 584 | R(96/8) | K(92/4) | 0.958 | helicase domain | ||
| NS4a | 56 | M(87/22) | V(78/13) | 0.916 | ||
| NS4b | 54 | I(86/22) | M(78/14) | 0.916 | ||
| 208 | L(100/0) | V(80/0) | 0.947 | |||
| NS5 | 20 | K(68/24) | R(76/32) | 0.916 | MT domain | near the GTO binding site |
| 31 | I(90/18) | V(82/10) | 0.926 | MT domain | near the GTO binding site | |
| 44 | R(96/7) | K(93/3) | 0.919 | MT domain | ||
| 113 | K(84/7) | R(93/16) | 0.916 | MT domain | near the active MT site | |
| 162 | K(75/22) | R(78/25) | 0.958 | MT domain | near the active MT site | |
| 226 | T(100/0) | S(100/0) | 1.000 | MT domain | near the RNA binding site 219 | |
| 260 | V(82/22) | T(78/14) | 0.920 | MT domain | ||
| 290 | E(99.6/0) | D(100/0.4) | 1.000 | extension structure | ||
| 404 | K(78/22) | R(78/22) | 0.958 | fingers subdomain | ||
| 590 | I(80/22) | V(78/20) | 0.958 | palm subdomain | ||
| 696 | H(78/22) | P(78/22) | 0.950 | inter-domain interface | binding the STAT2 protein | |
| 854 | K(96/0) | R(100/4) | 0.947 | thumb subdomains | ||
| 872 | K(96/4) | R(96/4) | 0.979 | thumb subdomains | ||
| 890 | D(99/0) | E(100/0) | 0.960 | thumb subdomains | ||
| Protein in the Strain SofjinKSY | Closely Related Atomic Structure from PDB | Similarity Degree between SofjinKSY and PDB Structure (%) | Structural Region Length (aa) | Coordinates of a Structural Region in SofjinKSY | Coordinates of a Structural Region in a Polyprotein |
|---|---|---|---|---|---|
| preM | 7qrf 1 | 96.88 | 79 | 6–84 | 118–196 |
| M | 7z51 2 | 88.00 | 74 | 94–167 | 206–279 |
| E | 7z51 | 95.36 | 494 | 1–494 | 281–774 |
| NS1 | 5gs6 3 | 42.12 | 351 | 2–352 | 778–1128 |
| NS2a | - 4 | - | - | - | - |
| NS2b | - | - | - | - | - |
| NS3 | 2whx 5 | 45.75 | 599 | 23–621 | 1512–2110 |
| NS4a | - | - | - | - | - |
| NS4b | - | - | - | - | - |
| NS5 | 4k6m 6 | 56.58 | 887 | 5–891 | 2516–3402 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondaryuk, A.N.; Kulakova, N.V.; Potapova, U.V.; Belykh, O.I.; Yudinceva, A.V.; Bukin, Y.S. Genomic Determinants Potentially Associated with Clinical Manifestations of Human-Pathogenic Tick-Borne Flaviviruses. Int. J. Mol. Sci. 2022, 23, 13404. https://doi.org/10.3390/ijms232113404
Bondaryuk AN, Kulakova NV, Potapova UV, Belykh OI, Yudinceva AV, Bukin YS. Genomic Determinants Potentially Associated with Clinical Manifestations of Human-Pathogenic Tick-Borne Flaviviruses. International Journal of Molecular Sciences. 2022; 23(21):13404. https://doi.org/10.3390/ijms232113404
Chicago/Turabian StyleBondaryuk, Artem N., Nina V. Kulakova, Ulyana V. Potapova, Olga I. Belykh, Anzhelika V. Yudinceva, and Yurij S. Bukin. 2022. "Genomic Determinants Potentially Associated with Clinical Manifestations of Human-Pathogenic Tick-Borne Flaviviruses" International Journal of Molecular Sciences 23, no. 21: 13404. https://doi.org/10.3390/ijms232113404
APA StyleBondaryuk, A. N., Kulakova, N. V., Potapova, U. V., Belykh, O. I., Yudinceva, A. V., & Bukin, Y. S. (2022). Genomic Determinants Potentially Associated with Clinical Manifestations of Human-Pathogenic Tick-Borne Flaviviruses. International Journal of Molecular Sciences, 23(21), 13404. https://doi.org/10.3390/ijms232113404