
Neuronal ApoE Regulates the Cell-to-Cell Transmission of α-Synuclein


Abstract
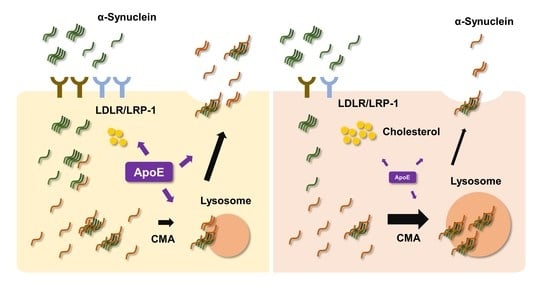
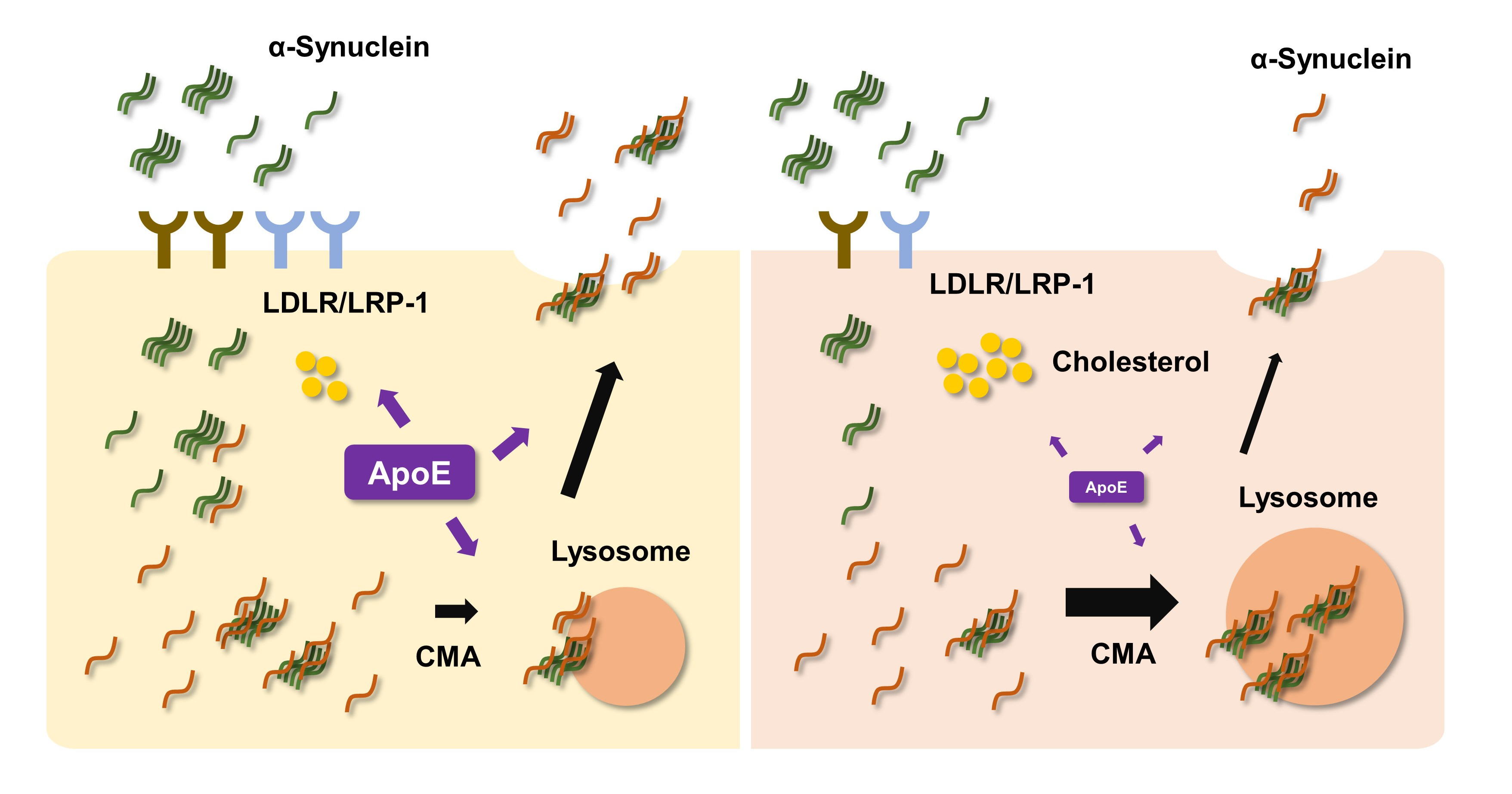{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
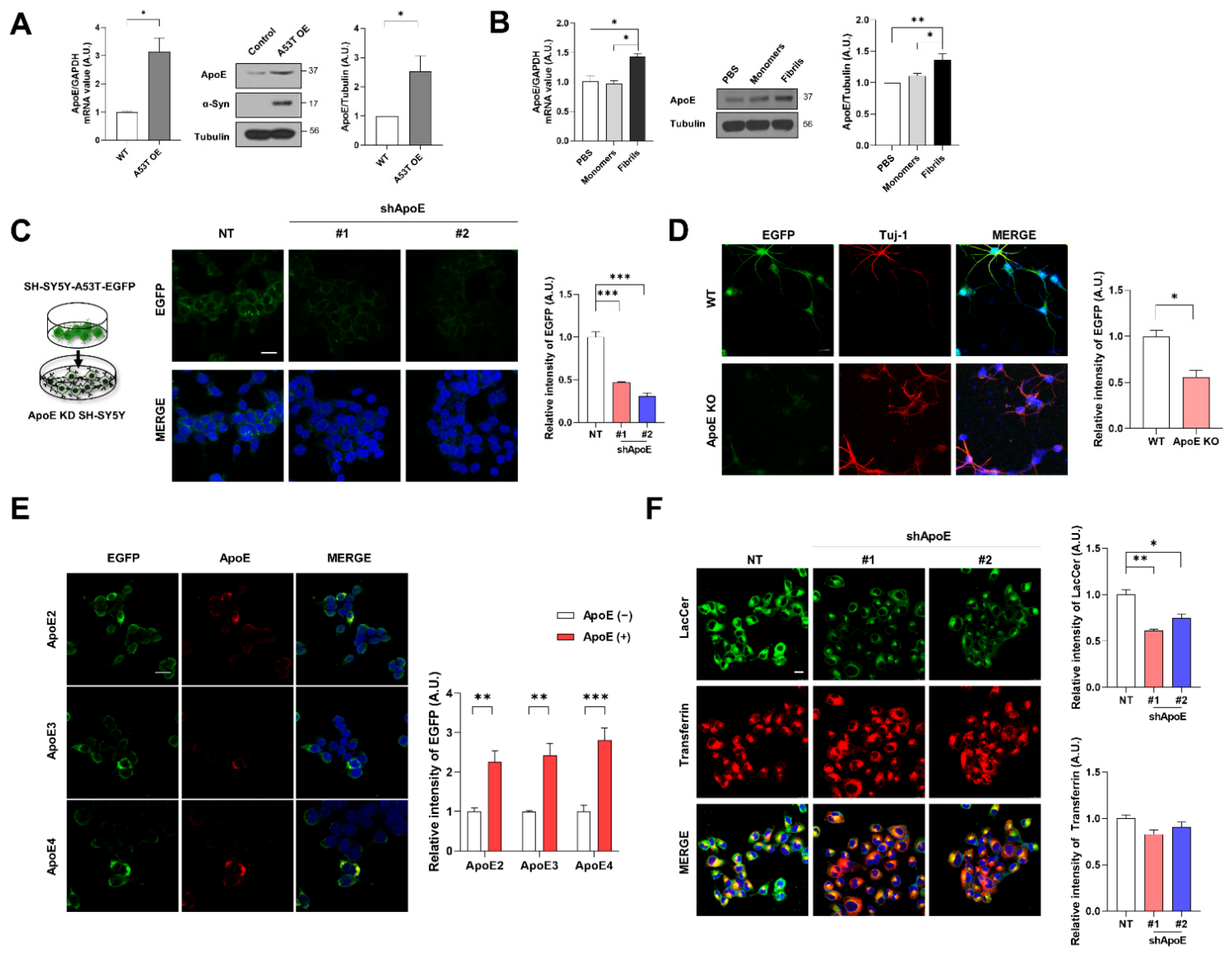
2.1. Neuronal ApoE Regulates the Uptake of α-Syn into Neurons
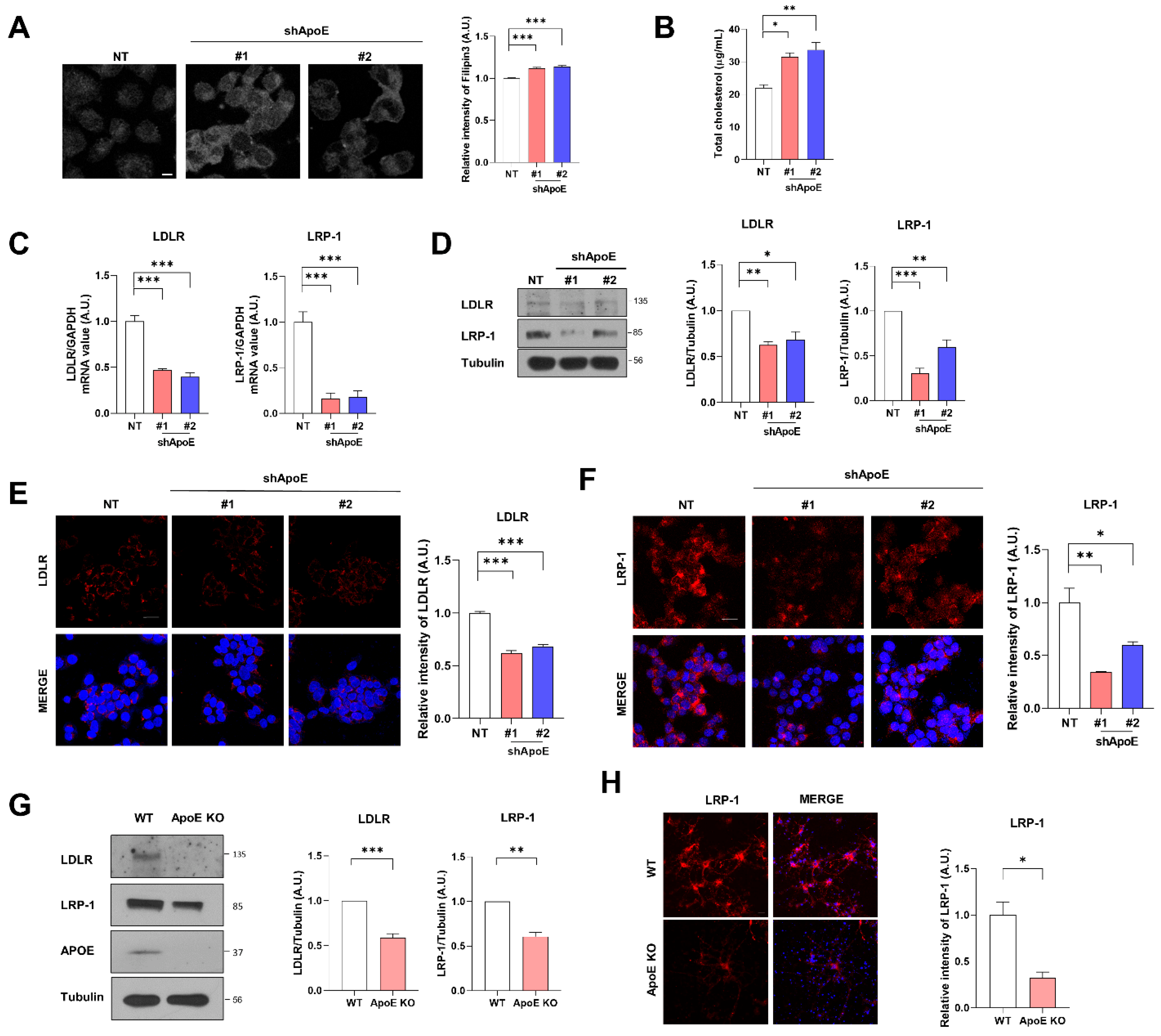
2.2. ApoE Deficiency Increases Intracellular Cholesterol Levels and Decreases Apoe Receptor Expression in Neurons
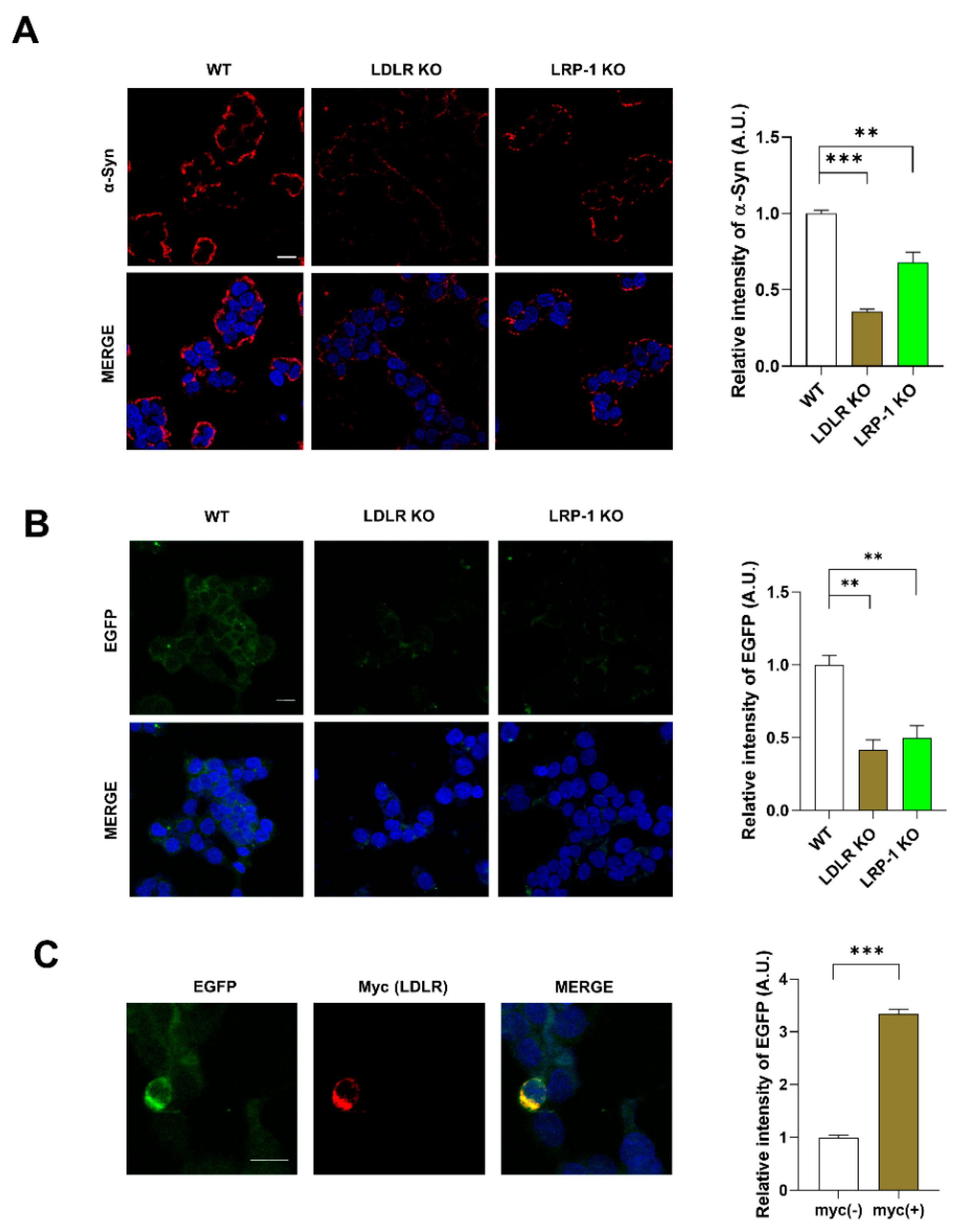
2.3. LDLR and LRP-1 Expressed in SH-SY5Y Cells Function as Receptors Responsible for α-Syn Uptake
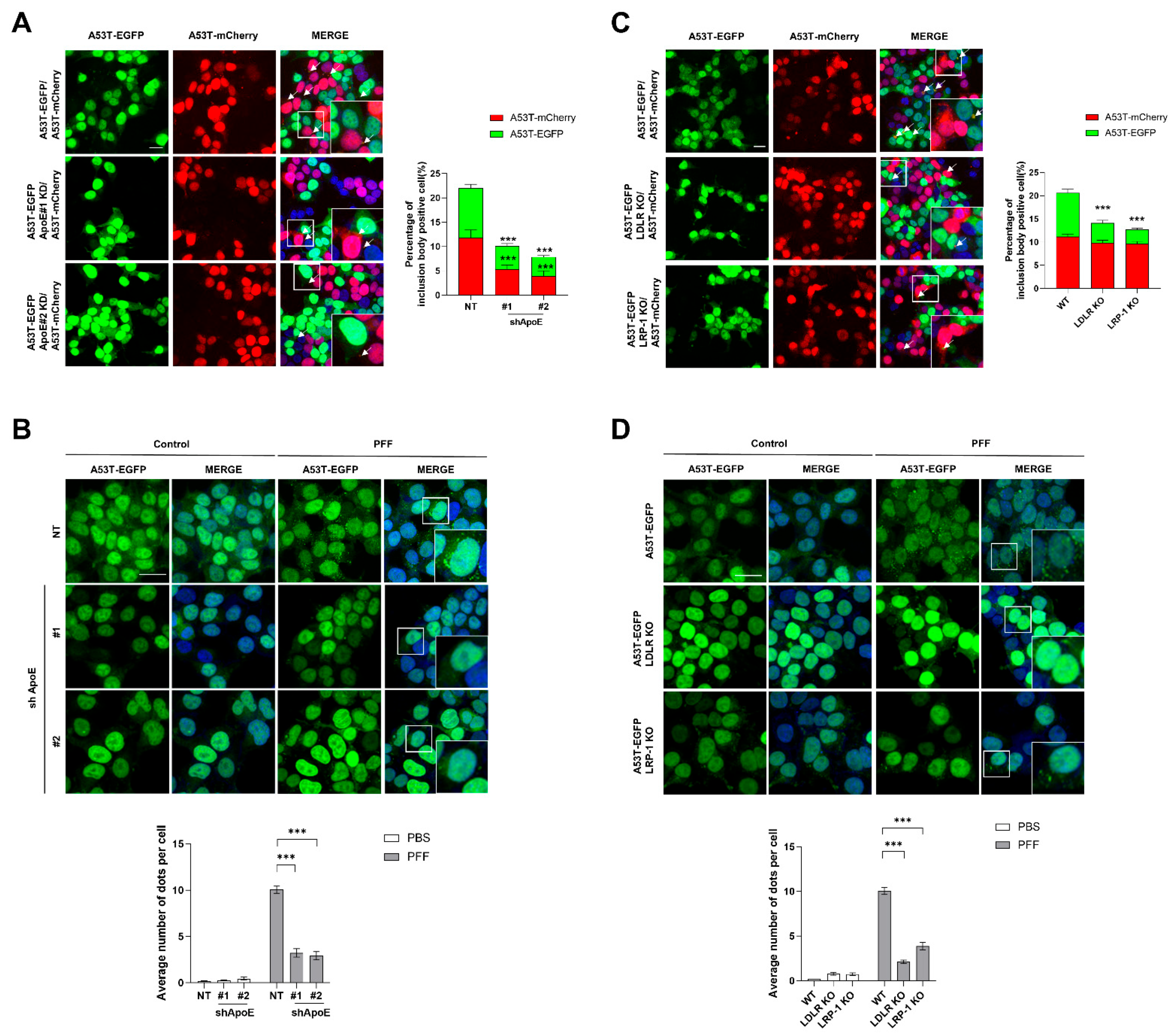
2.4. ApoE Deficiency in SH-SY5Y Cells Inhibits α-Syn Propagation
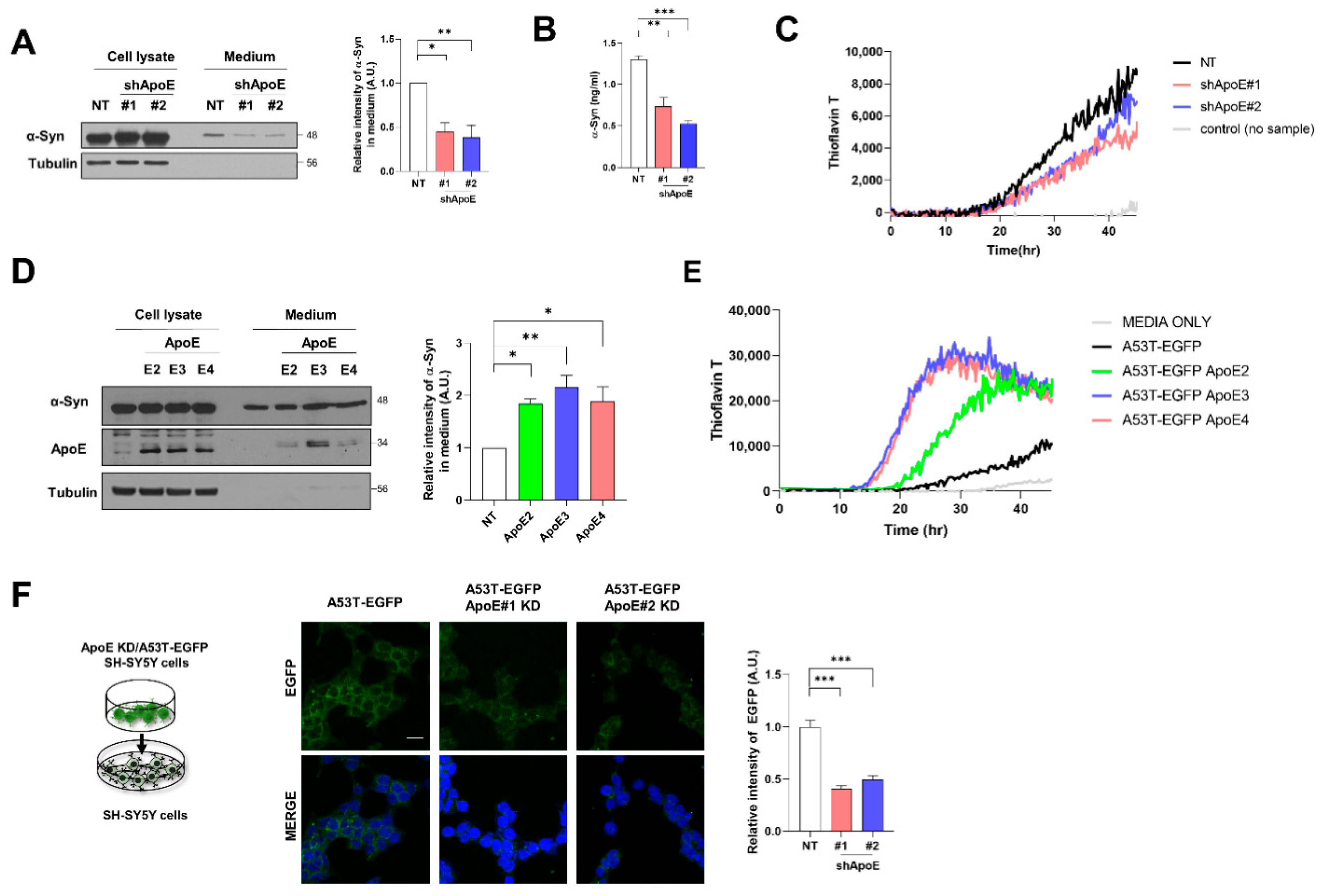
2.5. ApoE Deficiency Inhibits α-Syn Release in SH-SY5Y Cells
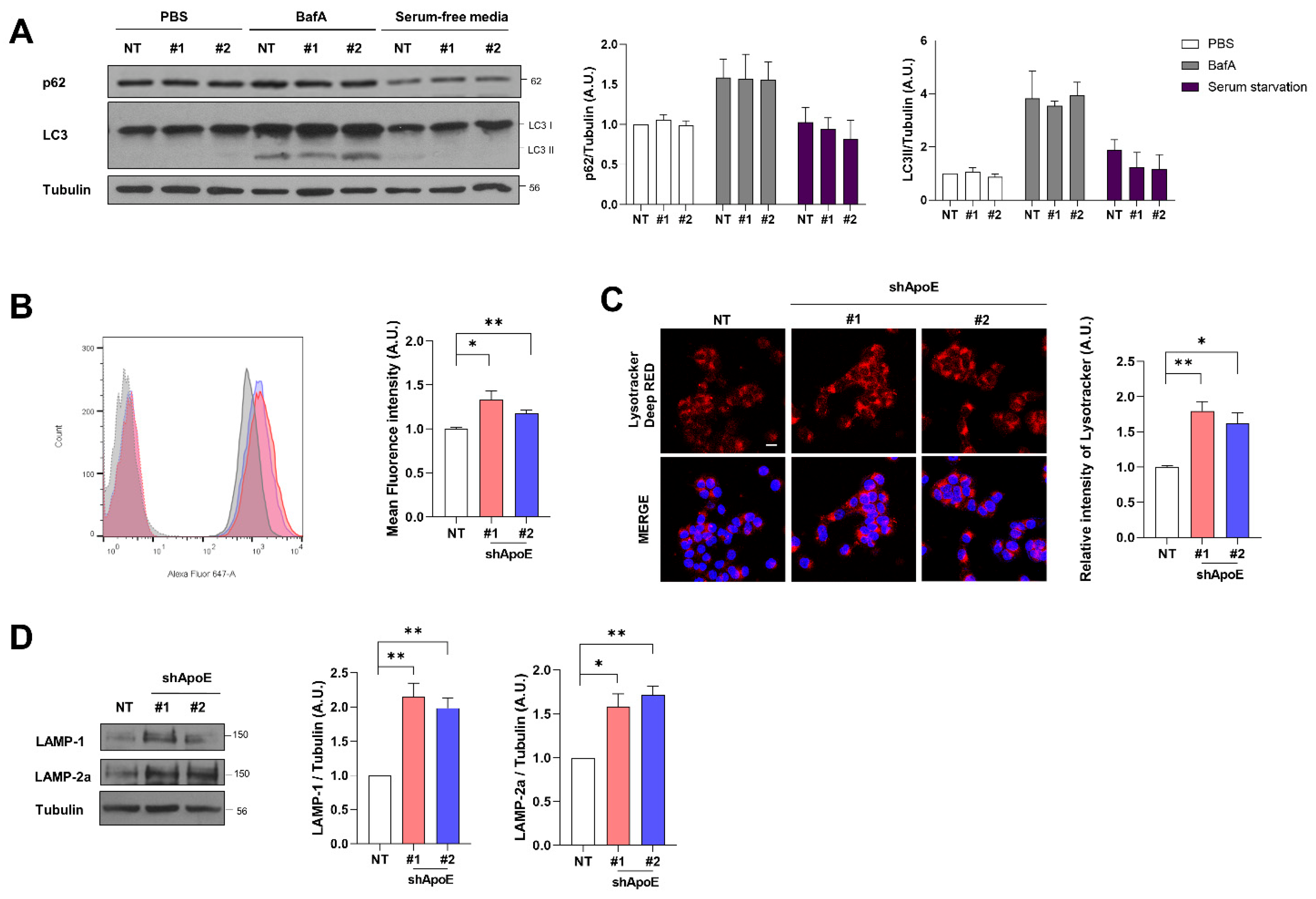
2.6. ApoE Deficiency Enhances Chaperone-Mediated Autophagy in SH-SY5Y Cells
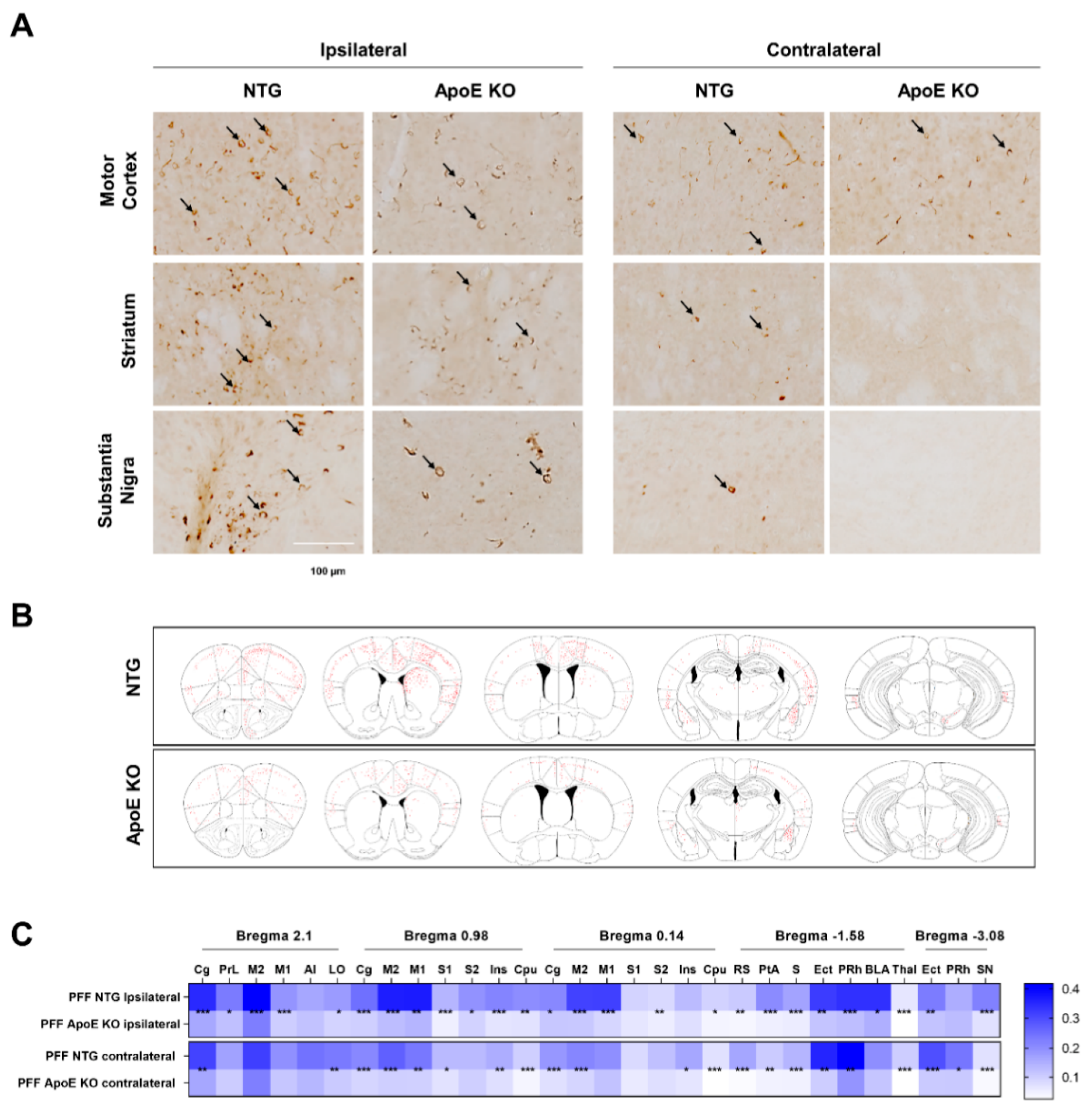
2.7. The Propagation of α-Syn Is Reduced in ApoE KO Mice
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Plasmids and Generation of Stable Cell Lines
4.3. Cell Culture
4.4. Western Blot
4.5. Confocal Microscopy
4.6. Dual Chamber and Coculture Assays
4.7. Cholesterol Measurement
4.8. Preparation of Conditioned Media from SH-SY5Y Cells
4.9. Measurement of α-Syn in Culture Media
4.10. α-Syn Real-Time Quaking-Induced Conversion (RT-QUIC) Assay
4.11. Animals and Intracerebral Injection
4.12. Immunohistochemistry
4.13. Quantitative pSer129 α-Syn Pathology
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCann, H.; Cartwright, H.; Halliday, G.M. Neuropathology of α-synuclein propagation and braak hypothesis. Mov. Disord. 2016, 31, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Eschbach, J.; Danzer, K.M. α-Synuclein in Parkinson’s disease: Pathogenic function and translation into animal models. Neuro-Degener. Dis. 2014, 14, 1–17. [Google Scholar] [CrossRef]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef]
