
TNF-α Plus IL-1β Induces Opposite Regulation of Cx43 Hemichannels and Gap Junctions in Mesangial Cells through a RhoA/ROCK-Dependent Pathway

 ,
,  , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
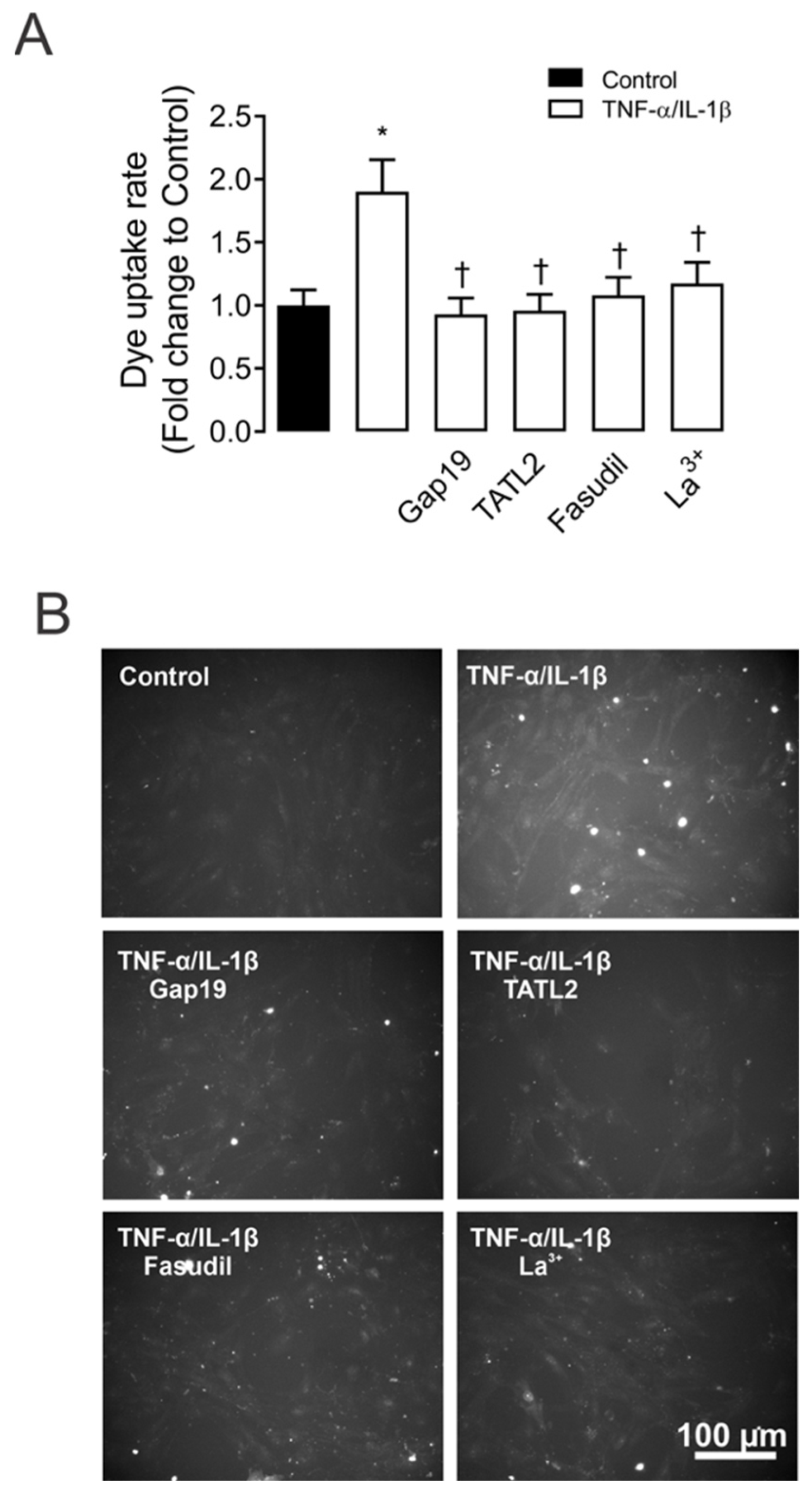
2.1. TNF-α/IL-1β Induced Activation of Cx43 HCs in Mesangial Cells Depends on RhoA/ROCK Pathway
2.2. TNF-α/IL-1β Reduces Intercellular Communication Mediated by GJs in Mesangial Cells
2.3. TNF-α/IL-1β Promotes Phosphorylation of MYPT and Increases the Amount of Cx43 in Mesangial Cells
2.4. Inhibition of RhoA/ROCK Prevents Increases in Lipid Peroxidation Responses Induced by TNF-α and IL-1β in Mesangial Cells
2.5. Inhibition of RhoA/ROCK Prevents Apoptosis and Cell Viability Induced by TNF-α and IL-1β in Primary Mesangial Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Experimental Animals and Isolation of Primary Glomerular MCs
4.3. Cell Cultures
4.4. Dye Uptake and Time-Lapse Fluorescence Imaging
4.5. Dye Coupling
4.6. Scrape Loading/Dye Diffusion Technique
4.7. Immunofluorescence
4.8. Western Blot Assays
4.9. Thiobarbituric Acid Reactive Substances (TBARS) Measurement
4.10. TUNEL Assay
4.11. Cell Viability
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AngII | Angiotensin II |
| AT1R | Angiotensin membrane G-protein-coupled receptors type I |
| AT2R | Angiotensin membrane G-protein-coupled receptors type II |
| CKD | Chronic kidney disease |
| Cx GJs | Connexin gap junctions |
| Cx HCs | Connexin hemichannels |
| Cx43 | Connexin 43 |
| ECM | Excess extracellular matrix |
| ESRD | End-stage renal disease |
| Etd+ | Ethidium |
| IFN-γ | Interferon-γ |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| iNOS | inducible Nitric oxide synthase |
| MCs | Mesangial Cells |
| MDA | Malondialdehyde |
| MM | Mesangial matrix |
| MYPT-1 | Myosin phosphatase target subunit 1 |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| OS | Oxidative stress |
| Panx1 | Pannexin-1 |
| RAS | Renin angiotensin system |
| Rho GTPase | Rho family of small GTPases |
| ROCK | Rho kinase |
| ROS | Reactive oxidative species |
| TBARS | Thiobarbituric reactive species |
| TNF-α | Tumor necrosis factor-α |
References
- Yang, Z.-J.; Wang, H.-R.; Wang, Y.-I.; Zhai, Z.-H.; Wang, L.-W.; Li, L.; Zhang, C.; Tang, L. Myricetin Attenuated Diabetes-Associated Kidney Injuries and Dysfunction via Regulating Nuclear Factor (Erythroid Derived 2)-Like 2 and Nuclear Factor-κB Signaling. Front. Pharmacol. 2019, 10, 647. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, J.; Yu, X.; Cheng, S.; Gan, H.; Xia, Y. Angiotensin II-Induced Early and Late Inflammatory Responses Through NOXs and MAPK Pathways. Inflammation 2017, 40, 154–165. [Google Scholar] [CrossRef]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.-J. Chronic kidney disease. Nat. Rev. Dis. Primers. 2017, 3, 17088. [Google Scholar] [CrossRef]
- Zhao, J.-H. Mesangial Cells and Renal Fibrosis. In Renal Fibrosis: Mechanisms and Therapies. Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 165–194. [Google Scholar]
- Kao, M.P.C.; Ang, D.S.C.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef]
