
FAK-Mediated Signaling Controls Amyloid Beta Overload, Learning and Memory Deficits in a Mouse Model of Alzheimer’s Disease




Abstract
1. Introduction
2. Results
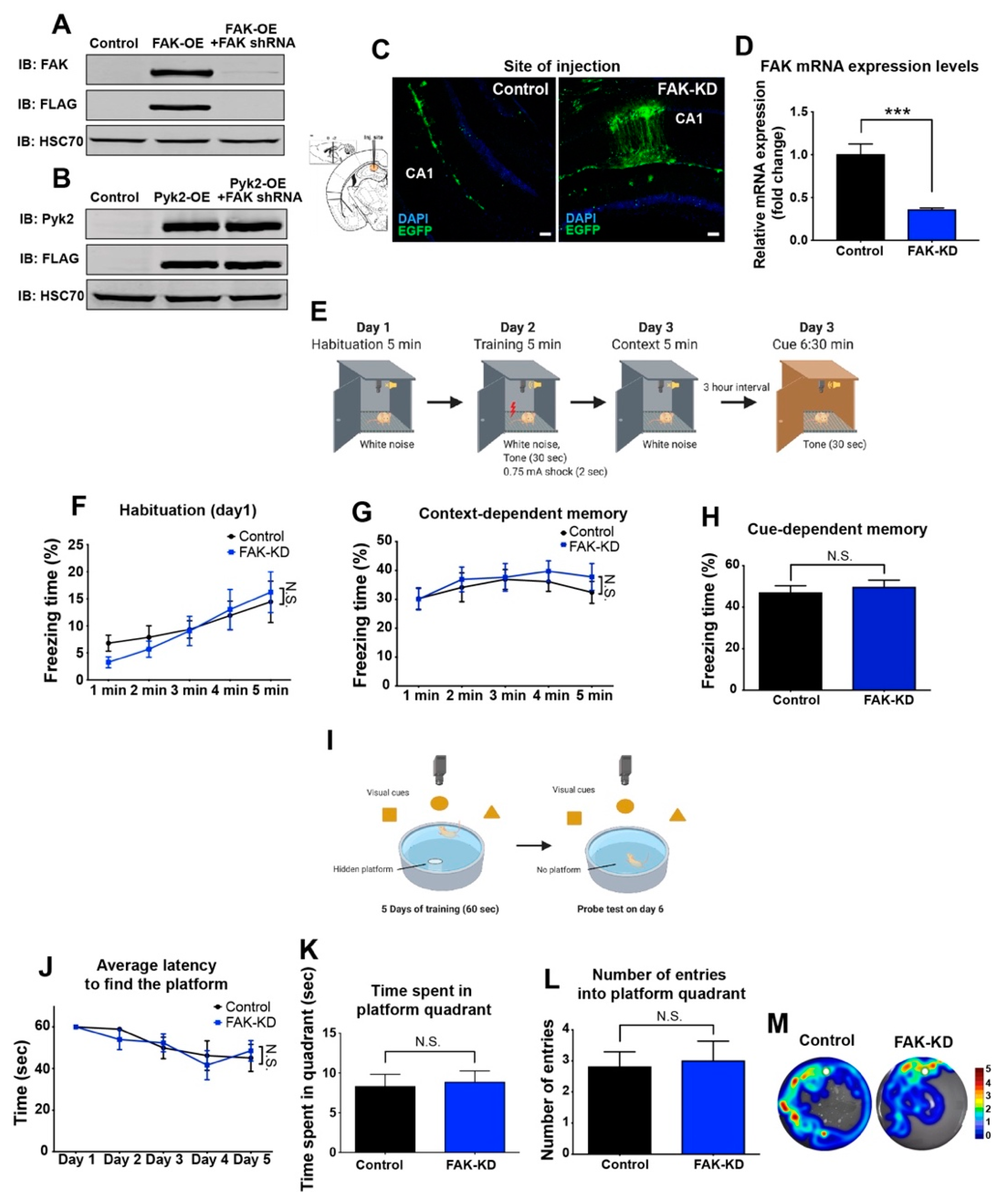
2.1. Hippocampal Depletion of FAK in 3xTg-AD Mice Does Not Significantly Affect Their Learning and Memory Capabilities
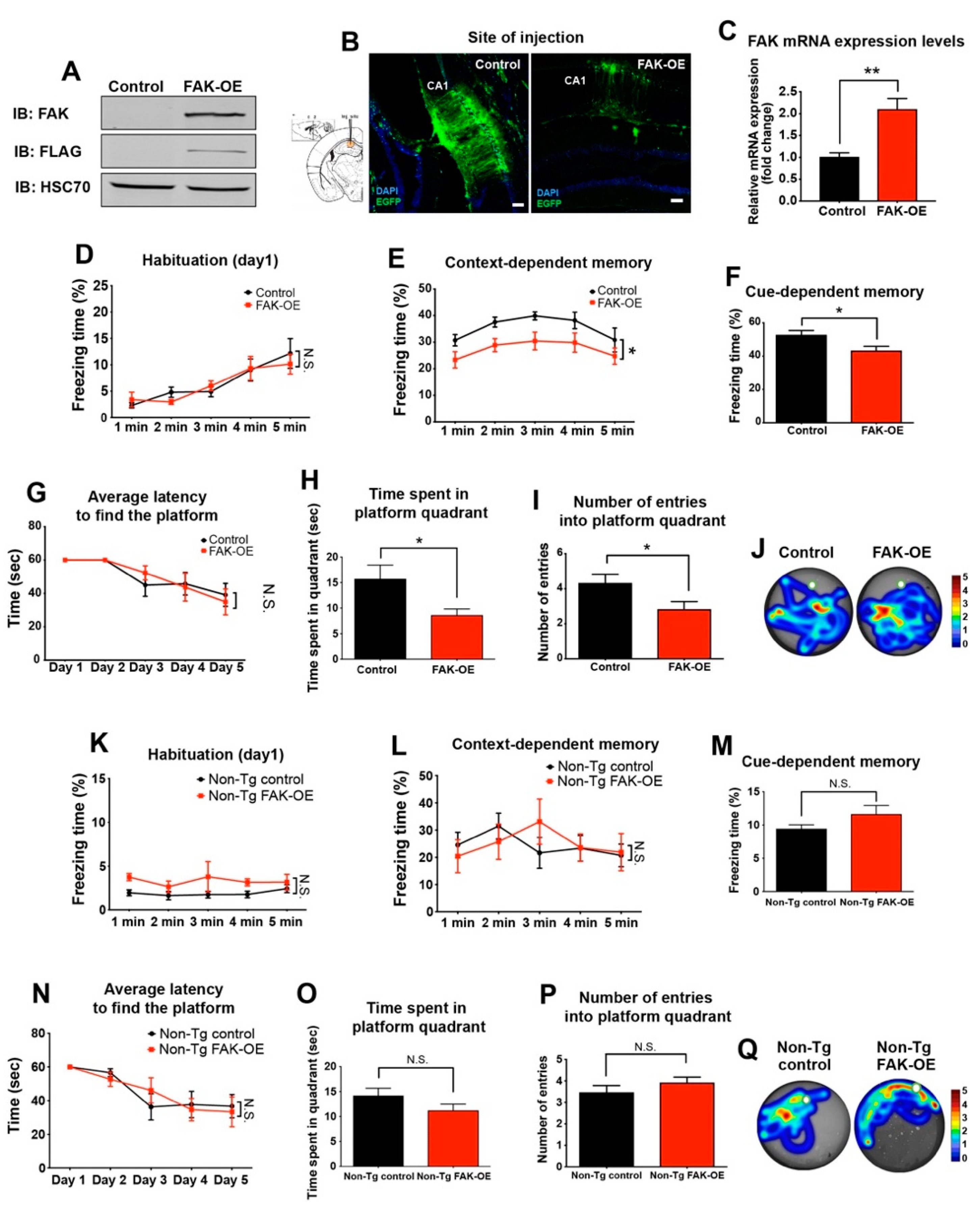
2.2. Overexpression of FAK in 3xTg-AD Mouse Hippocampus Leads to Significant Impairment in Learning and Memory
2.3. FAK Overexpression in Hippocampi of Non-Transgenic Aged Mice Does Not Affect Their Learning and Memory Capabilities
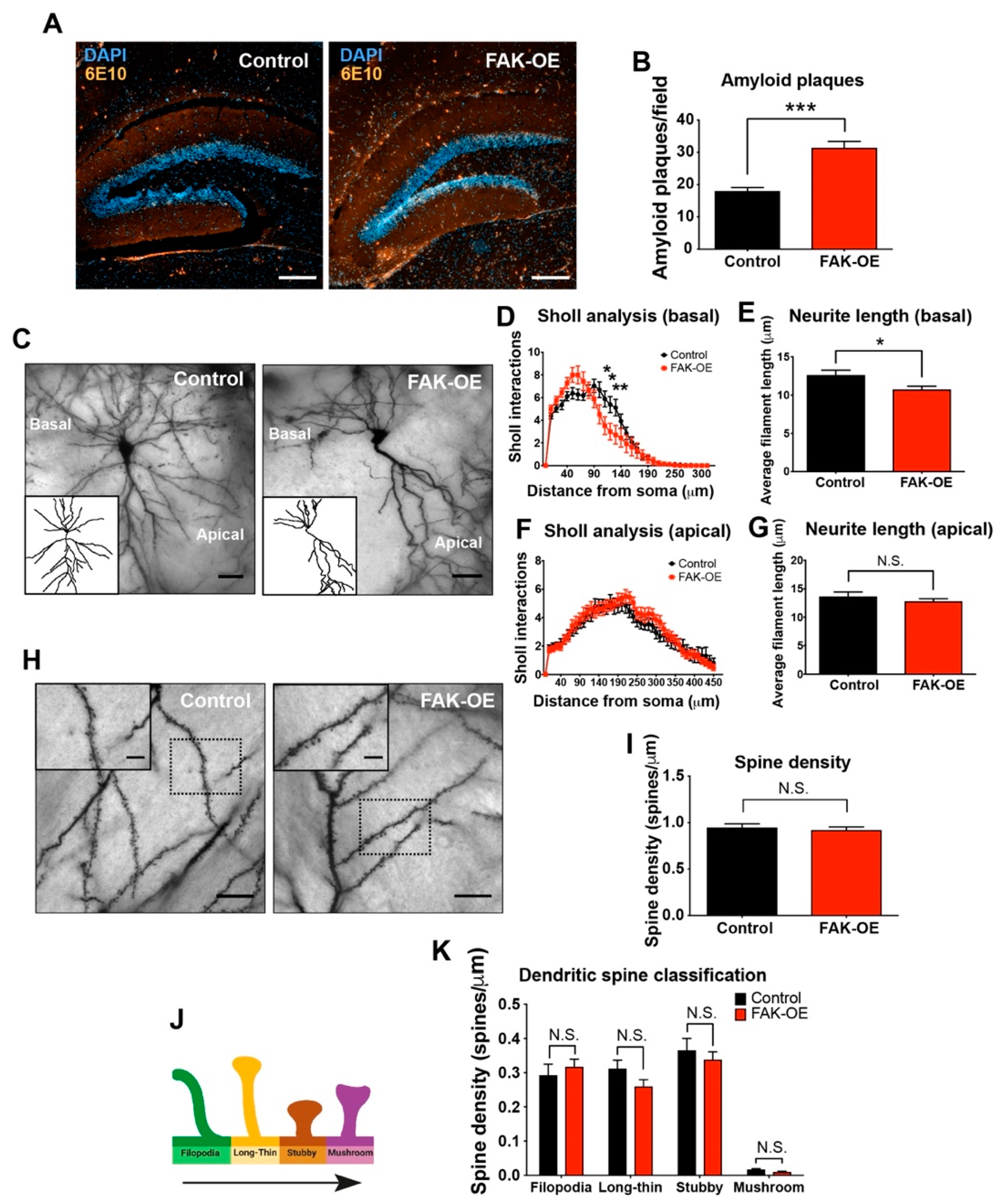
2.4. Hippocampal Overexpression of FAK Significantly Increases Amyloid Plaque Accumulation
2.5. FAK Overexpression Shows Reduced Arborization in Basal Regions of Hippocampus Neurons
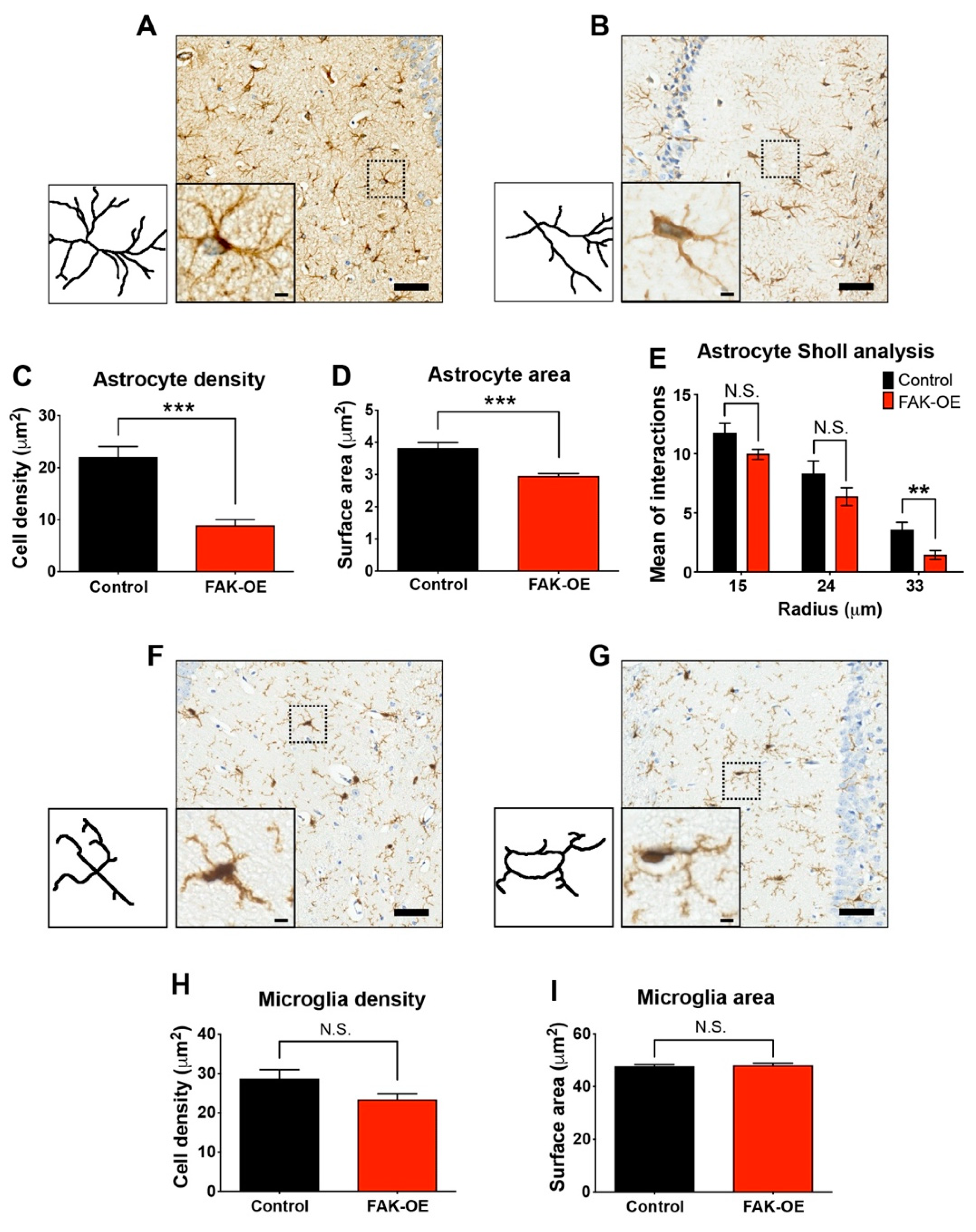
2.6. Hippocampal FAK Overexpression Significantly Reduces Astrogliosis
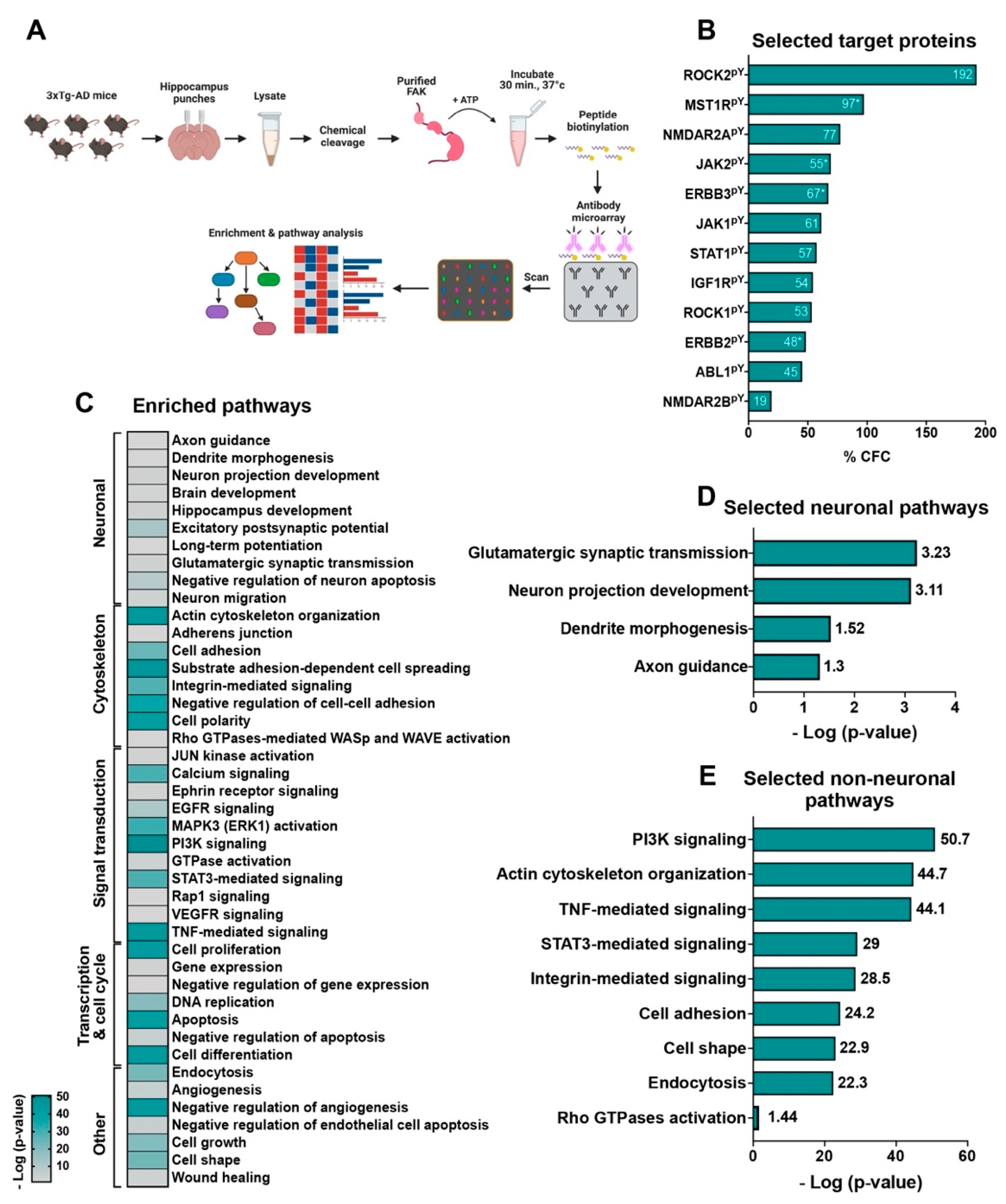
2.7. An in-Lysate Kinase Assay Reveals Novel Targets of FAK in 3xTg-AD Mouse Hippocampus
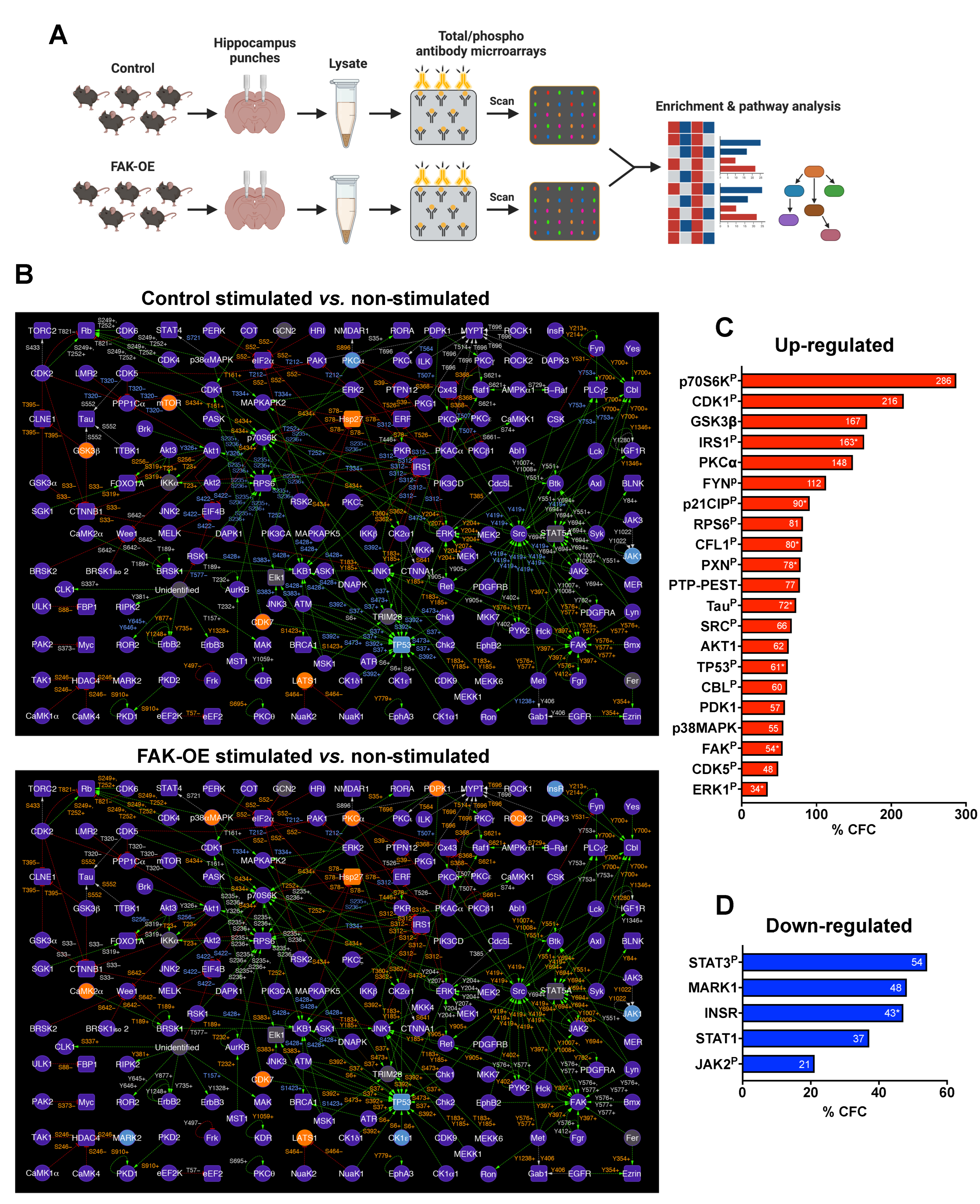
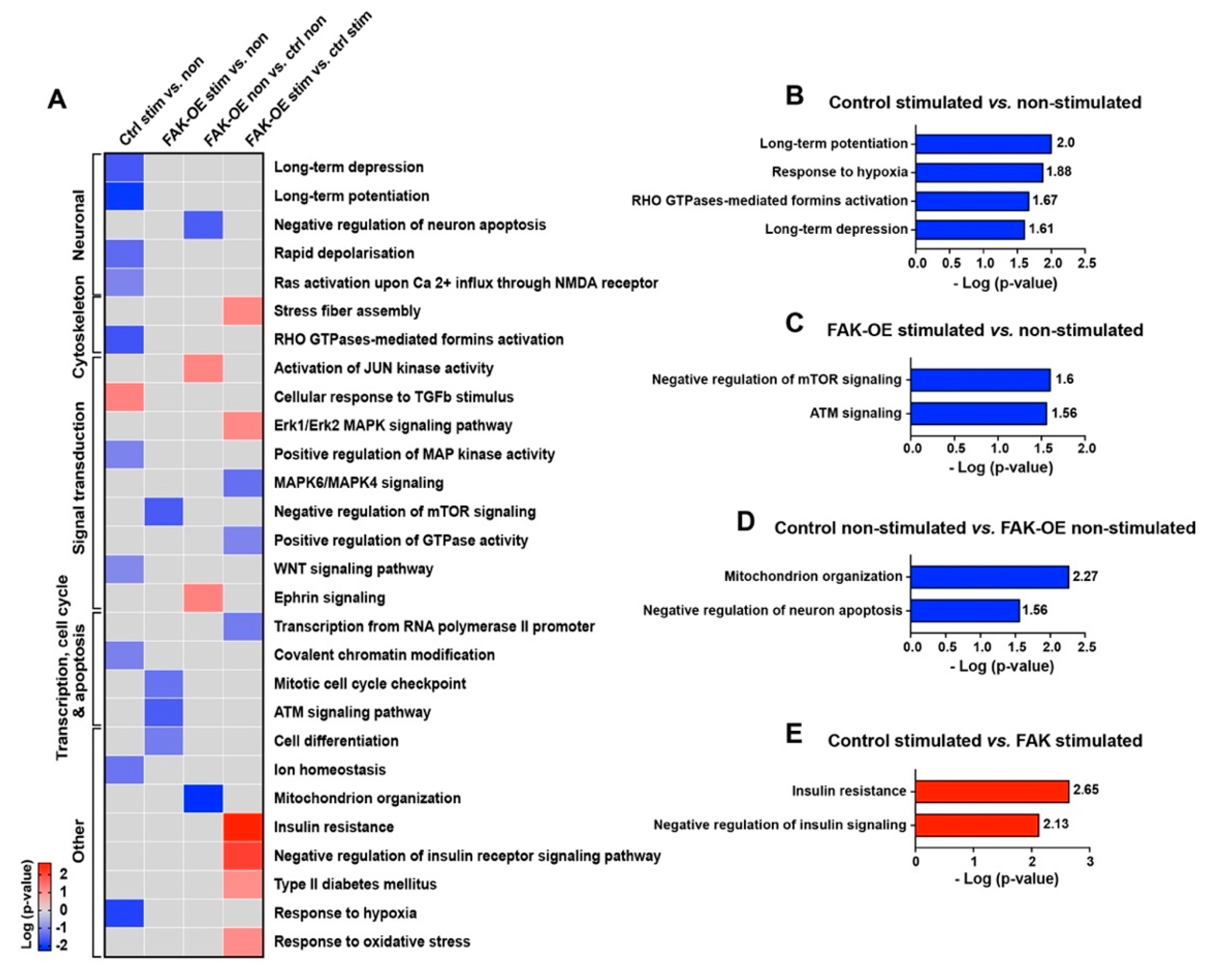
2.8. Overexpression of FAK Mediates AD-like Phenotypes in 3xTg-AD Mice by Controlling the PI3K and Insulin Signaling Pathways, Re-Entry into the Cell Cycle, and Neuronal Cell Death
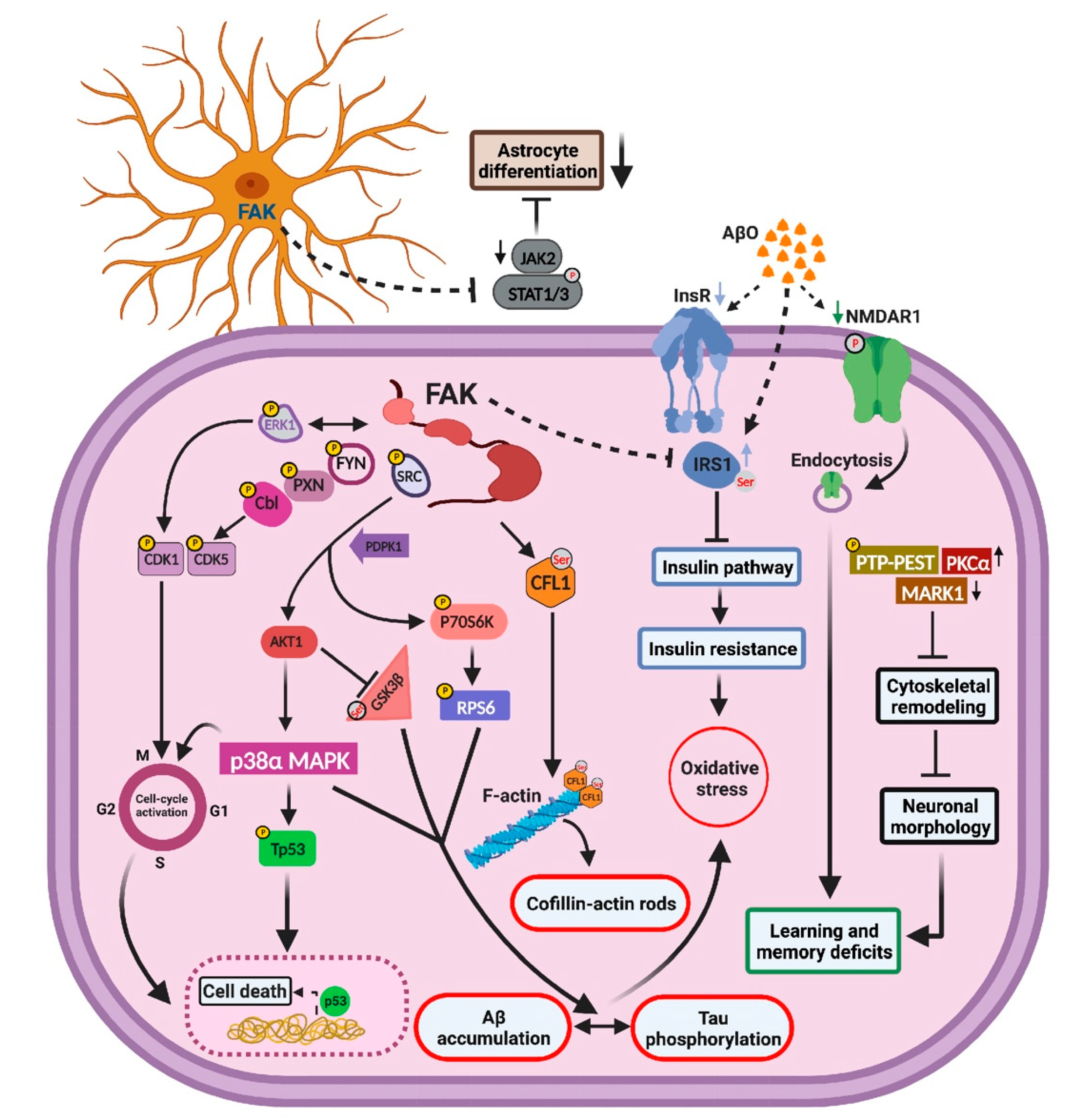
3. Discussion
3.1. Overexpression of FAK Leads to Decreased Basal Neurite Arborization
3.2. FAK Is Involved in the Hyperphosphorylation of Tau
3.3. FAK-Mediated Downregulation of JAK-STAT Pathway Leads to Reduced Astrogliosis
3.4. FAK Overexpression Regulates Cell Cycle Re-Entry and Consequent Neuronal Cell Death
3.5. Aberrations in Insulin Signaling and Insulin Resistance Are Resulted by FAK Overexpression
4. Materials and Methods
4.1. Animals
4.2. Quantitative Real-Time PCR (qRT-PCR)
4.3. Plasmids
4.4. Viral Vector Preparation
4.5. Viral Vector Delivery
4.6. Immunoblotting
4.7. Contextual and Cued Fear Conditioning
4.8. Morris Water Maze
4.9. Video Tracking Analysis
4.10. Brain Tissue Preparation and Immunofluorescence
4.11. Immunohistochemistry
4.12. Golgi-Cox Staining
4.13. Antibody Microarrays
4.14. In-Lysate Kinase Assay
4.15. Enrichment Analysis