- Vaquer-Alicea, J.; Diamond, M.I. Propagation of Protein Aggregation in Neurodegenerative Diseases. Annu. Rev. Biochem. 2019, 88, 785–810. [Google Scholar] [CrossRef]
- Choi, Y.R.; Park, S.J.; Park, S.M. Molecular events underlying the cell-to-cell transmission of α-synuclein. FEBS J. 2021, 288, 6593–6602. [Google Scholar] [CrossRef]
- Vitali, C.; Wellington, C.L.; Calabresi, L. HDL and cholesterol handling in the brain. Cardiovasc. Res. 2014, 103, 405–413. [Google Scholar] [CrossRef]
- Artiga, M.J.; Bullido, M.J.; Frank, A.; Sastre, I.; Recuero, M.; Garcia, M.A.; Lendon, C.L.; Han, S.W.; Morris, J.C.; Vazquez, J.; et al. Risk for Alzheimer’s disease correlates with transcriptional activity of the APOE gene. Hum. Mol. Genet. 1998, 7, 1887–1892. [Google Scholar] [CrossRef]
- Riddell, D.R.; Zhou, H.; Atchison, K.; Warwick, H.K.; Atkinson, P.J.; Jefferson, J.; Xu, L.; Aschmies, S.; Kirksey, Y.; Hu, Y.; et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci. 2008, 28, 11445–11453. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef]
- Wu, L.; Zhao, L. ApoE2 and Alzheimer’s disease: Time to take a closer look. Neural Regen. Res. 2016, 11, 412–413. [Google Scholar] [CrossRef]
- Gottschalk, W.K.; Mihovilovic, M.; Roses, A.D.; Chiba-Falek, O. The Role of Upregulated APOE in Alzheimer’s Disease Etiology. J. Alzheimers Dis. Parkinsonism 2016, 6, 209. [Google Scholar] [CrossRef]
- Zareparsi, S.; Kaye, J.; Camicioli, R.; Grimslid, H.; Oken, B.; Litt, M.; Nutt, J.; Bird, T.; Schellenberg, G.; Payami, H. Modulation of the age at onset of Parkinson’s disease by apolipoprotein E genotypes. Ann. Neurol. 1997, 42, 655–658. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Kakita, A.; Hayashi, S.; Okuizumi, K.; Onodera, O.; Tanaka, H.; Ishikawa, A.; Tsuji, S.; Takahashi, H. Apolipoprotein E epsilon4 allele and progression of cortical Lewy body pathology in Parkinson’s disease. Acta Neuropathol. 1998, 95, 450–454. [Google Scholar] [CrossRef]