- Tucker, P.S.; Scanlan, A.; Dalbo, V.J. Chronic Kidney Disease Influences Multiple Systems: Describing the Relationship between Oxidative Stress, Inflammation, Kidney Damage, and Concomitant Disease. Oxidative Med. Cell. Longev. 2015, 2015, 60803. [Google Scholar] [CrossRef]
- Alique, M.; Sánchez-López, E.; Rayego-Mateos, S.; Egido, J.; Ortiz, A.; Ruiz-Ortega, M. Angiotensin II, via angiotensin receptor type 1/nuclear factor-κB activation, causes a synergistic effect on interleukin-1-β-induced inflammatory responses in cultured mesangial cells. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 23–32. [Google Scholar] [CrossRef]
- Zhang, F.; Sun, D.; Chen, J.; Guan, N.; Huo, X.; Xi, H. Simvastatin attenuates angiotensin II-induced inflammation and oxidative stress in human mesangial cells. Mol. Med. Rep. 2015, 11, 1246–1251. [Google Scholar] [CrossRef]
- Gómez, G.I.; Fernández, P.; Velarde, V.; Sáez, J.C. Angiotensin II-Induced Mesangial Cell Damage Is Preceded by Cell Membrane Permeabilization Due to Upregulation of Non-Selective Channels. Int. J. Mol. Sci. 2018, 19, 957. [Google Scholar] [CrossRef]
- Gómez, G.I.; Velarde, V. Boldine Improves Kidney Damage in the Goldblatt 2K1C Model Avoiding the Increase in TGF-β. Int. J. Mol. Sci. 2018, 19, 1864. [Google Scholar] [CrossRef]
- Gómez, G.I.; Velarde, V.; Sáez, J.C. Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage. Int. J. Mol. Sci. 2019, 20, 4408. [Google Scholar] [CrossRef]
- Da Silva Novaes, A.; Ribeiro, R.S.; Pereira, L.G.; Borges, F.T.; Boim, M.A. Intracrine action of angiotensin II in mesangial cells: Subcellular distribution of angiotensin II receptor subtypes AT1 and AT2. Mol. Cell. Biochem. 2018, 448, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Anders, H.-J.; Gaikwad, A.B. Fiend and friend in the renin angiotensin system: An insight on acute kidney injury. Biomed. Pharmacother. 2019, 110, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Gómez, G.I.; Velarde, V.; Sáez, J.C. Connexin-Based Channels and RhoA/ROCK Pathway in Angiotensin II-Induced Kidney Damage. In Selected Chapters from the Renin—Angiotensin System; IntechOpen: London, UK, 2020. [Google Scholar]
- Rupérez, M.; Sánchez-López, E.; Blanco-Colio, L.M.; Esteban, V.; Rodríguez-Vita, J.; Plaza, J.J.; Egido, J.; Ruiz-Ortega, M. The Rho-kinase pathway regulates angiotensin II-induced renal damage. Kidney Int. 2005, 68, S39–S45. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Rolli-Derkinderen, M.; Loufrani, L.; Bourgé, A.; Henrion, D.; Sabourin, L.; Loirand, G.; Pacaud, P. Ste20-Related Kinase SLK Phosphorylates Ser188 of RhoA to Induce Vasodilation in Response to Angiotensin II Type 2 Receptor Activation. Circ. Res. 2008, 102, 1265–1274. [Google Scholar] [CrossRef]
- Rao, J.; Ye, Z.; Tang, H.; Wang, C.; Peng, H.; Lai, W.; Li, Y.; Huang, W.; Lou, T. The RhoA/ROCK Pathway Ameliorates Adhesion and Inflammatory Infiltration Induced by AGEs in Glomerular Endothelial Cells. Sci. Rep. 2017, 7, 39727. [Google Scholar] [CrossRef]
- Ye, Q.; Zhao, S.; Zhang, Y.; Su, Y.-M.; Chen, M.; Zhao, J.; Jia, G.-Z.; Han, B.-M.; Jiang, J.-T. Activation of the RhoA/ROCK pathway contributes to renal fibrosis in offspring rats induced by maternal exposure to di-n-butyl phthalate. Toxicology 2020, 443, 152573. [Google Scholar] [CrossRef]