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Asociation. Alzheimer’s disease facts and figures. Alzheimers Dement 2021, 17, 327–406. [Google Scholar]
- Dos Santos Picanco, L.C.; Ozela, P.F.; de Fatima de Brito Brito, M.; Pinheiro, A.A.; Padilha, E.C.; Braga, F.S.; de Paula da Silva, C.H.T.; Dos Santos, C.B.R.; Rosa, J.M.C.; da Silva Hage-Melim, L.I. Alzheimer’s disease: A review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr. Med. Chem. 2018, 25, 3141–3159. [Google Scholar] [CrossRef]
- Polis, B.; Gil-Henn, H. Commentary on Giralt et al.: PTK2B/Pyk2 overexpression improves a mouse model of Alzheimer’s disease. Exp. Neurol. 2019, 311, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Scarpini, E.; Scheltens, P.; Feldman, H. Treatment of Alzheimer’s disease: Current status and new perspectives. Lancet Neurol. 2003, 2, 539–547. [Google Scholar] [CrossRef]
- Burgaya, F.; Girault, J.A. Cloning of focal adhesion kinase, pp125FAK, from rat brain reveals multiple transcripts with different patterns of expression. Brain Res. Mol. Brain Res. 1996, 37, 63–73. [Google Scholar] [CrossRef]
- Grant, S.G.; Karl, K.A.; Kiebler, M.A.; Kandel, E.R. Focal adhesion kinase in the brain: Novel subcellular localization and specific regulation by Fyn tyrosine kinase in mutant mice. Genes Dev. 1995, 9, 1909–1921. [Google Scholar] [CrossRef]
- Toutant, M.; Studler, J.M.; Burgaya, F.; Costa, A.; Ezan, P.; Gelman, M.; Girault, J.A. Autophosphorylation of Tyr397 and its phosphorylation by Src-family kinases are altered in focal-adhesion-kinase neuronal isoforms. Biochem. J. 2000, 348 Pt 1, 119–128. [Google Scholar] [CrossRef]
- Koleske, A.J. Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 2013, 14, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Pontrello, C.G.; DeFea, K.A.; Reichardt, L.F.; Ethell, I.M. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J. Neurosci. 2009, 29, 8129–8142. [Google Scholar] [CrossRef]
- Eyermann, C.; Czaplinski, K.; Colognato, H. Dystroglycan promotes filopodial formation and process branching in differentiating oligodendroglia. J. Neurochem. 2012, 120, 928–947. [Google Scholar] [CrossRef]