- Huang, X.; Chen, P.C.; Poole, C. APOE-[epsilon]2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology 2004, 62, 2198–2202. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Bol, J.G.; Van Haastert, E.S.; Rozemuller, A.J.; Bu, G.; Drukarch, B.; Hoozemans, J.J. Apolipoprotein E and LRP1 Increase Early in Parkinson’s Disease Pathogenesis. Am. J. Pathol. 2011, 179, 2152–2156. [Google Scholar] [CrossRef]
- Paslawski, W.; Zareba-Paslawska, J.; Zhang, X.; Hölzl, K.; Wadensten, H.; Shariatgorji, M.; Janelidze, S.; Hansson, O.; Forsgren, L.; Andrén, P.E.; et al. α-synuclein− lipoprotein interactions and elevated ApoE level in cerebrospinal fluid from Parkinson’s disease patients. Proc. Natl. Acad. Sci. USA 2019, 116, 15226–15235. [Google Scholar] [CrossRef]
- Gallardo, G.; Schluter, O.M.; Sudhof, T.C. A molecular pathway of neurodegeneration linking α-synuclein to ApoE and Aβ peptides. Nat. Neurosci. 2008, 11, 301–308. [Google Scholar] [CrossRef]
- Davis, A.A.; Inman, C.E.; Wargel, Z.M.; Dube, U.; Freeberg, B.M.; Galluppi, A.; Haines, J.N.; Dhavale, D.D.; Miller, R.; Choudhury, F.A.; et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 2020, 12, eaay3069. [Google Scholar] [CrossRef]
- Zhao, N.; Attrebi, O.N.; Ren, Y.; Qiao, W.; Sonustun, B.; Martens, Y.A.; Meneses, A.D.; Li, F.; Shue, F.; Zheng, J.; et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 2020, 12, eaay1809. [Google Scholar] [CrossRef]
- Jin, Y.; Li, F.; Sonoustoun, B.; Kondru, N.C.; Martens, Y.A.; Qiao, W.; Heckman, M.G.; Ikezu, T.C.; Li, Z.; Burgess, J.D.; et al. APOE4 exacerbates α-synuclein seeding activity and contributes to neurotoxicity in Alzheimer’s disease with Lewy body pathology. Acta Neuropathol. 2022, 143, 641–662. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333. [Google Scholar] [CrossRef]
- Xu, P.T.; Schmechel, D.; Qiu, H.L.; Herbstreith, M.; Rothrock-Christian, T.; Eyster, M.; Roses, A.D.; Gilbert, J.R. Sialylated human apolipoprotein E (apoEs) is preferentially associated with neuron-enriched cultures from APOE transgenic mice. Neurobiol. Dis. 1999, 6, 63–75. [Google Scholar] [CrossRef]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef]