- Baba, I.; Egi, Y.; Utsumi, H.; Kakimoto, T.; Suzuki, K. Inhibitory effects of fasudil on renal interstitial fibrosis induced by unilateral ureteral obstruction. Mol. Med. Rep. 2015, 12, 8010–8020. [Google Scholar] [CrossRef]
- Sáez, J.C.; Contreras-Duarte, S.; Gómez, G.I.; Labra, V.C.; Santibañez, C.A.; Gajardo-Gómez, R.; Avendaño, B.C.; Díaz, E.F.; Montero, T.D.; Velarde, V.; et al. Connexin 43 Hemichannel Activity Promoted by Pro-Inflammatory Cytokines and High Glucose Alters Endothelial Cell Function. Front. Immunol. 2018, 9, 1899. [Google Scholar] [CrossRef]
- Prakoura, N.; Kavvadas, P.; Chadjichristos, C.E. Connexin 43: A New Therapeutic Target Against Chronic Kidney Disease. Cell. Physiol. Biochem. 2018, 49, 985. [Google Scholar] [CrossRef]
- Kurtz, A. Renal Connexins and Blood Pressure. Biochim Biophys Acta 2012, 1818, 1903–1908. [Google Scholar] [CrossRef][Green Version]
- Hanner, F.; Sorensen, C.M.; Holstein-Rathlou, N.-H.; Peti-Peterdi, J. Connexins and the Kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1143–R1155. [Google Scholar] [CrossRef]
- Nakazawa, K.; Liu, M.; Inoue, K.; Ohno, Y. Potent inhibition by trivalent cations of ATP-gated channels. Eur. J. Pharmacol. 1997, 325, 237–243. [Google Scholar] [CrossRef]
- Gomez, G.; Falcon, R.V.; Maturana, C.J.; Labra, V.C.; Salgado, N.; Rojas, C.A.; Oyarzún, J.E.; Cerpa, W.; Quintanilla, R.A.; Orellana, J. Heavy Alcohol Exposure Activates Astroglial Hemichannels and Pannexons in the Hippocampus of Adolescent Rats: Effects on Neuroinflammation and Astrocyte Arborization. Front. Cell. Neurosci. 2018, 12, 472. [Google Scholar] [CrossRef] [PubMed]
- Kushiyama, T.; Oda, T.; Yamamoto, K.; Higashi, K.; Watanabe, A.; Takechi, H.; Uchida, T.; Oshima, N.; Sakurai, Y.; Miura, S.; et al. Protective Effects of Rho Kinase Inhibitor Fasudil on Rats with Chronic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2013, 304, F1325–F1334. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Sáez, P.J.; Sáez, J.C.; Giaume, C. Cx43 Hemichannels and Gap Junction Channels in Astrocytes Are Regulated Oppositely by Proinflammatory Cytokines Released from Activated Microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Orellana, J.A.; Hernández, D.E.; Ezan, P.; Velarde, V.; Bennett, M.V.L.; Giaume, C.; Sáez, J.C. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 2010, 58, 329–343. [Google Scholar] [CrossRef]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine Prevents Renal Alterations in Diabetic Rats. J. Diabetes Res. 2013, 2013, 593672. [Google Scholar] [CrossRef]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef]
- Matoba, K.; Kawanami, D.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Rho-kinase mediates TNF-α-induced MCP-1 expression via p38 MAPK signaling pathway in mesangial cells. Biochem. Biophys. Res. Commun. 2010, 402, 725–730. [Google Scholar] [CrossRef]
- González, H.E.; Eugenín, E.A.; Garcés, G.; Solís, N.; Pizarro, M.; Accatino, L.; Sáez, J.C. Regulation of Hepatic Connexins in Cholestasis: Possible Involvement of Kupffer Cells and Inflammatory Mediators. Am. J. Physiol.-Gastrointest. Liver Physiol. 2002, 282, G991–G1001. [Google Scholar] [CrossRef]