- Monje, F.J.; Kim, E.J.; Pollak, D.D.; Cabatic, M.; Li, L.; Baston, A.; Lubec, G. Focal adhesion kinase regulates neuronal growth, synaptic plasticity and hippocampus-dependent spatial learning and memory. Neurosignals 2012, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Shin, M.R.; Sesti, F. Oxidation of KCNB1 channels in the human brain and in mouse model of Alzheimer’s disease. Cell Death Dis. 2018, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.; Scales, T.; Clark, B.R.; Gibb, G.; Reynolds, C.H.; Kellie, S.; Bird, I.N.; Varndell, I.M.; Sheppard, P.W.; Everall, I.; et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: Involvement of Src family protein kinases. J. Neurosci. 2002, 22, 10–20. [Google Scholar] [CrossRef]
- Xu, Y.X.; Wang, H.Q.; Yan, J.; Sun, X.B.; Guo, J.C.; Zhu, C.Q. Antibody binding to cell surface amyloid precursor protein induces neuronal injury by deregulating the phosphorylation of focal adhesion signaling related proteins. Neurosci. Lett. 2009, 465, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bisht, B.; Dey, C.S. Focal adhesion kinase negatively regulates neuronal insulin resistance. Biochim. Biophys. Acta 2012, 1822, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Grace, E.A.; Busciglio, J. Aberrant activation of focal adhesion proteins mediates fibrillar amyloid beta-induced neuronal dystrophy. J. Neurosci. 2003, 23, 493–502. [Google Scholar] [CrossRef]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Tinkle, B.T.; Bieberich, C.J.; Haudenschild, C.C.; Jay, G. The Alzheimer’s A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat. Genet. 1995, 9, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Moechars, D.; Dewachter, I.; Lorent, K.; Reverse, D.; Baekelandt, V.; Naidu, A.; Tesseur, I.; Spittaels, K.; Haute, C.V.; Checler, F.; et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999, 274, 6483–6492. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Koleske, A.J. Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu. Rev. Neurosci. 2010, 33, 349–378. [Google Scholar] [CrossRef]
- Brzdak, P.; Wojcicka, O.; Zareba-Koziol, M.; Minge, D.; Henneberger, C.; Wlodarczyk, J.; Mozrzymas, J.W.; Wojtowicz, T. Synaptic potentiation at basal and apical dendrites of hippocampal pyramidal neurons involves activation of a distinct set of extracellular and intracellular molecular cues. Cereb. Cortex 2019, 29, 283–304. [Google Scholar] [CrossRef]