- Aoki, K.; Uchihara, T.; Sanjo, N.; Nakamura, A.; Ikeda, K.; Tsuchiya, K.; Wakayama, Y. Increased expression of neuronal apolipoprotein E in human brain with cerebral infarction. Stroke 2003, 34, 875–880. [Google Scholar] [CrossRef]
- Boschert, U.; Merlo-Pich, E.; Higgins, G.; Roses, A.D.; Catsicas, S. Apolipoprotein E expression by neurons surviving excitotoxic stress. Neurobiol. Dis. 1999, 6, 508–514. [Google Scholar] [CrossRef]
- Andrews-Zwilling, Y.; Bien-Ly, N.; Xu, Q.; Li, G.; Bernardo, A.; Yoon, S.Y.; Zwilling, D.; Yan, T.X.; Chen, L.; Huang, Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 2010, 30, 13707–13717. [Google Scholar] [CrossRef]
- Knoferle, J.; Yoon, S.Y.; Walker, D.; Leung, L.; Gillespie, A.K.; Tong, L.M.; Bien-Ly, N.; Huang, Y. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci. 2014, 34, 14069–14078. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.R.; Cha, S.H.; Kang, S.J.; Kim, J.B.; Jou, I.; Park, S.M. Prion-like Propagation of α-Synuclein Is Regulated by the FcgammaRIIB-SHP-1/2 Signaling Pathway in Neurons. Cell Rep. 2018, 22, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.R.; Kim, J.B.; Kang, S.J.; Noh, H.R.; Jou, I.; Joe, E.H.; Park, S.M. The dual role of c-src in cell-to-cell transmission of α-synuclein. EMBO Rep. 2020, 21, e48950. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, J.S.; Park, J.Y.; Suh, Y.H.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum. Mol. Genet. 2013, 22, 4805–4817. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Apoprotein E and Reverse Cholesterol Transport. Int. J. Mol. Sci. 2018, 19, 3497. [Google Scholar] [CrossRef]
- Jin, U.; Park, S.J.; Park, S.M. Cholesterol Metabolism in the Brain and Its Association with Parkinson’s Disease. Exp. Neurobiol. 2019, 28, 554–567. [Google Scholar] [CrossRef]
- Li, R.; Hao, J.; Fujiwara, H.; Xu, M.; Yang, S.; Dai, S.; Long, Y.; Swaroop, M.; Li, C.; Vu, M.; et al. Analytical Characterization of Methyl-beta-Cyclodextrin for Pharmacological Activity to Reduce Lysosomal Cholesterol Accumulation in Niemann-Pick Disease Type C1 Cells. Assay Drug Dev. Technol. 2017, 15, 154–166. [Google Scholar] [CrossRef]
