
Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 2: Nitrogen-Coordinated Complexes of Cobalt




Abstract
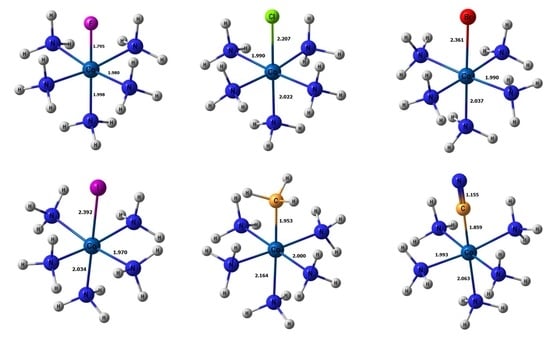
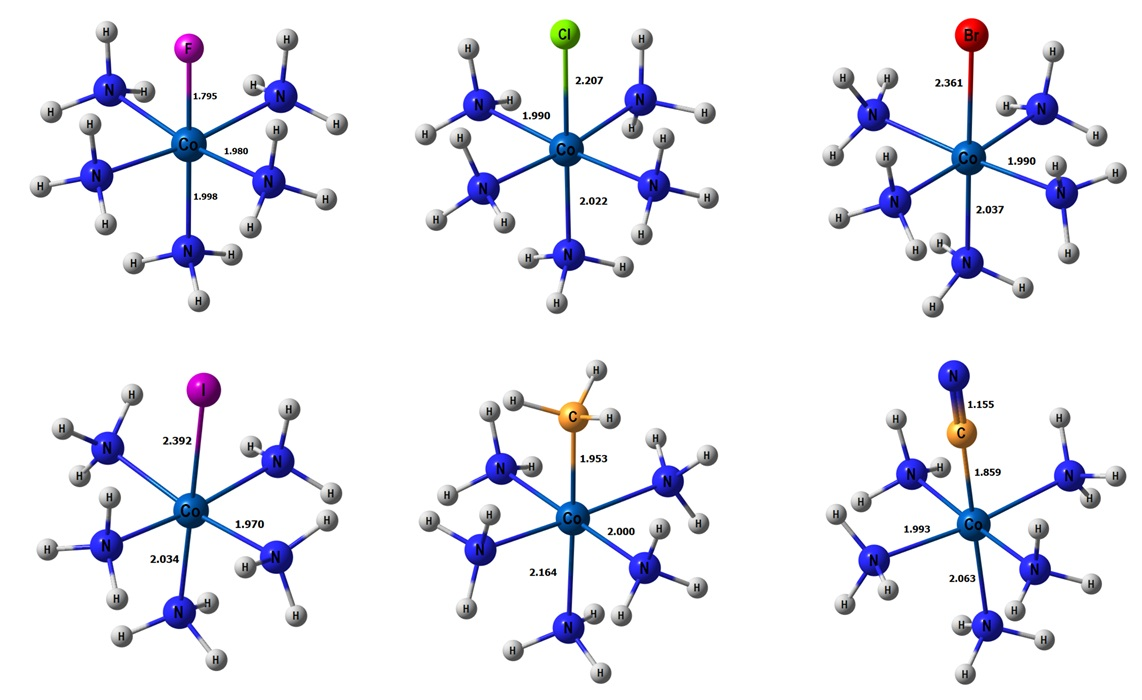
1. Introduction
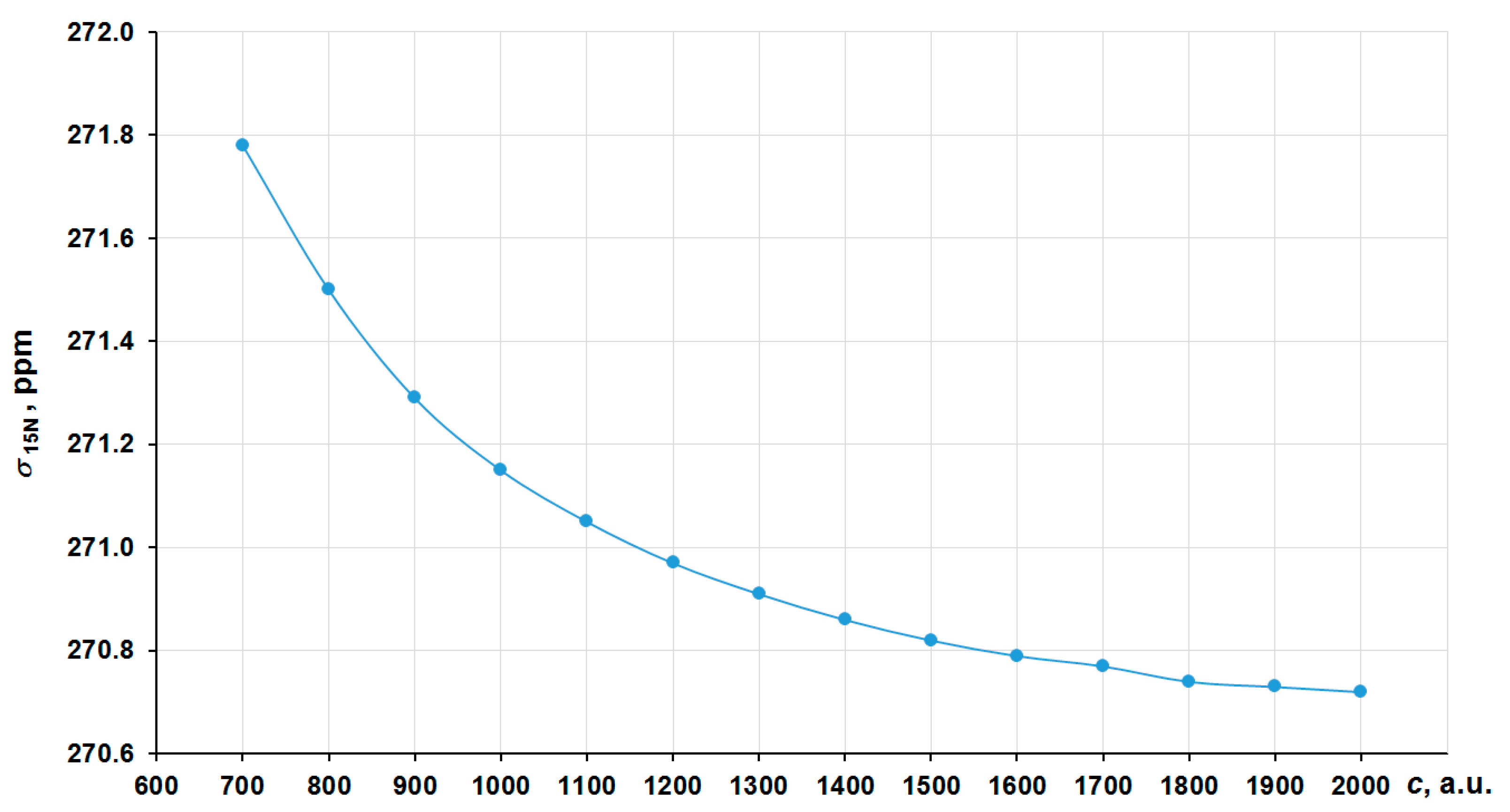
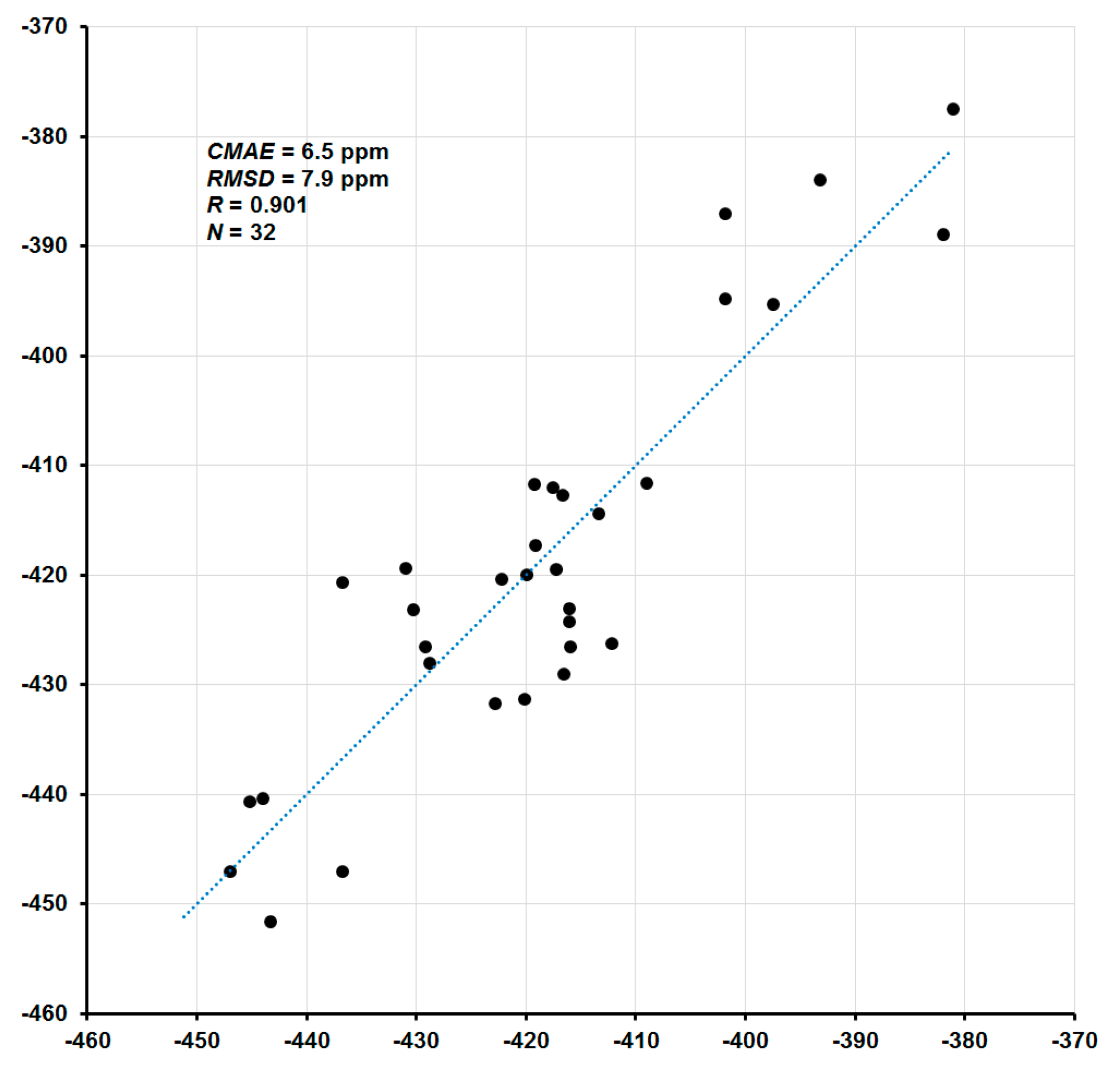
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeşilela, O.Z.; Mutlu, A.; Darcan, C.; Büyükgüngör, O. Syntheses, structural characterization and antimicrobial activities of novel cobalt-pyrazine-2,3-dicarboxylate complexes with N-donor ligands. J. Mol. Struct. 2010, 964, 39–46. [Google Scholar] [CrossRef]
- Mishra, A.; Kaushik, N.K.; Verma, A.K.; Gupta, R. Synthesis, characterization and antibacterial activity of cobalt(III) complexes with pyridine–amide ligands. Eur. J. Med. Chem. 2008, 43, 2189–2196. [Google Scholar] [CrossRef] [PubMed]
- Shalash, A.M.; Abu Ali, H.I. Synthesis, crystallographic, spectroscopic studies and biological activity of new cobalt(II) complexes with bioactive mixed sulindac and nitrogen-donor ligands. Chem. Cent. J. 2017, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Miodragović, D.U.; Bogdanović, G.A.; Miodragović, Z.M.; Radulović, M.D.; Novaković, S.B.; Kaluderović, G.N.; Kozłowski, H. Interesting coordination abilities of antiulcer drug famotidine and antimicrobial activity of drug and its cobalt(III) complex. J. Inorg. Biochem. 2006, 100, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.-Y.; Xu, M.; Zhu, C.-G.; Wei, Y.-J.; Wang, F.-W. Synthesis, Structures, and Antimicrobial Studies of Cobalt(III) Complexes with Tridentate Schiff Bases. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2012, 42, 109–114. [Google Scholar] [CrossRef]
- Dorkova, P.; Pantcheva, I.N.; Sheldrick, W.S.; Mayer-Figge, H.; Petrova, R.; Mitewa, M. Synthesis, structure and antimicrobial activity of manganese(II) and cobalt(II) complexes of the polyether ionophore antibiotic Sodium Monensin A. J. Inorg. Biochem. 2008, 102, 26–32. [Google Scholar] [CrossRef]
- Witanowski, M.; Webb, G.A. Nitrogen NMR, 1st ed.; Plenum Press: New York, NY, USA, 1973. [Google Scholar] [CrossRef]
- Chan, J.C.C.; Au-Yeung, S.C.F. Cobalt-59 NMR spectroscopy. Ann. Rep. NMR Spectrosc. 2000, 41, 1–54. [Google Scholar] [CrossRef]
- Jensen, F. Introduction to Computational Chemistry; John Wiley and Sons Ltd.: Chichester, UK, 2007; p. 624. [Google Scholar]
- Sauer, S.P.A. Molecular Electromagnetism: A Computational Chemistry Approach; University Press: Oxford, UK, 2012; p. 322. [Google Scholar]
- Krivdin, L.B. Computational 1H and 13C NMR in structural and stereochemical studies. Magn. Reson. Chem. 2022, 60, 733–828. [Google Scholar] [CrossRef]
- Rusakova, I.L.; Rusakov, Y.Y. Quantum chemical calculations of 77Se and 125Te nuclear magnetic resonance spectral parameters and their structural applications. Magn. Reson. Chem. 2021, 59, 359–407. [Google Scholar] [CrossRef]
- Rusakova, I.L. Quantum Chemical Approaches to the Calculation of NMR Parameters: From Fundamentals to Recent Advances. Magnetochemistry 2022, 8, 50. [Google Scholar] [CrossRef]
- Samultsev, D.O.; Semenov, V.A.; Krivdin, L.B. On the accuracy of the GIAO-DFT calculation of 15N NMR chemical shifts of the nitrogen-containing heterocycles–a gateway to better agreement with experiment at lower computational cost. Magn. Reson. Chem. 2014, 52, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Semenov, V.A.; Samultsev, D.O.; Krivdin, L.B. Calculation of 15N NMR chemical shifts in a diversity of nitrogen-containing compounds using composite method approximation at the DFT, MP2, and CCSD levels. J. Phys. Chem. A 2019, 123, 8417–8426. [Google Scholar] [CrossRef] [PubMed]