- Guerriero, F.; Sgarlata, C.; Francis, M.; Maurizi, N.; Faragli, A.; Perna, S.; Rondanelli, M.; Rollone, M.; Ricevuti, G. Neuroinflammation, immune system and Alzheimer disease: Searching for the missing link. Aging Clin. Exp. Res. 2017, 29, 821–831. [Google Scholar] [CrossRef]
- Ehrengruber, M.U.; Hennou, S.; Bueler, H.; Naim, H.Y.; Deglon, N.; Lundstrom, K. Gene transfer into neurons from hippocampal slices: Comparison of recombinant Semliki Forest Virus, adenovirus, adeno-associated virus, lentivirus, and measles virus. Mol. Cell. Neurosci. 2001, 17, 855–871. [Google Scholar] [CrossRef]
- Swanger, S.A.; Mattheyses, A.L.; Gentry, E.G.; Herskowitz, J.H. ROCK1 and ROCK2 inhibition alters dendritic spine morphology in hippocampal neurons. Cell. Logist. 2015, 5, e1133266. [Google Scholar] [CrossRef]
- Henderson, B.W.; Gentry, E.G.; Rush, T.; Troncoso, J.C.; Thambisetty, M.; Montine, T.J.; Herskowitz, J.H. Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer’s disease and ROCK1 depletion reduces amyloid-beta levels in brain. J. Neurochem. 2016, 138, 525–531. [Google Scholar] [CrossRef]
- Herskowitz, J.H.; Feng, Y.; Mattheyses, A.L.; Hales, C.M.; Higginbotham, L.A.; Duong, D.M.; Montine, T.J.; Troncoso, J.C.; Thambisetty, M.; Seyfried, N.T.; et al. Pharmacologic inhibition of ROCK2 suppresses amyloid-beta production in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 19086–19098. [Google Scholar] [CrossRef]
- Herskowitz, J.H.; Seyfried, N.T.; Gearing, M.; Kahn, R.A.; Peng, J.; Levey, A.I.; Lah, J.J. Rho kinase II phosphorylation of the lipoprotein receptor LR11/SORLA alters amyloid-beta production. J. Biol. Chem. 2011, 286, 6117–6127. [Google Scholar] [CrossRef]
- Jing, Z.; Caltagarone, J.; Bowser, R. Altered subcellular distribution of c-Abl in Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 17, 409–422. [Google Scholar] [CrossRef]
- Xiao, L.; Chen, D.; Hu, P.; Wu, J.; Liu, W.; Zhao, Y.; Cao, M.; Fang, Y.; Bi, W.; Zheng, Z.; et al. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. J. Neurosci. 2011, 31, 9611–9619. [Google Scholar] [CrossRef]