- Castanho, M.A.; Coutinho, A.; Prieto, M.J. Absorption and fluorescence spectra of polyene antibiotics in the presence of cholesterol. J. Biol. Chem. 1992, 267, 204–209. [Google Scholar] [CrossRef]
- Zhang, L.; Reue, K.; Fong, L.G.; Young, S.G.; Tontonoz, P. Feedback regulation of cholesterol uptake by the LXR-IDOL-LDLR axis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2541–2546. [Google Scholar] [CrossRef]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Woerman, A.L.; Stohr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Vascellari, S.; Orru, C.D.; Caughey, B. Real-Time Quaking- Induced Conversion Assays for Prion Diseases, Synucleinopathies, and Tauopathies. Front. Aging Neurosci. 2022, 14, 853050. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is α-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Dekroon, R.M.; Armati, P.J. Synthesis and processing of apolipoprotein E in human brain cultures. Glia 2001, 33, 298–305. [Google Scholar] [CrossRef]
- Tesseur, I.; Van Dorpe, J.; Spittaels, K.; Van den Haute, C.; Moechars, D.; Van Leuven, F. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am. J. Pathol. 2000, 156, 951–964. [Google Scholar] [CrossRef]
- Harris, F.M.; Tesseur, I.; Brecht, W.J.; Xu, Q.; Mullendorff, K.; Chang, S.; Wyss-Coray, T.; Mahley, R.W.; Huang, Y. Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer’s disease. J. Biol. Chem. 2004, 279, 3862–3868. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, Z. ApoE and Neurodegenerative Diseases in Aging. Adv. Exp. Med. Biol. 2018, 1086, 77–92. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Konings, S.C.; Torres-Garcia, L.; Martinsson, I.; Gouras, G.K. Astrocytic and Neuronal Apolipoprotein E Isoforms Differentially Affect Neuronal Excitability. Front. Neurosci. 2021, 15, 734001. [Google Scholar] [CrossRef]