- Latypov, S.K.; Kondrashova, S.A.; Polyancev, F.M.; Sinyashin, O.G. Quantum chemical calculations of 31P NMR chemical shifts in nickel complexes: Scope and limitations. Organometallics 2020, 39, 1413–1422. [Google Scholar] [CrossRef]
- Payard, P.-A.; Perego, L.A.; Grimaud, L.; Ciofini, I. A DFT protocol for the prediction of 31P NMR chemical shifts of phosphine ligands in first-row transition-metal complexes. Organometallics 2020, 39, 3121–3130. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Akman, F.; Vasilieva, N.Y.; Issaoui, N.; Malyar, Y.N.; Kondrasenko, A.A.; Borovkova, V.S.; Miroshnikova, A.V.; Kazachenko, A.S.; Al-Dossary, O.; et al. Catalytic sulfation of betulin with sulfamic acid: Experiment and DFT calculation. Int. J. Mol. Sci. 2022, 23, 1602. [Google Scholar] [CrossRef]
- Chan, J.C.C.; Au-Yeung, S.C.F.; Wilson, P.J.; Webb, G.A. SOS-DFPT-IGLO calculations of 59Co NMR shielding parameters of hexacoordinated diamagnetic Co(III) complexes. J. Mol. Struct. (THEOCHEM) 1996, 365, 125–130. [Google Scholar] [CrossRef]
- Malkin, V.G.; Malkina, O.L.; Salahub, D.R. Calculations of NMR shielding constants by uncoupled density functional theory. Chem. Phys. Lett. 1993, 204, 80–86. [Google Scholar] [CrossRef]
- Kofod, P. 14N NMR of amminecobalt(III) compounds. Magn. Reson. Chem. 2003, 41, 531–534. [Google Scholar] [CrossRef]
- Fung, B.M.; Wei, S.C. Nitrogen-14 Chemical Shifts in Cobalt(III) Complexes. J. Magn. Reson. 1970, 3, 1–4. [Google Scholar] [CrossRef]
- Odaka, Y.; Yamasaki, A.; Watabe, M. 15N NMR spectra of cobalt(III) complexes containing 15NO2-. Bull. Chem. Soc. Jpn. 1985, 58, 3407–3408. [Google Scholar] [CrossRef]
- Yoshiaki, N.; Masayuki, M.; Keiichi, K.; Yoshimasa, K.; Yuzo, Y. Nitrogen-15 NMR Chemical Shifts in Metal–Ammine Complexes. II. Cobalt(III) Ammine and Amine Complexes. Bull. Chem. Soc. Jpn. 1989, 62, 2455–2460. [Google Scholar] [CrossRef]
- Samultsev, D.O.; Semenov, V.A.; Krivdin, L.B. Four-component relativistic calculations of NMR shielding constants of the transition metal complexes. Part 1: Pentaammines of cobalt, rhodium and iridium. Magn. Reson. Chem. 2022, 60, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Nomura, Y.; Takeuchi, Y. Substituent effects in aromatic proton nmr spectra. III substituent effects caused by halogens. Tetrahedron Lett. 1969, 10, 639–642. [Google Scholar] [CrossRef]
- Schreckenbach, G.; Ziegler, T. Density functional calculations of NMR chemical shifts and ESR g-tensors. Theor. Chem. Acc. 1998, 99, 71–82. [Google Scholar] [CrossRef]
- Bühl, M.; Kaupp, M.; Malkina, O.L.; Malkin, V.G. The DFT route to NMR chemical shifts. J. Comput. Chem. 1999, 20, 91–105. [Google Scholar] [CrossRef]
- Chan, J.C.C.; Au-Yeung, S.C.F. A comparative study of the calculation of 59Co NMR shielding constants of hexacoordinated diamagnetic Co(III) complexes using DFT-IGLO and DFT-GIAO methods. J. Mol. Struct. (THEOCHEM) 1997, 393, 93–96. [Google Scholar] [CrossRef]
- Chan, J.C.C.; Au-Yeung, S.C.F. Density Functional Study of 59Co Chemical Shielding Tensors Using Gauge-Including Atomic Orbitals. J. Phys. Chem. A 1997, 101, 3637–3640. [Google Scholar] [CrossRef]