- Hanson, J.E.; Ma, K.; Elstrott, J.; Weber, M.; Saillet, S.; Khan, A.S.; Simms, J.; Liu, B.; Kim, T.A.; Yu, G.Q.; et al. GluN2A NMDA receptor enhancement improves brain oscillations, synchrony, and cognitive functions in dravet syndrome and Alzheimer’s disease models. Cell Rep. 2020, 30, 381–396.e4. [Google Scholar] [CrossRef]
- Sohrabi, M.; Floden, A.M.; Manocha, G.D.; Klug, M.G.; Combs, C.K. IGF-1Rinhibitor ameliorates neuroinflammation in an Alzheimer’s disease transgenic mouse model. Front. Cell. Neurosci. 2020, 14, 200. [Google Scholar] [CrossRef] [PubMed]
- Britsch, S.; Li, L.; Kirchhoff, S.; Theuring, F.; Brinkmann, V.; Birchmeier, C.; Riethmacher, D. The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1, are essential for development of the sympathetic nervous system. Genes Dev. 1998, 12, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.J.; Her, G.M.; Hu, M.K.; Chen, Y.W.; Tung, Y.T.; Wu, P.Y.; Hsu, W.M.; Lee, H.; Jin, L.W.; Hwang, S.L.; et al. ErbB2 regulates autophagic flux to modulate the proteostasis of APP-CTFs in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3129–E3138. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Yamada, M.; Aiso, S. Targeting the JAK2/STAT3 axis in Alzheimer’s disease. Expert Opin. Ther. Targets 2009, 13, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The role of Cdk5 in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L. Role of focal adhesion kinase in integrin signaling. Int. J. Biochem. Cell Biol. 1997, 29, 1085–1096. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Eng, C.H.; Schlaepfer, D.D.; Marcantonio, E.E.; Gundersen, G.G. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science 2004, 303, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123 Pt 7, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 2014, 10 (Suppl. 1), S26–S32. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Chami, B.; Steel, A.J.; De La Monte, S.M.; Sutherland, G.T. The rise and fall of insulin signaling in Alzheimer’s disease. Metab. Brain Dis. 2016, 31, 497–515. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dey, C.S. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol. Biol. Cell 2012, 23, 3882–3898. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Jha, S.K.; Jha, N.K.; Kar, R.; Ambasta, R.K.; Kumar, P. p38 MAPK and PI3K/AKT signalling cascades in Parkinson’s disease. Int. J. Mol. Cell. Med. 2015, 4, 67–86. [Google Scholar]