- Haefliger, J.-A.; Krattinger, N.; Martín, D.; Pedrazzini, T.; Capponi, A.; Döring, B.; Plum, A.; Charollais, A.; Willecke, K.; Meda, P. Connexin43-dependent mechanism modulates renin secretion and hypertension. J. Clin. Investig. 2006, 116, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Même, W.; Calvo, C.; Froger, N.; Ezan, P.; Amigou, E.; Koulakoff, A.; Giaume, C. Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: Potentiation by β-amyloid. FASEB J. 2006, 20, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Long, H.; Fei, C.; Yu, Y. Oxoglaucine Mediates Ca 2+ Influx and Activates Autophagy to Alleviate Osteoarthritis through the TRPV5/Calmodulin/CAMK-II Pathway. Br. J. Pharmacol. 2021, 178, 2931–2947. [Google Scholar] [CrossRef]
- Peng, F.; Wu, D.; Gao, B.; Ingram, A.J.; Zhang, B.; Chorneyko, K.; McKenzie, R.; Krepinsky, J.C. RhoA/Rho-Kinase Contribute to the Pathogenesis of Diabetic Renal Disease. Diabetes 2008, 57, 1683–1692. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, S.S.; Chen, Y.; Ahokas, R.A.; Sun, Y. Kidney Fibrosis in Hypertensive Rats: Role of Oxidative Stress. Am. J. Nephrol. 2008, 28, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Sánchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Sáez, J.C. Connexin 43 Hemichannels Mediate the Ca 2+ Influx Induced by Extracellular Alkalinization. Am. J. Physiol.-Cell Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.C.; Figueroa, V.; Zoghbi, M.; Saéz, J.C.; Reuss, L.; Altenberg, G.A. Permeation of Calcium through Purified Connexin 26 Hemichannels. J. Biol. Chem. 2012, 287, 40826–40834. [Google Scholar] [CrossRef]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Stockand, J.D.; Sansom, S.C. Glomerular Mesangial Cells: Electrophysiology and Regulation of Contraction. Physiol. Rev. 1998, 78, 723–744. [Google Scholar] [CrossRef]
- Schlöndorff, D.; Banas, B. The Mesangial Cell Revisited: No Cell Is an Island. J. Am. Soc. Nephrol. 2009, 20, 1179–1187. [Google Scholar] [CrossRef]
- Vaughan, M.R.; Quaggin, S.E. How Do Mesangial and Endothelial Cells Form the Glomerular Tuft? J. Am. Soc. Nephrol. 2008, 19, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R.; Beavan, L.A.; McCarthy, K. Glomerular matrix: Synthesis, turnover and role in mesangial expansion. Kidney Int. 1994, 45, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.M.; Wahab, N.A. Extracellular Matrix Metabolism in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1358–1373. [Google Scholar] [CrossRef]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004, 572, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Bahrami, L.; Castillo, A.; Majid, D.S.A. TNF-α Type 2 Receptor Mediates Renal Inflammatory Response to Chronic Angiotensin II Administration with High Salt Intake in Mice. Am. J. Physiol. Ren. Physiol. 2013, 304, F991–F999. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.; Ferdinandy, P.; Beyer, E.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef]