- Zalocusky, K.A.; Najm, R.; Taubes, A.L.; Hao, Y.; Yoon, S.Y.; Koutsodendris, N.; Nelson, M.R.; Rao, A.; Bennett, D.A.; Bant, J.; et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. α-Synuclein (αSyn) Preformed Fibrils Induce Endogenous αSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef] [PubMed]
- Dupont-Wallois, L.; Soulié, C.; Sergeant, N.; Wavrant-de Wrieze, N.; Chartier-Harlin, M.C.; Delacourte, A.; Caillet-Boudin, M.L. ApoE synthesis in human neuroblastoma cells. Neurobiol. Dis. 1997, 4, 356–364. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leduc, V.; Jasmin-Belanger, S.; Poirier, J. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med. 2010, 16, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Sehayek, E.; Shefer, S.; Nguyen, L.B.; Ono, J.G.; Merkel, M.; Breslow, J.L. Apolipoprotein E regulates dietary cholesterol absorption and biliary cholesterol excretion: Studies in C57BL/6 apolipoprotein E knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 3433–3437. [Google Scholar] [CrossRef]
- Bonacina, F.; Coe, D.; Wang, G.; Longhi, M.P.; Baragetti, A.; Moregola, A.; Garlaschelli, K.; Uboldi, P.; Pellegatta, F.; Grigore, L.; et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat. Commun. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, W.; Ren, Y.; Fu, Y.; Martens, Y.A.; Shue, F.; Davis, M.D.; Wang, X.; Chen, K.; Li, F.; et al. Apolipoprotein E regulates lipid metabolism and α-synuclein pathology in human iPSC-derived cerebral organoids. Acta Neuropathol. 2021, 142, 807–825. [Google Scholar] [CrossRef]
- Simons, K.; Ehehalt, R. Cholesterol, lipid rafts, and disease. J. Clin. Investig. 2002, 110, 597–603. [Google Scholar] [CrossRef]
- Gonias, S.L.; Campana, W.M. LDL receptor-related protein-1: A regulator of inflammation in atherosclerosis, cancer, and injury to the nervous system. Am. J. Pathol. 2014, 184, 18–27. [Google Scholar] [CrossRef]
- Liu, W.; Mani, S.; Davis, N.R.; Sarrafzadegan, N.; Kavathas, P.B.; Mani, A. Mutation in EGFP domain of LDL receptor-related protein 6 impairs cellular LDL clearance. Circ. Res. 2008, 103, 1280–1288. [Google Scholar] [CrossRef]