- Lee, J.K.; Kim, N.J. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef]
- Zhu, X.; Rottkamp, C.A.; Boux, H.; Takeda, A.; Perry, G.; Smith, M.A. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Meimaridou, E.; Tavassoli, M.; Melino, G.; Lovestone, S.; Killick, R. p53 is upregulated in Alzheimer’s disease and induces tau phosphorylation in HEK293a cells. Neurosci. Lett. 2007, 418, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Barone, E.; Di Domenico, F.; Butterfield, D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta 2016, 1862, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Frasca, G.; Carbonaro, V.; Merlo, S.; Copani, A.; Sortino, M.A. Integrins mediate beta-amyloid-induced cell-cycle activation and neuronal death. J. Neurosci. Res. 2008, 86, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Caltagarone, J.; Jing, Z.; Bowser, R. Focal adhesions regulate Abeta signaling and cell death in Alzheimer’s disease. Biochim. Biophys. Acta 2007, 1772, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.; Nievergall, E.; Gegenbauer, K.; Llerena, C.; Atapattu, L.; Halle, M.; Tremblay, M.L.; Janes, P.W.; Lackmann, M. PTP-PEST controls EphA3 activation and ephrin-induced cytoskeletal remodelling. J. Cell Sci. 2016, 129, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Bernstein, B.W. Actin dynamics and cofilin-actin rods in alzheimer disease. Cytoskeleton 2016, 73, 477–497. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.E.; Woo, J.A. Cofilin, a master node regulating cytoskeletal pathogenesis in Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 72 (Suppl. 1), S131–S144. [Google Scholar] [CrossRef] [PubMed]
- Kwan, V.; Meka, D.P.; White, S.H.; Hung, C.L.; Holzapfel, N.T.; Walker, S.; Murtaza, N.; Unda, B.K.; Schwanke, B.; Yuen, R.K.C.; et al. DIXDC1 phosphorylation and control of dendritic morphology are impaired by rare genetic variants. Cell Rep. 2016, 17, 1892–1904. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alfonso, S.I.; Callender, J.A.; Hooli, B.; Antal, C.E.; Mullin, K.; Sherman, M.A.; Lesne, S.E.; Leitges, M.; Newton, A.C.; Tanzi, R.E.; et al. Gain-of-function mutations in protein kinase Calpha (PKCalpha) may promote synaptic defects in Alzheimer’s disease. Sci. Signal. 2016, 9, ra47. [Google Scholar] [CrossRef] [PubMed]