- Toubas, J.; Beck, S.; Pageaud, A.-L.; Huby, A.-C.; Mael-Ainin, M.; Dussaule, J.-C.; Chatziantoniou, C.; Chadjichristos, C.E. Alteration of Connexin Expression Is an Early Signal for Chronic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F24–F32. [Google Scholar] [CrossRef]
- Johansen, D.; Cruciani, V.; Sundset, R.; Ytrehus, K.; Mikalsen, S.-O. Ischemia Induces Closure of Gap Junctional Channels and Opening of Hemichannels in Heart-derived Cells and Tissue. Cell. Physiol. Biochem. 2011, 28, 103–114. [Google Scholar] [CrossRef]
- Lucero, C.M.; Andrade, D.C.; Toledo, C.; Díaz, H.S.; Pereyra, K.V.; Diaz-Jara, E.; Schwarz, K.G.; Marcus, N.J.; Retamal, M.A.; Quintanilla, R.A.; et al. Cardiac remodeling and arrhythmogenesis are ameliorated by administration of Cx43 mimetic peptide Gap27 in heart failure rats. Sci. Rep. 2020, 10, 6878. [Google Scholar] [CrossRef]
- Orellana, J.; Von Bernhardi, R.; Giaume, C.; Sáez, J.C. Glial hemichannels and their involvement in aging and neurodegenerative diseases. Rev. Neurosci. 2012, 23, 163–177. [Google Scholar] [CrossRef]
- Crespo Yanguas, S.; da Silva, T.; Pereira, I.; Willebrords, J.; Maes, M.; Sayuri Nogueira, M.; Alves de Castro, I.; Leclercq, I.; Romualdo, G.; Barbisan, L.; et al. TAT-Gap19 and Carbenoxolone Alleviate Liver Fibrosis in Mice. Int. J. Mol. Sci. 2018, 19, 817. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The Diverse and Complex Roles of NF-ΚB Subunits in Cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Alonso, F.; Krattinger, N.; Mazzolai, L.; Simon, A.; Waeber, G.; Meda, P.; Haefliger, J.-A. An angiotensin II- and NF-κB-dependent mechanism increases connexin 43 in murine arteries targeted by renin-dependent hypertension. Cardiovasc. Res. 2010, 87, 166–176. [Google Scholar] [CrossRef]
- Zhao, Y.; Rivieccio, M.A.; Lutz, S.; Scemes, E.; Brosnan, C.F. The TLR3 ligand polyI:C downregulates connexin 43 expression and function in astrocytes by a mechanism involving the NF-κB and PI3 kinase pathways. Glia 2006, 54, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Hiramatsu, N.; Zhu, Y.; Morioka, T.; Takeda, M.; Oite, T.; Kitamura, M. Nitric Oxide-Mediated Regulation of Connexin43 Expression and Gap Junctional Intercellular Communication in Mesangial Cells. J. Am. Soc. Nephrol. 2005, 16, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Cortés, C.J.; Reuss, L.; Bennett, M.V.L.; Sáez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef]
- Lillo, M.; Himelman, E.; Shirokova, N.; Xie, L.-H.; Fraidenraich, D.; Contreras, J.E. S-nitrosylation of connexin43 hemichannels elicits cardiac stress–induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight 2019, 4, e130091. [Google Scholar] [CrossRef]
- Vergara, L.; Bao, X.; Cooper, M.; Bello-Reuss, E.; Reuss, L. Gap-junctional Hemichannels Are Activated by ATP Depletion in Human Renal Proximal Tubule Cells. J. Membr. Biol. 2003, 196, 173–184. [Google Scholar] [CrossRef]
- Hills, C.E.; Bland, R.; Wheelans, D.C.; Bennett, J.; Ronco, P.M.; Squires, P.E. Glucose-Evoked Alterations in Connexin43-Mediated Cell-to-Cell Communication in Human Collecting Duct: A Possible Role in Diabetic Nephropathy. Am. J. Physiol. Ren. Physiol. 2006, 291, F1045–F1051. [Google Scholar] [CrossRef][Green Version]