- Tuck, B.J.; Miller, L.V.C.; Katsinelos, T.; Smith, A.E.; Wilson, E.L.; Keeling, S.; Cheng, S.; Vaysburd, M.J.; Knox, C.; Tredgett, L.; et al. Cholesterol determines the cytosolic entry and seeded aggregation of tau. Cell Rep. 2022, 39, 110776. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, K.L.; Ruan, X.Z.; Liu, B.C. Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int. J. Biol. Sci. 2016, 12, 569–579. [Google Scholar] [CrossRef]
- Rebeck, G.W.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef]
- Moestrup, S.K.; Gliemann, J.; Pallesen, G. Distribution of the α 2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 1992, 269, 375–382. [Google Scholar] [CrossRef]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef]
- Kim, J.; Castellano, J.M.; Jiang, H.; Basak, J.M.; Parsadanian, M.; Pham, V.; Mason, S.M.; Paul, S.M.; Holtzman, D.M. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A β clearance. Neuron 2009, 64, 632–644. [Google Scholar] [CrossRef]
- Sui, Y.T.; Bullock, K.M.; Erickson, M.A.; Zhang, J.; Banks, W.A. A synuclein is transported into and out of the brain by the blood-brain barrier. Peptides 2014, 62, 197–202. [Google Scholar] [CrossRef]
- Kruger, R.; Vieira-Saecker, A.M.; Kuhn, W.; Berg, D.; Muller, T.; Kuhnl, N.; Fuchs, G.A.; Storch, A.; Hungs, M.; Woitalla, D.; et al. Increased susceptibility to sporadic Parkinson’s disease by a certain combined α-synuclein/apolipoprotein E genotype. Ann. Neurol. 1999, 45, 611–617. [Google Scholar] [CrossRef]
- Fantini, J.; Carlus, D.; Yahi, N. The fusogenic tilted peptide (67-78) of α-synuclein is a cholesterol binding domain. Biochim. Et Biophys. Acta 2011, 1808, 2343–2351. [Google Scholar] [CrossRef]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of α-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Allsop, D. α-Synuclein Interacts with Lipoproteins in Plasma. J. Mol. Neurosci. 2017, 63, 165–172. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Fagan, A.M.; Mackey, B.; Tenkova, T.; Sartorius, L.; Paul, S.M.; Bales, K.; Ashe, K.H.; Irizarry, M.C.; Hyman, B.T. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann. Neurol. 2000, 47, 739–747. [Google Scholar] [CrossRef]