- Keeler, A.B.; Schreiner, D.; Weiner, J.A. Protein kinase C phosphorylation of a gamma-protocadherin C-terminal lipid binding domain regulates focal adhesion kinase inhibition and dendrite arborization. J. Biol. Chem. 2015, 290, 20674–20686. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, B.F.; Ataie-Kachoie, P.; Pourgholami, M.H.; Morris, D.L. p70 Ribosomal protein S6 kinase (Rps6kb1): An update. J. Clin. Pathol. 2014, 67, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; Bjorkdahl, C.; Zhang, H.; Zhou, X.; Winblad, B. p70 S6 kinase and tau in Alzheimer’s disease. J. Alzheimers Dis. 2008, 14, 385–392. [Google Scholar] [CrossRef] [PubMed]
- An, W.L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Talboom, J.S.; Shaw, D.M.; Turner, D.; Ma, L.; Messina, A.; Huang, Z.; Wu, J.; Oddo, S. Reducing ribosomal protein S6 kinase 1 expression improves spatial memory and synaptic plasticity in a mouse model of Alzheimer’s disease. J. Neurosci. 2015, 35, 14042–14056. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ge, W.; Martinowich, K.; Becker-Catania, S.; Coskun, V.; Zhu, W.; Wu, H.; Castro, D.; Guillemot, F.; Fan, G.; et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 2005, 8, 616–625. [Google Scholar] [CrossRef]
- Li, S.B.; Du, D.; Hasan, M.T.; Kohr, G. D4 receptor activation differentially modulates hippocampal basal and apical dendritic synapses in freely moving mice. Cereb. Cortex 2016, 26, 647–655. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- McCall, M.A.; Gregg, R.G.; Behringer, R.R.; Brenner, M.; Delaney, C.L.; Galbreath, E.J.; Zhang, C.L.; Pearce, R.A.; Chiu, S.Y.; Messing, A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc. Natl. Acad. Sci. USA 1996, 93, 6361–6366. [Google Scholar] [CrossRef]
- Barzilai, A.; Biton, S.; Shiloh, Y. The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair 2008, 7, 1010–1027. [Google Scholar] [CrossRef]
- Coppede, F.; Migliore, L. DNA damage and repair in Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 36–47. [Google Scholar] [CrossRef]
- Shen, X.; Chen, J.; Li, J.; Kofler, J.; Herrup, K. Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro 2016, 3, ENEURO.0124-15.2016. [Google Scholar] [CrossRef]