- Godbout, N.; Oldfield, E. Density Functional Study of Cobalt-59 Nuclear Magnetic Resonance Chemical Shifts and Shielding Tensor Elements in Co(III) Complexes. J. Am. Chem. Soc. 1997, 119, 8065–8069. [Google Scholar] [CrossRef]
- Xu, X.-P.; Au-Yeung, S.C.F. A DFT and 59Co Solid-State NMR Study of the Chemical Shielding Property and Electronic Interaction in the Metalloporphyrin System. J. Am. Chem. Soc. 2000, 122, 6468–6475. [Google Scholar] [CrossRef]
- Grigoleit, S.; Bühl, M. Computational 59Co NMR Spectroscopy: Beyond Static Molecules. J. Chem. Theory Comput. 2005, 1, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Ooms, K.J.; Bernard, G.M.; Kadziola, A.; Kofod, P.; Wasylishen, R.E. Solid-state 13C and 59Co NMR spectroscopy of 13C-methylcobalt(III) complexes with amine ligands. Phys. Chem. Chem. Phys. 2009, 11, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Senn, F.; Zlatar, M.; Gruden-Pavlovic, M.; Daul, C. Computational analysis of tris(1,2-ethanediamine) cobalt(III) complex ion: Calculation of the 59Co shielding tensor using LF-DFT. Monatsh. Chem.–Chem. Month. 2011, 142, 593–597. [Google Scholar] [CrossRef]
- Kutzelnigg, W. Theory of magnetic susceptibilities and NMR chemical shifts in terms of localized quantities. Isr. J. Chem. 1980, 19, 193–200. [Google Scholar] [CrossRef]
- Schindler, M.; Kutzelnigg, W. Theory of magnetic susceptibilities and NMR chemical shifts in terms of localized quantities. II. Application to some simple molecules. J. Chem. Phys. 1982, 76, 1919–1933. [Google Scholar] [CrossRef]
- Ditchfield, R. Molecular orbital theory of magnetic shielding and magnetic susceptibility. J. Chem. Phys. 1972, 56, 5688–5693. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. I. A gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Malli, G.; Froese, C. Nuclear magnetic shielding constants calculated from numerical Hartree-Fock wave functions. Int. J. Quantum. Chem. 1967, 1, 95–98. [Google Scholar] [CrossRef]
- Wolff, S.K.; Ziegler, T.; van Lenthe, E.; Baerends, E.J. Density functional calculations of nuclear magnetic shieldings using the zeroth-order regular approximation (ZORA) for relativistic effects: ZORA nuclear magnetic resonance. J. Chem. Phys. 1999, 110, 7689–7698. [Google Scholar] [CrossRef]
- Dyall, K.G. Relativistic and nonrelativistic finite nucleus optimized double zeta basis sets for the 4p, 5p and 6p elements. Theor. Chem. Acc. 1998, 99, 366–371. [Google Scholar] [CrossRef]
- Dyall, K.G. Relativistic and nonrelativistic finite nucleus optimized triple-zeta basis sets for the 4p, 5p and 6p elements. Theor. Chem. Acc. 2002, 108, 335–340. [Google Scholar] [CrossRef]
- Jensen, F. Basis set convergence of nuclear magnetic shielding constants calculated by density functional methods. J. Chem. Theory Comp. 2008, 4, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Jensen, F. Polarization consistent basis sets: Principles. J. Chem. Phys. 2001, 115, 9113–9125. [Google Scholar] [CrossRef]
- Jensen, F. Polarization consistent basis sets. II. Estimating the Kohn-Sham basis set limit. J. Chem. Phys. 2002, 116, 7372–7379. [Google Scholar] [CrossRef]
- Gauss, J.; Stanton, J.F. Electron-correlated methods for the calculation of NMRchemical shifts. In Calculation of NMR and EPR Parameters, Theory and Applications, 1st ed.; Kaupp, M., Bühl, M., Malkin, V.G., Eds.; WILEY-CH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004; Chapter 8; pp. 123–139. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Liu, W.; Kutzelnigg, W. Comparison of restricted, unrestricted, inverse, and dual kinetic balances for four-component relativistic calculations. Theor. Chem. Acc. 2011, 129, 423–436. [Google Scholar] [CrossRef]