- Cliff, C.L.; Williams, B.M.; Chadjichristos, C.E.; Mouritzen, U.; Squires, P.E.; Hills, C.E. Connexin 43: A Target for the Treatment of Inflammation in Secondary Complications of the Kidney and Eye in Diabetes. Int. J. Mol. Sci. 2022, 23, 600. [Google Scholar] [CrossRef]
- Price, G.W.; Chadjichristos, C.E.; Kavvadas, P.; Tang, S.C.; Yiu, W.H.; Green, C.R.; Potter, J.A.; Siamantouras, E.; Squires, P.E.; Hills, C.E. Blocking Connexin-43 mediated hemichannel activity protects against early tubular injury in experimental chronic kidney disease. Cell Commun. Signal. 2020, 18, 79. [Google Scholar] [CrossRef]
- Jin, L.; Ying, Z.; Hilgers, R.H.P.; Yin, J.; Zhao, X.; Imig, J.; Webb, R.C. Increased RhoA/Rho-Kinase Signaling Mediates Spontaneous Tone in Aorta from Angiotensin II-Induced Hypertensive Rats. J. Pharmacol. Exp. Ther. 2006, 318, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.A.; Bower, H.S.; Baxter, G.F. Rho Kinase Activation Plays a Major Role as a Mediator of Irreversible Injury in Reperfused Myocardium. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H2598–H2606. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rikitake, Y.; Kim, H.-H.; Huang, Z.; Seto, M.; Yano, K.; Asano, T.; Moskowitz, M.A.; Liao, J.K. Inhibition of Rho Kinase (ROCK) Leads to Increased Cerebral Blood Flow and Stroke Protection. Stroke 2005, 36, 2251–2257. [Google Scholar] [CrossRef]
- Mueller, B.K.; Mack, H.; Teusch, N. Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discov. 2005, 4, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Honjo, M.; Tanihara, H.; Inatani, M.; Kido, N.; Sawamura, T.; Yue, B.Y.; Narumiya, S.; Honda, Y. Effects of Rho-Associated Protein Kinase Inhibitor Y-27632 on Intraocular Pressure and Outflow Facility. Investig. Ophthalmol. Vis. Sci. 2001, 42, 137–144. [Google Scholar]
- Xie, X.; Peng, J.; Chang, X.; Huang, K.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Activation of RhoA/ROCK regulates NF-κB signaling pathway in experimental diabetic nephropathy. Mol. Cell. Endocrinol. 2013, 369, 86–97. [Google Scholar] [CrossRef]
- Lai, A.; Frishman, W.H. Rho-Kinase Inhibition in the Therapy of Cardiovascular Disease. Cardiol. Rev. 2005, 13, 285–292. [Google Scholar] [CrossRef]
- Mong, P.Y.; Petrulio, C.; Kaufman, H.L.; Wang, Q. Activation of Rho Kinase by TNF-α Is Required for JNK Activation in Human Pulmonary Microvascular Endothelial Cells. J. Immunol. 2008, 180, 550–558. [Google Scholar] [CrossRef]
- Price, G.W.; Potter, J.A.; Williams, B.M.; Cliff, C.L.; Squires, P.E.; Hills, C.E. Connexin-mediated cell communication in the kidney: A potential therapeutic target for future intervention of diabetic kidney disease? Exp. Physiol. 2020, 105, 219–229. [Google Scholar] [CrossRef]
- Burnstock, G.; Arnett, T.R.; Orriss, I. Purinergic signalling in the musculoskeletal system. Purinergic Signal. 2013, 9, 541–572. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.V.; Passos, D.F.; Leal, D.B.R.; Morsch, V.M.; Schetinger, M.R.C. ATP signaling and NTPDase in Systemic Lupus Erythematosus (SLE). Immunobiology 2019, 224, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, W.; Braet, K.; Cabooter, L.; Leybaert, L. Tumour necrosis factor alpha inhibits purinergic calcium signalling in blood-brain barrier endothelial cells. J. Neurochem. 2003, 88, 411–421. [Google Scholar] [CrossRef]