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; De Felice, F.G. Insulin resistance in Alzheimer’s disease. Front. Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Rosoklija, G.B.; Petrushevski, V.M.; Stankov, A.; Dika, A.; Jakovski, Z.; Pavlovski, G.; Davcheva, N.; Lipkin, R.; Schnieder, T.; Scobie, K.; et al. Reliable and durable Golgi staining of brain tissue from human autopsies and experimental animals. J. Neurosci. Methods 2014, 230, 20–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yue, L.; Sam, C.; Arora, N.; Winkler, D.F.H.; Pelech, S. Antibody microarray and immunoblotting analyses of the EGF signaling phosphorylation network in human A431 epidermoid carcinoma cells. Clin. Proteom. Bioinform. 2017, 2, 1–10. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| FAK Fwd | 5′-CGT GAA GCC TTT TCA AGG AG-3′ |
| FAK Rev | 5′-GCA CCT TCT CCT CCT CCA G-3′ |
| HPRT Fwd | 5′-GCA GTA CAG CCC CAA AAT GG-3′ |
| HPRT Rev | 5′-GGT CCT TTT CAC CAG CAA GCT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, B.; Srikanth, K.D.; Sneh, T.; Yue, L.; Pelech, S.; Elliott, E.; Gil-Henn, H. FAK-Mediated Signaling Controls Amyloid Beta Overload, Learning and Memory Deficits in a Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 9055. https://doi.org/10.3390/ijms23169055
Saleh B, Srikanth KD, Sneh T, Yue L, Pelech S, Elliott E, Gil-Henn H. FAK-Mediated Signaling Controls Amyloid Beta Overload, Learning and Memory Deficits in a Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(16):9055. https://doi.org/10.3390/ijms23169055
Chicago/Turabian StyleSaleh, Bisan, Kolluru D. Srikanth, Tal Sneh, Lambert Yue, Steven Pelech, Evan Elliott, and Hava Gil-Henn. 2022. "FAK-Mediated Signaling Controls Amyloid Beta Overload, Learning and Memory Deficits in a Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 16: 9055. https://doi.org/10.3390/ijms23169055
APA StyleSaleh, B., Srikanth, K. D., Sneh, T., Yue, L., Pelech, S., Elliott, E., & Gil-Henn, H. (2022). FAK-Mediated Signaling Controls Amyloid Beta Overload, Learning and Memory Deficits in a Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 23(16), 9055. https://doi.org/10.3390/ijms23169055