- Visscher, L. Magnetic Balance and Explicit Diamagnetic Expressions for Nuclear Magnetic Resonance Shielding Tensors. Adv. Quantum Chem. 2005, 48, 369–381. [Google Scholar] [CrossRef]
- Olejniczak, M.; Bast, R.; Saue, T.; Pecul, M. A simple scheme for magnetic balance in four-component relativistic Kohn−Sham calculations of nuclear magnetic resonance shielding constants in a Gaussian basis. J. Chem. Phys. 2012, 136, 014108–014111. [Google Scholar] [CrossRef]
- Iliaš, M.; Saue, T.; Enevoldsen, T.; Jensen, H.J.A. Gauge origin independent calculations of nuclear magnetic shieldings in relativistic four-component theory. J. Chem. Phys. 2009, 131, 124119. [Google Scholar] [CrossRef]
- Visscher, L.; Aerts, J.C.; Visser, O.; Nieuwpoort, W.C. Kinetic balance in contracted basis sets for relativistic calculations. Int. J. Quantum Chem. 1991, 40, 131–139. [Google Scholar] [CrossRef]
- Hartman, J.D.; Monaco, S.; Schatschneider, B.; Beran, G.J.O. Fragment-based 13C nuclear magnetic resonance chemical shift predictions in molecular crystals: An alternative to planewavemethods. J. Chem. Phys. 2015, 143, 102809. [Google Scholar] [CrossRef]
- Rusakov, Y.Y.; Rusakova, I.L. What Most Affects the Accuracy of 125Te NMR Chemical Shift Calculations. J. Phys. Chem. A 2020, 124, 6714–6725. [Google Scholar] [CrossRef]
- Semenov, V.A.; Samultsev, D.O.; Krivdin, L.B. The 1H and 13C NMR chemical shifts of strychnos alkaloids revisited at the DFT level. Magn. Reson. Chem. 2020, 58, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Vícha, J.; Novotný, J.; Komorovsky, S.; Straka, M.; Kaupp, M.; Marek, R. Relativistic Heavy-Neighbor-Atom Effects on NMR Shifts: Concepts and Trends Across the Periodic Table. Chem. Rev. 2020, 120, 7065–7103. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, M.; Malkina, O.L.; Malkin, V.G.; Pyykkö, P. How Do Spin-Orbit-Induced Heavy-Atom Effects on NMR Chemical Shifts Function? Validation of a Simple Analogy to Spin-Spin Coupling by Density Functional Theory (DFT) Calculations on Some Iodo Compounds. Chem. Eur. J. 1998, 4, 118–126. [Google Scholar] [CrossRef]
- Vícha, J.; Komorovsky, S.; Repisky, M.; Marek, R.; Straka, M. Relativistic Spin−Orbit Heavy Atom on the Light Atom NMR Chemical Shifts: General Trends Across the Periodic Table Explained. J. Chem. Theory Comput. 2018, 14, 3025–3039. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.K.; Ziegler, T. Calculation of DFT-GIAO NMR Shifts with the Inclusion of Spin-Orbit Coupling. J. Chem. Phys. 1998, 109, 895–905. [Google Scholar] [CrossRef]
- Sutter, K.; Autschbach, J. Computational Study and Molecular Orbital Analysis of NMR Shielding, Spin−Spin Coupling, and Electric Field Gradients of Azido Platinum Complexes. J. Am. Chem. Soc. 2012, 134, 13374–13385. [Google Scholar] [CrossRef]
- Vícha, J.; Straka, M.; Munzarová, M.L.; Marek, R. Mechanism of Spin−Orbit Effects on the Ligand NMR Chemical Shift in Transition-Metal Complexes: Linking NMR to EPR. J. Chem. Theory Comput. 2014, 10, 1489–1499. [Google Scholar] [CrossRef]
- Vícha, J.; Foroutan-Nejad, C.; Pawlak, T.; Munzarová, M.L.; Straka, M.; Marek, R. Understanding the Electronic Factors Responsible for Ligand Spin−Orbit NMR Shielding in Transition-Metal Complexes. J. Chem. Theory Comput. 2015, 11, 1509–1517. [Google Scholar] [CrossRef]