- Choi, Y.R.; Kang, S.J.; Kim, J.M.; Lee, S.J.; Jou, I.; Joe, E.H.; Park, S.M. FcγRIIB mediates the inhibitory effect of aggregated α-synuclein on microglial phagocytosis. Neurobiol. Dis. 2015, 83, 90–99. [Google Scholar] [CrossRef]
- Hudry, E.; Dashkoff, J.; Roe, A.D.; Takeda, S.; Koffie, R.M.; Hashimoto, T.; Scheel, M.; Spires-Jones, T.; Arbel-Ornath, M.; Betensky, R.; et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 2013, 5, 212ra161. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783. [Google Scholar] [CrossRef]
- Henderson, M.X.; Cornblath, E.J.; Darwich, A.; Zhang, B.; Brown, H.; Gathagan, R.J.; Sandler, R.M.; Bassett, D.S.; Trojanowski, J.Q.; Lee, V.M.Y. Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 2019, 22, 1248–1257. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, 5th ed.; Academic Press: Cambridge, MA, USA, 2019; 375p. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.-J.; Kim, S.-J.; Noh, H.R.; Kim, B.J.; Kim, J.-B.; Jin, U.; Park, S.A.; Park, S.M. Neuronal ApoE Regulates the Cell-to-Cell Transmission of α-Synuclein. Int. J. Mol. Sci. 2022, 23, 8311. https://doi.org/10.3390/ijms23158311
Kang S-J, Kim S-J, Noh HR, Kim BJ, Kim J-B, Jin U, Park SA, Park SM. Neuronal ApoE Regulates the Cell-to-Cell Transmission of α-Synuclein. International Journal of Molecular Sciences. 2022; 23(15):8311. https://doi.org/10.3390/ijms23158311
Chicago/Turabian StyleKang, Seo-Jun, Soo-Jeong Kim, Hye Rin Noh, Beom Jin Kim, Jae-Bong Kim, Uram Jin, Sun Ah Park, and Sang Myun Park. 2022. "Neuronal ApoE Regulates the Cell-to-Cell Transmission of α-Synuclein" International Journal of Molecular Sciences 23, no. 15: 8311. https://doi.org/10.3390/ijms23158311
APA StyleKang, S.-J., Kim, S.-J., Noh, H. R., Kim, B. J., Kim, J.-B., Jin, U., Park, S. A., & Park, S. M. (2022). Neuronal ApoE Regulates the Cell-to-Cell Transmission of α-Synuclein. International Journal of Molecular Sciences, 23(15), 8311. https://doi.org/10.3390/ijms23158311