- Lohman, A.W.; Leskov, I.L.; Butcher, J.; Johnstone, S.; Stokes, T.A.; Begandt, D.; DeLalio, L.; Best, A.K.; Penuela, S.; Leitinger, N.; et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat. Commun. 2015, 6, 7965. [Google Scholar] [CrossRef]
- Sathanoori, R.; Swärd, K.; Olde, B.; Erlinge, D. The ATP Receptors P2X7 and P2X4 Modulate High Glucose and Palmitate-Induced Inflammatory Responses in Endothelial Cells. PLoS ONE 2015, 10, e0125111. [Google Scholar] [CrossRef]
- Schulz, R.; Heusch, G. Connexin 43 and ischemic preconditioning. Cardiovasc. Res. 2004, 62, 335–344. [Google Scholar] [CrossRef]
- Kim, H.-J.; Kim, M.-J.; Mostafa, M.N.; Park, J.-H.; Choi, H.-S.; Kim, Y.-S.; Choi, E.-K. RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity. Int. J. Mol. Sci. 2020, 21, 1255. [Google Scholar] [CrossRef]
- Anderson, S.C.; Stone, C.; Tkach, L.; SundarRaj, N. Rho and Rho-Kinase (ROCK) Signaling in Adherens and Gap Junction Assembly in Corneal Epithelium. Investig. Ophthalmol. Vis. Sci. 2002, 43, 978–986. [Google Scholar]
- Langevin, H.M.; Fujita, T.; Bouffard, N.A.; Takano, T.; Koptiuch, C.; Badger, G.J.; Nedergaard, M. Fibroblast cytoskeletal remodeling induced by tissue stretch involves ATP signaling. J. Cell. Physiol. 2013, 228, 1922–1926. [Google Scholar] [CrossRef]
- Ponsaerts, R.; D’Hondt, C.; Hertens, F.; Parys, J.; Leybaert, L.; Vereecke, J.; Himpens, B.; Bultynck, G. RhoA GTPase Switch Controls Cx43-Hemichannel Activity through the Contractile System. PLoS ONE 2012, 7, e42074. [Google Scholar] [CrossRef]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium Sensitization of Smooth Muscle Mediated by a Rho-Associated Protein Kinase in Hypertension. Nature 1997, 389, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Shimokawa, H.; Matoba, T.; Kandabashi, T.; Satoh, S.; Hiroki, J.; Kaibuchi, K.; Takeshita, A. Involvement of Rho-Kinase in Hypertensive Vascular Disease: A Novel Therapeutic Target in Hypertension. FASEB J. 2001, 15, 1062–1064. [Google Scholar]
- Perona, R.; Montaner, S.; Saniger, L.; Sánchez-Pérez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Tergaonkar, V. Roles of NF-κB in health and disease: Mechanisms and therapeutic potential. Clin. Sci. 2009, 116, 451–465. [Google Scholar] [CrossRef]
- Salva, E.; Turan, S.; Akbuğa, J. Inhibition of Glomerular Mesangial Cell Proliferation by siPDGF-B- and siPDGFR-β-Containing Chitosan Nanoplexes. AAPS PharmSciTech 2017, 18, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Menè, P.; Stoppacciaro, A. Isolation and Propagation of Glomerular Mesangial Cells. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: Totowa, NJ, USA, 2009; Volume 466, pp. 1–15. [Google Scholar]
- Wilson, H.M.; Stewart, K.N. Glomerular Epithelial and Mesangial Cell Culture and Characterization. In Human Cell Culture Protocols; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 187–201. [Google Scholar]
- Wang, H.; Sheng, J.; He, H.; Chen, X.; Li, J.; Tan, R.; Wang, L.; Lan, H.-Y. A Simple and Highly Purified Method for Isolation of Glomeruli from the Mouse Kidney. Am. J. Physiol. Ren. Physiol. 2019, 317, F1217–F1223. [Google Scholar] [CrossRef]