- Vícha, J.; Marek, R.; Straka, M. High-Frequency 1H NMR Chemical Shifts of SnII and PbII Hydrides Induced by Relativistic Effects: Quest for PbII Hydrides. Inorg. Chem. 2016, 55, 10302–10309. [Google Scholar] [CrossRef]
- Smiles, D.E.; Wu, G.; Hrobarik, P.; Hayton, T.W. Use of 77Se and 125Te NMR Spectroscopy to Probe Covalency of the ActinideChalcogen Bonding in [Th(En){N(SiMe3)2}3]− (E = Se, Te; N = 1,2) and Their Oxo-Uranium(VI) Congeners. J. Am. Chem. Soc. 2016, 138, 814–825. [Google Scholar] [CrossRef]
- Novotný, J.; Vícha, J.; Bora, P.L.; Repisky, M.; Straka, M.; Komorovsky, S.; Marek, R. Linking the Character of the Metal–Ligand Bond to the Ligand NMR Shielding in Transition-Metal Complexes: NMR Contributions from Spin–Orbit Coupling. J. Chem. Theory Comput. 2017, 13, 3586–3601. [Google Scholar] [CrossRef] [PubMed]
- Hrobárik, P.; Hrobárikova, V.; Meier, F.; Repiský, M.; Komorovský, S.; Kaupp, M. Relativistic Four-Component DFT Calculations of 1H NMR Chemical Shifts in Transition-Metal Hydride Complexes: Unusual High-Field Shifts Beyond the Buckingham-Stephens Model. J. Phys. Chem. A 2011, 115, 5654–5659. [Google Scholar] [CrossRef] [PubMed]
- Hrobárik, P.; Hrobárikova, V.; Greif, A.H.; Kaupp, M. Giant Spin-Orbit Effects on NMR Shifts in Diamagnetic Actinide Complexes: Guiding the Search of Uranium(VI) Hydride Complexes in the Correct Spectral Range. Angew. Chem. Int. Ed. 2012, 51, 10884–10888. [Google Scholar] [CrossRef] [PubMed]
- Greif, A.H.; Hrobárik, P.; Kaupp, M. Insights into trans-Ligand and Spin-Orbit Effects on Electronic Structure and Ligand NMR Shifts in Transition-Metal Complexes. Chem. Eur. J. 2017, 23, 9790–9803. [Google Scholar] [CrossRef] [PubMed]
- Greif, A.H.; Hrobárik, P.; Hrobárikova, V.; Arbuznikov, A.V.; Autschbach, J.; Kaupp, M. A Relativistic Quantum-Chemical Analysis of the trans Influence on 1H NMR Hydride Shifts in Square-Planar Platinum(II) Complexes. Inorg. Chem. 2015, 54, 7199–7208. [Google Scholar] [CrossRef] [PubMed]
- Bagno, A.; Saielli, G. Relativistic DFT calculations of the NMR properties and reactivity of transition metal methane σ-complexes: Insights on C–H bond activation. Phys. Chem. Chem. Phys. 2011, 13, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Morales, Y.; Schreckenbach, G.; Ziegler, T. Origin of the Hydridic 1H NMR Chemical Shift in Low-Valent Transition-Metal Hydrides. Organometallics 1996, 15, 3920–3923. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; de Menezes, S.M.C.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties andconventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 795–1818. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; de Menezes, S.M.C.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008). Pure Appl. Chem. 2008, 80, 59–84. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B. GAUSSIAN 09; Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009; Available online: http://www.gaussian.com (accessed on 1 September 2022).
- Semenov, V.A.; Samultsev, D.O.; Rusakova, I.L.; Krivdin, L.B. Computational multinuclear NMR of platinum complexes: A relativistic four-component study. J. Phys. Chem. A 2019, 123, 4908–4920. [Google Scholar] [CrossRef]
- Jensen, H.J.A.; Bast, R.; Saue, T.; Visscher, L. DIRAC 16. Revision 2016. Available online: http://www.diracprogram.org (accessed on 1 September 2022).
- Mao, Y.; Horn, P.R.; Mardirossian, N.; Head-Gordon, T.; Skylaris, C.-K.; Head-Gordon, M. Approaching the basis set limit for DFT calculations using an environment-adapted minimal basis with perturbation theory: Formulation, proof of concept, and a pilot implementation. J. Chem. Phys. 2016, 145, 044109. [Google Scholar] [CrossRef] [PubMed]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef] [PubMed]
- Lutnæs, O.B.; Helgaker, T.; Jaszuński, M. Spin–spin coupling constants and triplet instabilities in Kohn–Sham theory. Mol. Phys. 2010, 108, 2579–2590. [Google Scholar] [CrossRef]
- Labanowski, J.K.; Andxelm, J.W. Density Function Methods in Chemistry; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Adamo, C.; Barone, V. Toward chemical accuracy in the computation of NMR shieldings: The PBE0 model. Chem. Phys. Lett. 1998, 298, 113–119. [Google Scholar] [CrossRef]
- de Giovanetti, M.; Bitencourt, L.F.F.; Cormanich, R.; Sauer, S.P.A. On the Unexpected Accuracy of the M06L Functional in the Calculation of 1JFC Spin–Spin Coupling Constants. J. Chem. Thery Comput. 2021, 17, 7712–7723. [Google Scholar] [CrossRef]
- Manninen, P.; Lantto, P.; Vaara, J.; Ruud, K. Perturbational ab initio calculations of relativistic contributions to nuclear magnetic resonance shielding tensors. J. Chem. Phys. 2003, 119, 2623–2637. [Google Scholar] [CrossRef]
- Manninen, P.; Ruud, K.; Lantto, P.; Vaara, J. Leading-order relativistic effects on nuclear magnetic resonance shielding tensors. J. Chem. Phys. 2005, 122, 114107. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd. | Formula | σGP | Δsolv | Δrel | σtot | δtot1 | δexp2 |
|---|---|---|---|---|---|---|---|
| trans | |||||||
| 1 | [Co(NH3)5F]2+ | 283.8 | 3.2 | 3.8 | 290.8 | −426.2 | −451.2 |
| 2 | [Co(NH3)5Cl]2+ | 271.6 | −11.7 | 11.1 | 271.0 | −408.2 | −434.9 |
| 3 | [Co(NH3)5Br]2+ | 266.2 | −2.8 | 13.7 | 277.1 | −413.8 | - |
| 4 | [Co(NH3)5I]2+ | 256.5 | 1.6 | 15.3 | 273.4 | −410.4 | - |
| 5 | [Co(NH3)5CH3]2+ | 249.5 | 1.7 | −1.6 | 249.6 | −388.9 | −382.0 |
| 6 | [Co(NH3)5CN]2+ | 257.1 | 2.2 | −11.8 | 247.5 | −387.0 | −401.8 |
| [Co(NH3)5CN]2+ | −174.8 | 63.2 | 10.0 | −101.6 | −70.9 | - | |
| 7 | [Co(NH3)5NO2]2+ | 273.0 | 3.5 | 12.2 | 288.7 | −424.3 | −416.1 |
| [Co(NH3)5NO2]2+ | −239.0 | −34.6 | 65.0 | −208.7 | 26.0 | - | |
| 8 | [Co(NH3)5OH]2+ | 273.5 | 2.7 | 15.0 | 291.2 | −426.6 | −429.2 |
| 9 | [Co(NH3)5N3]2+ | 269.6 | 5.1 | 12.8 | 287.5 | −423.2 | −430.3 |
| [Co(NH3)5(N=N=N)]2+ | 210.6 | 2.6 | −11.3 | 201.9 | −345.7 | - | |
| [Co(NH3)5(N=N=N)]2+ | −17.1 | −5.5 | −0.7 | −23.4 | −141.7 | - | |
| [Co(NH3)5(N=N=N)]2+ | 33.0 | 50.2 | −5.9 | 77.3 | −232.8 | - | |
| 10 | [Co(NH3)5CO3]+ | 270.0 | 9.0 | 0.4 | 279.4 | −415.9 | −437.5 |
| 11 | [Co(NH3)5H2O]3+ | 298.2 | −1.5 | 17.1 | 313.8 | −447.0 | −447.0 |
| 12 | cis-[Co(NH3)4(NO2)2]+ | 269.8 | 4.2 | 17.2 | 291.2 | −426.5 | −416.0 |
| cis-[Co(NH3)4(NO2)2]+ | −254.0 | −25.3 | 61.2 | −218.1 | 34.6 | - | |
| 13 | cis-[Co(NH3)4CO3]+ | 267.1 | 6.0 | 11.6 | 284.7 | −420.6 | −436.7 |
| 14 | cis-[Co(NH3)4(H2O)Cl]+ to H2O | 282.9 | −0.3 | 24.2 | 306.8 | −440.7 | −445.2 |
| cis-[Co(NH3)4(H2O)Cl]+ to Cl | 262.7 | 4.8 | 15.8 | 283.3 | −419.4 | −431.0 | |
| 15 | cis-[Co(NH3)4(H2O)2]3+ | 287.4 | −1.2 | 20.3 | 306.5 | −440.4 | −444.0 |
| 16 | mer-[Co(NH3)3(NO2)3]3+ | 264.7 | 6.2 | 20.0 | 290.9 | −426.2 | −412.2 |
| mer-[Co(NH3)3(NO2)3]3+ to NO2− | −290.2 | −6.6 | 38.9 | −257.9 | 70.6 | - | |
| mer-[Co(NH3)3(NO2)3]3+ to NH3 | −262.9 | −18.7 | 51.2 | −230.4 | 45.7 | - | |
| 17 | fac-[Co(NH3)3(NO2)3]3+ | 245.8 | 1.9 | 12.9 | 260.6 | −398.8 | - |
| fac-[Co(NH3)3(NO2)3]3+ | −282.6 | −18.6 | 49.9 | −251.3 | 64.6 | - | |
| 18 | mer-[Co(NH3)3(H2O)3]3+ | 281.8 | −1.2 | 38.3 | 318.9 | −451.6 | −443.3 |
| 19 | fac-[Co(NH3)3(H2O)3]3+ | 287.4 | −1.5 | 28.5 | 314.4 | −447.5 | - |