- Orellana, J.A.; Sáez, P.J.; Cortés-Campos, C.; Elizondo, R.J.; Shoji, K.F.; Contreras-Duarte, S.; Figueroa, V.; Velarde, V.; Jiang, J.X.; Nualart, F.; et al. Glucose Increases Intracellular Free Ca(2+) in Tanycytes via ATP Released through Connexin 43 Hemichannels. Glia 2012, 60, 53–68. [Google Scholar] [CrossRef]
- Contreras, J.E.; Sánchez, H.A.; Eugenín, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.L.; Sáez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef]
- Gajardo-Gómez, R.; Labra, V.C.; Maturana, C.J.; Shoji, K.F.; Santibañez, C.A.; Sáez, J.C.; Giaume, C.; Orellana, J. Cannabinoids prevent the amyloid β-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia 2017, 65, 122–137. [Google Scholar] [CrossRef]
- Dydowiczová, A.; Brózman, O.; Babica, P.; Sovadinová, I. Improved multiparametric scrape loading-dye transfer assay for a simultaneous high-throughput analysis of gap junctional intercellular communication, cell density and viability. Sci. Rep. 2020, 10, 730. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Ramanathan, L.; Das, N.P.; Li, Q.-T. Studies on lipid oxidation in fish phospholipid liposomes. Biol. Trace Elem. Res. 1994, 40, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Niu, C.; Zhang, X.; Dong, M. β-Ecdysterone protects SH-SY5Y cells against β-amyloid-induced apoptosis via c-Jun N-terminal kinase- and Akt-associated complementary pathways. Lab. Investig. 2018, 98, 489–499. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucero, C.M.; Marambio-Ruiz, L.; Balmazabal, J.; Prieto-Villalobos, J.; León, M.; Fernández, P.; Orellana, J.A.; Velarde, V.; Sáez, J.C.; Gómez, G.I. TNF-α Plus IL-1β Induces Opposite Regulation of Cx43 Hemichannels and Gap Junctions in Mesangial Cells through a RhoA/ROCK-Dependent Pathway. Int. J. Mol. Sci. 2022, 23, 10097. https://doi.org/10.3390/ijms231710097
Lucero CM, Marambio-Ruiz L, Balmazabal J, Prieto-Villalobos J, León M, Fernández P, Orellana JA, Velarde V, Sáez JC, Gómez GI. TNF-α Plus IL-1β Induces Opposite Regulation of Cx43 Hemichannels and Gap Junctions in Mesangial Cells through a RhoA/ROCK-Dependent Pathway. International Journal of Molecular Sciences. 2022; 23(17):10097. https://doi.org/10.3390/ijms231710097
Chicago/Turabian StyleLucero, Claudia M., Lucas Marambio-Ruiz, Javiera Balmazabal, Juan Prieto-Villalobos, Marcelo León, Paola Fernández, Juan A. Orellana, Victoria Velarde, Juan C. Sáez, and Gonzalo I. Gómez. 2022. "TNF-α Plus IL-1β Induces Opposite Regulation of Cx43 Hemichannels and Gap Junctions in Mesangial Cells through a RhoA/ROCK-Dependent Pathway" International Journal of Molecular Sciences 23, no. 17: 10097. https://doi.org/10.3390/ijms231710097
APA StyleLucero, C. M., Marambio-Ruiz, L., Balmazabal, J., Prieto-Villalobos, J., León, M., Fernández, P., Orellana, J. A., Velarde, V., Sáez, J. C., & Gómez, G. I. (2022). TNF-α Plus IL-1β Induces Opposite Regulation of Cx43 Hemichannels and Gap Junctions in Mesangial Cells through a RhoA/ROCK-Dependent Pathway. International Journal of Molecular Sciences, 23(17), 10097. https://doi.org/10.3390/ijms231710097