| 20 | cis-[Co(NH3)2(NO2)4]− | 232.8 | 2.4 | 12.4 | 247.6 | −387.1 | - |
| cis-[Co(NH3)2(NO2)4]− to NO2− | −303.8 | −4.1 | 41.3 | −266.6 | 78.5 | - | |
| cis-[Co(NH3)2(NO2)4]− to NH3 | −279.7 | −15.3 | 44.0 | −251.0 | 64.4 | - | |
| 21 | cis-[Co(NH3)2(CO3)2]− | 254.7 | 8.5 | 50.6 | 313.8 | −447.0 | −436.7 |
| 22 | cis,mer-[Co(NH3)2(NO2)3CH3]− to NO2− | 238.4 | 5.2 | 31.5 | 275.1 | −411.9 | - |
| cis,mer-[Co(NH3)2(NO2)3CH3]− to CH3 | 226.9 | 1.2 | −10.9 | 217.2 | −359.5 | - | |
| cis,mer-[Co(NH3)2(NO2)3CH3]− to NO2 | −337.2 | −7.4 | 73.8 | −270.8 | 82.3 | - | |
| cis,mer-[Co(NH3)2(NO2)3CH3]− to NH3 | −304.2 | −20.4 | 43.8 | −280.8 | 91.3 | - | |
| 23 | cis,fac-[Co(NH3)2(NO2)3CH3]− | 240.5 | 5.5 | 18.3 | 264.3 | −402.2 | - |
| cis,fac-[Co(NH3)2(NO2)3CH3]− to CH3 | −334.8 | −12.4 | 17.0 | −330.2 | 136.1 | - | |
| cis,fac-[Co(NH3)2(NO2)3CH3]− to NH3 | −305.9 | −19.0 | 44.4 | −280.5 | 91.1 | - | |
| 24 | trans,mer-[Co(NH3)2(NO2)3CH3]− to NO2− | −322.0 | −7.3 | 39.3 | −290.0 | 99.7 | - |
| trans,mer-[Co(NH3)2(NO2)3CH3]− to CH3 | −318.8 | −9.1 | 17.8 | −310.1 | 117.9 | - | |
| cis | |||||||
| 1 | [Co(NH3)5F]2+ | 256.1 | −1.2 | 20.3 | 275.2 | −412.0 | −417.5 |
| 2 | [Co(NH3)5Cl]2+ | 260.3 | −0.4 | 24.1 | 284 | −420.0 | −419.9 |
| 3 | [Co(NH3)5Br]2+ | 264.0 | 10.4 | 25.5 | 299.9 | −434.4 | - |
| 4 | [Co(NH3)5I]2+ | 258.1 | 3.9 | 24.2 | 286.2 | −422.0 | - |
| 5 | [Co(NH3)5CH3]2+ | 268.9 | −0.3 | 24.3 | 292.9 | −428.1 | −428.8 |
| 6 | [Co(NH3)5CN]2+ | 271.0 | 1.0 | −19.2 | 252.8 | −391.8 | −425.9 |
| 7 | [Co(NH3)5NO2]2+ | 261.3 | 3.9 | 22.2 | 287.4 | −423.1 | −416.1 |
| 8 | [Co(NH3)5OH]2+ | 255.7 | −1.0 | 20.1 | 274.8 | −411.7 | −419.2 |
| 9 | [Co(NH3)5N3]2+ | 261.0 | 0.9 | 22.5 | 284.4 | −420.4 | −422.2 |
| 10 | [Co(NH3)5CO3]+ | 254.1 | 0.3 | 26.6 | 281.0 | −417.3 | −419.1 |
| 11 | [Co(NH3)5H2O]3+ | 271.0 | −1.3 | 26.8 | 296.5 | −431.3 | −420.1 |
| 12 | cis-[Co(NH3)4(NO2)2]+ | 239.1 | 2.3 | 14.7 | 256.1 | −394.7 | −401.8 |
| 13 | cis-[Co(NH3)4CO3]+ | 256.8 | −1.3 | 20.4 | 275.9 | −412.7 | −416.7 |
| 14 | cis-[Co(NH3)4(H2O)Cl]+ | 260.4 | −0.9 | 23.9 | 283.4 | −419.5 | −417.2 |
| 15 | cis-[Co(NH3)4(H2O)2]3+ | 266.4 | −1.1 | 28.7 | 294.0 | −429.1 | −416.6 |
| 16 | mer-[Co(NH3)3(NO2)3]3+ | 228.4 | 2.1 | 13.7 | 244.2 | −384.0 | −393.2 |
| 18 | mer-[Co(NH3)3(H2O)3]3+ | 261.9 | −1.4 | 17.3 | 277.8 | −414.4 | −413.4 |
| 24 | trans,mer-[Co(NH3)2(NO2)3CH3]− | 237.6 | 2.6 | 16.5 | 256.7 | −395.3 | −397.5 |
| 25 | [Co(NH3)6]3+ | 271.7 | −0.9 | 26.1 | 296.9 | −431.7 | −422.8 |
| 26 | trans-[Co(NH3)4(NO2)2]+ | 251.9 | 2.5 | 20.3 | 274.7 | −411.6 | −409.0 |
| trans-[Co(NH3)4(NO2)2]+ | −279.2 | −9.6 | 34.1 | −254.7 | 67.8 | - | |
| 27 | trans-[Co(NH3)2(NO2)4]− | 223.9 | 0.8 | 12.3 | 237.0 | −377.5 | −381.1 |
| trans-[Co(NH3)2(NO2)4]− | −291.2 | −1.6 | 40.1 | −252.7 | 65.9 | - | |
| Cmpd. | Formula | σnr | σrel | Δrelσ 1 | Δsolvσ 2 | σtot 3 | δnr 4 | δrel 5 | Δrelδ 6 | Δsolvδ 7 | δtot 8 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | [Co(NH3)5F]2+ | −10,394.4 | −10,039.7 | 354.7 | −221.1 | −15,963.4 | 3675.1 | 3550.2 | −124.8 | 230.9 | 3781.1 |
| 2 | [Co(NH3)5Cl]2+ | −14,224.8 | −13,813.3 | 411.5 | −351.9 | −15,479.7 | 7505.4 | 7323.8 | −181.6 | 361.7 | 7685.5 |
| 3 | [Co(NH3)5Br]2+ | −15,554.1 | −15,102.6 | 451.5 | −496.5 | −15,560.0 | 8834.8 | 8613.1 | −221.7 | 506.3 | 9119.4 |
| 4 | [Co(NH3)5I]2+ | −15,124.0 | −14,715.5 | 408.5 | −403.4 | −13,126.6 | 8404.7 | 8226.0 | −178.7 | 413.2 | 8639.2 |
| 5 | [Co(NH3)5CH3]2+ | −14,331.6 | −13,440.4 | 891.2 | −132.8 | −13,538.3 | 7612.3 | 6951.0 | −661.3 | 142.6 | 7093.6 |
| 6 | [Co(NH3)5CN]2+ | −12,300.9 | −11,850.6 | 450.3 | 120.1 | −13,199.3 | 5581.6 | 5361.1 | −220.4 | −110.2 | 5250.9 |
| 7 | [Co(NH3)5NO2]2+ | −15,244.7 | −14,681.2 | 563.5 | −207.4 | −14,586.5 | 8525.4 | 8191.7 | −333.6 | 217.2 | 8409.0 |
| 8 | [Co(NH3)5OH]2+ | −16,051.5 | −15,397.3 | 654.2 | −198.4 | −15,265.4 | 9332.1 | 8907.8 | −424.3 | 208.2 | 9116.0 |
| 9 | [Co(NH3)5N3]2+ | −15,959.8 | −15,200.1 | 759.7 | −279.8 | −15,103.9 | 9240.5 | 8710.7 | −529.8 | 289.6 | 9000.3 |
| 10 | [Co(NH3)5CO3]+ | −16,649.2 | −15,473.8 | 1175.4 | −180.3 | −15,008.2 | 9929.9 | 8984.4 | −945.5 | 190.1 | 9174.5 |
| 11 | [Co(NH3)5H2O]3+ | −19,128.1 | −18,236.5 | 891.6 | −274.8 | −18,393.9 | 12,408.8 | 11,747.0 | −661.8 | 284.6 | 12,031.6 |
| 12 | cis-[Co(NH3)4(NO2)2]+ | −14,130.2 | −13,662.1 | 468.1 | −209.8 | −13,621.5 | 7410.9 | 7172.7 | −238.2 | 219.7 | 7392.3 |
| 13 | cis-[Co(NH3)4CO3]+ | −15,914.4 | −15,255.3 | 659.1 | −418.3 | −15,249.1 | 9195.0 | 8765.8 | −429.3 | 428.2 | 9194.0 |
| 14 | cis-[Co(NH3)4(H2O)Cl]+ | −17,032.5 | −16,333.1 | 699.3 | −242.1 | −16,271.4 | 10,313.1 | 9843.7 | −469.5 | 251.9 | 10,095.6 |
| 15 | cis-[Co(NH3)4(H2O)2]3+ | −18,696.1 | −17,945.5 | 750.6 | −252.9 | −17,942.9 | 11,976.8 | 11,456.0 | −520.8 | 262.7 | 11,718.7 |
| 16 | mer-[Co(NH3)3(NO2)3]3+ | −13,349.6 | −12,947.8 | 401.8 | −7.1 | −12,806.9 | 6630.3 | 6458.4 | −171.9 | 17.0 | 6475.4 |
| 17 | fac-[Co(NH3)3(NO2)3]3+ | −13,740.3 | −13,375.0 | 365.3 | −50.4 | −13,279.2 | 7021.0 | 6885.6 | −135.5 | 60.2 | 6945.8 |
| 18 | mer-[Co(NH3)3(H2O)3]3+ | −21,251.8 | −19,978.9 | 1272.8 | −311.7 | −20,157.4 | 14,532.5 | 13,489.5 | −1043.0 | 321.5 | 13,810.9 |
| 19 | fac-[Co(NH3)3(H2O)3]3+ | −19,478.0 | −18,746.8 | 731.2 | −242.4 | −18,809.3 | 12,758.7 | 12,257.4 | −501.3 | 252.2 | 12,509.6 |
| 20 | cis-[Co(NH3)2(NO2)4]− | −13,551.3 | −13,204.7 | 346.7 | 105.0 | −13,015.0 | 6832.0 | 6715.2 | −116.8 | −95.2 | 6620.0 |
| 21 | cis-[Co(NH3)2(CO3)2]− | −18,115.3 | −16,493.3 | 1622.0 | −209.6 | −16,275.4 | 11,396.0 | 10,003.8 | −1392.1 | 219.4 | 10,223.2 |
| 22 | cis,mer-[Co(NH3)2(NO2)3CH3]− | −12,948.1 | −12,188.7 | 759.4 | −63.5 | −12,219.8 | 6228.8 | 5699.2 | −529.6 | 73.4 | 5772.6 |
| 23 | cis,fac-[Co(NH3)2(NO2)3CH3]− | −12,684.5 | −12,287.6 | 396.9 | 221.1 | −12,057.4 | 5965.2 | 5798.1 | −167.1 | −211.3 | 5586.8 |
| 24 | trans,mer-[Co(NH3)2(NO2)3CH3]− | −11,844.1 | −11,475.9 | 368.2 | 132.2 | −11,372.5 | 5124.8 | 4986.5 | −138.3 | −122.3 | 4864.1 |
| 25 | [Co(NH3)6]3+ | −15,961.7 | −15,470.5 | 491.2 | −232.2 | −15,667.9 | 9242.4 | 8981.1 | −261.4 | 242.0 | 9223.1 |
| 26 | trans-[Co(NH3)4(NO2)2]+ | −14,042.9 | −13,548.9 | 494.0 | 67.4 | −13,283.8 | 7323.5 | 7059.4 | −264.1 | −57.6 | 7001.9 |
| 27 | trans-[Co(NH3)2(NO2)4]− | −13,131.6 | −12,790.0 | 341.6 | 46.7 | −12,664.6 | 6412.3 | 6300.5 | −111.7 | −36.9 | 6263.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samultsev, D.O.; Semenov, V.A.; Rusakova, I.L.; Krivdin, L.B. Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 2: Nitrogen-Coordinated Complexes of Cobalt. Int. J. Mol. Sci. 2022, 23, 13178. https://doi.org/10.3390/ijms232113178
Samultsev DO, Semenov VA, Rusakova IL, Krivdin LB. Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 2: Nitrogen-Coordinated Complexes of Cobalt. International Journal of Molecular Sciences. 2022; 23(21):13178. https://doi.org/10.3390/ijms232113178
Chicago/Turabian StyleSamultsev, Dmitry O., Valentin A. Semenov, Irina L. Rusakova, and Leonid B. Krivdin. 2022. "Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 2: Nitrogen-Coordinated Complexes of Cobalt" International Journal of Molecular Sciences 23, no. 21: 13178. https://doi.org/10.3390/ijms232113178
APA StyleSamultsev, D. O., Semenov, V. A., Rusakova, I. L., & Krivdin, L. B. (2022). Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 2: Nitrogen-Coordinated Complexes of Cobalt. International Journal of Molecular Sciences, 23(21), 13178. https://doi.org/10.3390/ijms232113178